
Crosstalk Between nNOS/NO and COX-2 Enhances Interferon-Gamma-Stimulated Melanoma Progression

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Reagents, and Antibodies
2.2. Reverse Phase Protein Array and the Cancer Proteome Atlas Analysis
2.3. Isolation of Protein and Western Blotting Analysis
2.4. Detection of PD-L1 Expression Levels Using Flow Cytometry and Confocal Microscopy
2.5. Detection of Intracellular Nitric Oxide Levels by Flow Cytometry
2.6. Detection of PGE2 Level by LC-MS/MS
2.7. MTT Colorimetric Assay
2.8. Detection of PGE2 Levels in Tumors Using a Syngeneic Murine Melanoma Mouse Model
2.9. Evaluation of In Vivo Antitumor Activities Using a Human Melanoma Xenograft Mouse Model
2.10. Statistical Analyses
3. Results
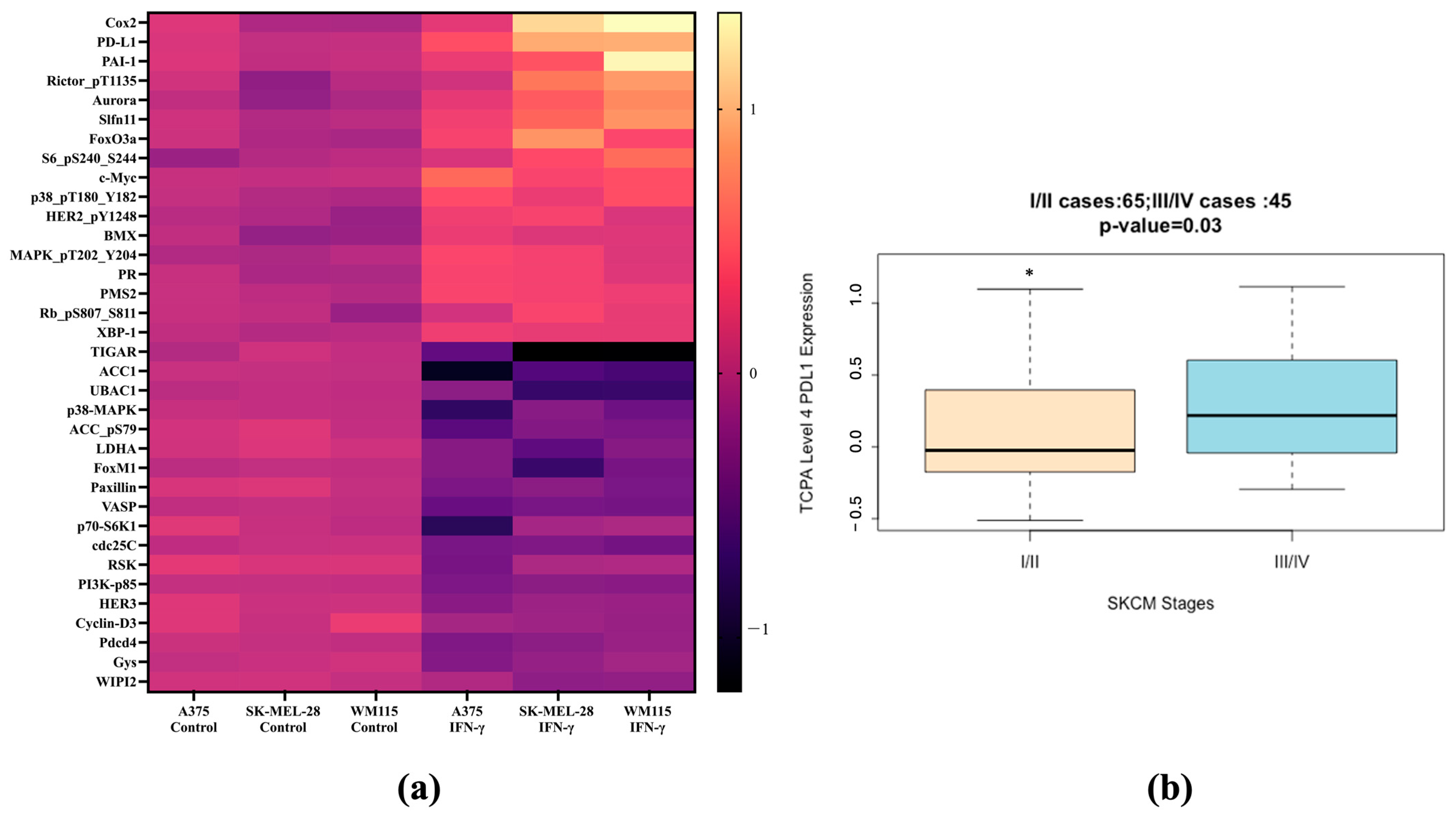
3.1. PD-L1 and COX-2 Identified as Differentially Expressed Proteins Predictive of IFN-γ Treatment in Melanoma
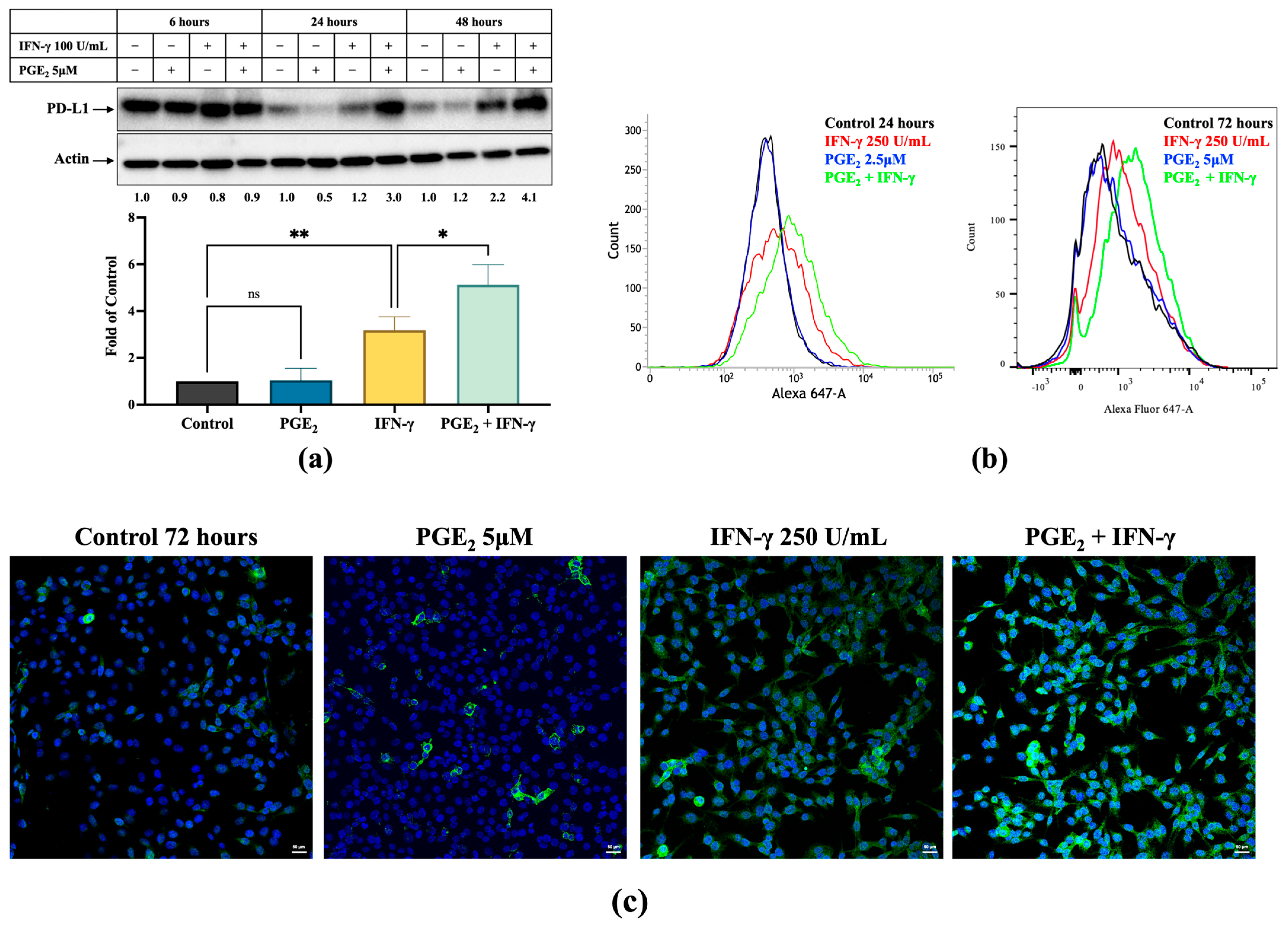
3.2. Induction of PD-L1 Expression by IFN-γ Was Increased by PGE2 and Inhibited by Celecoxib
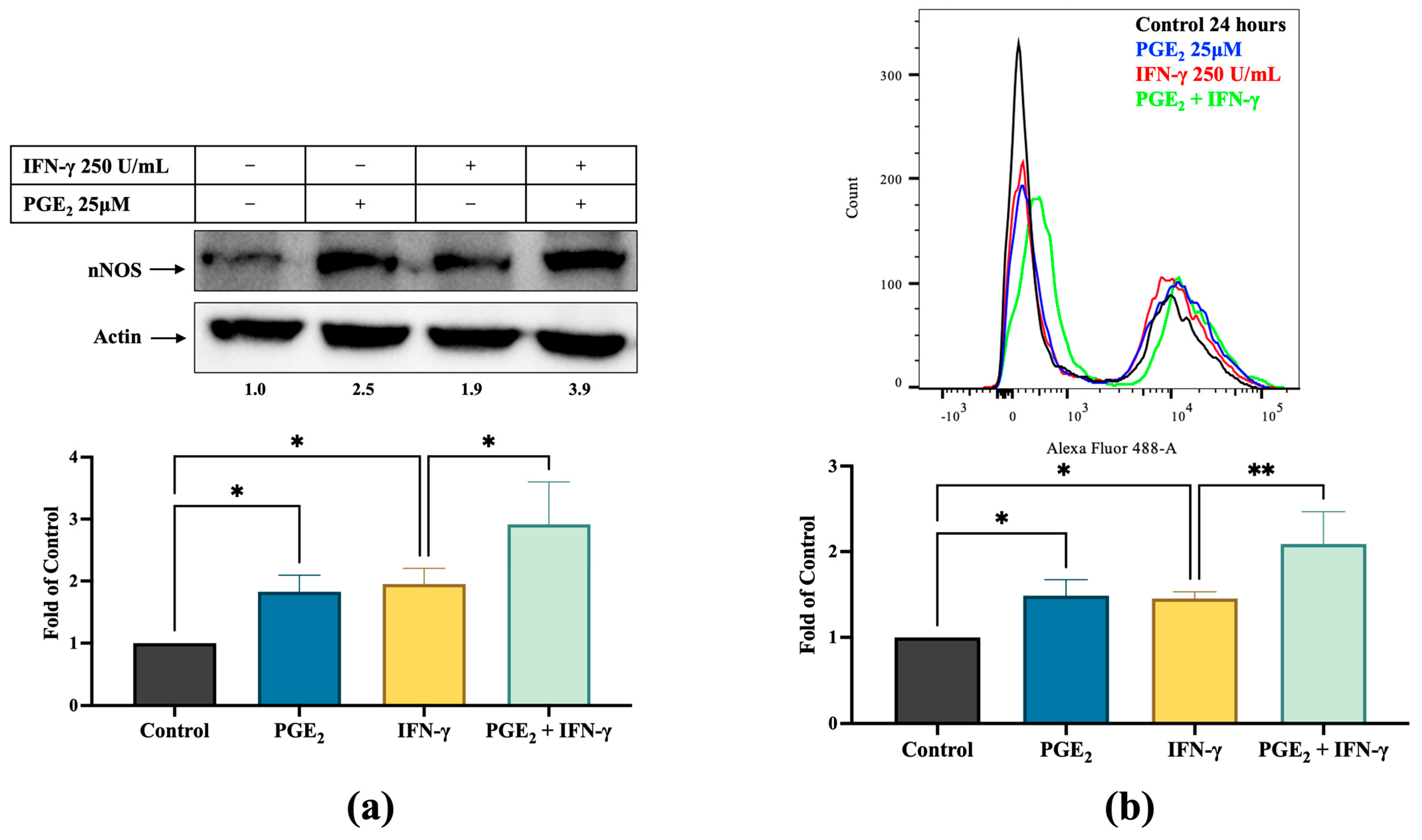
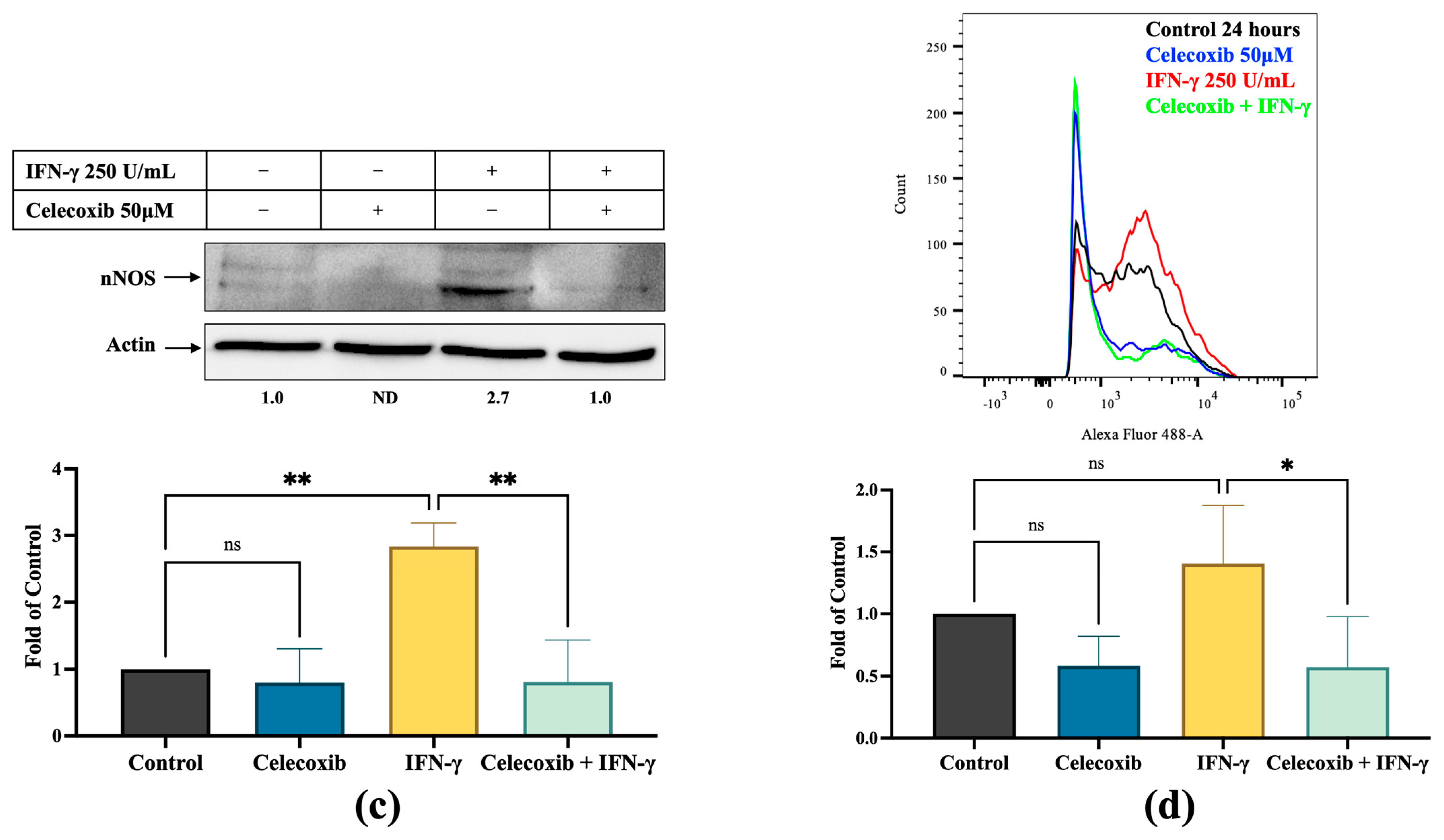
3.3. PGE2 Induces nNOS Expression in Melanoma Cells, Which Is Inhibited by Celecoxib
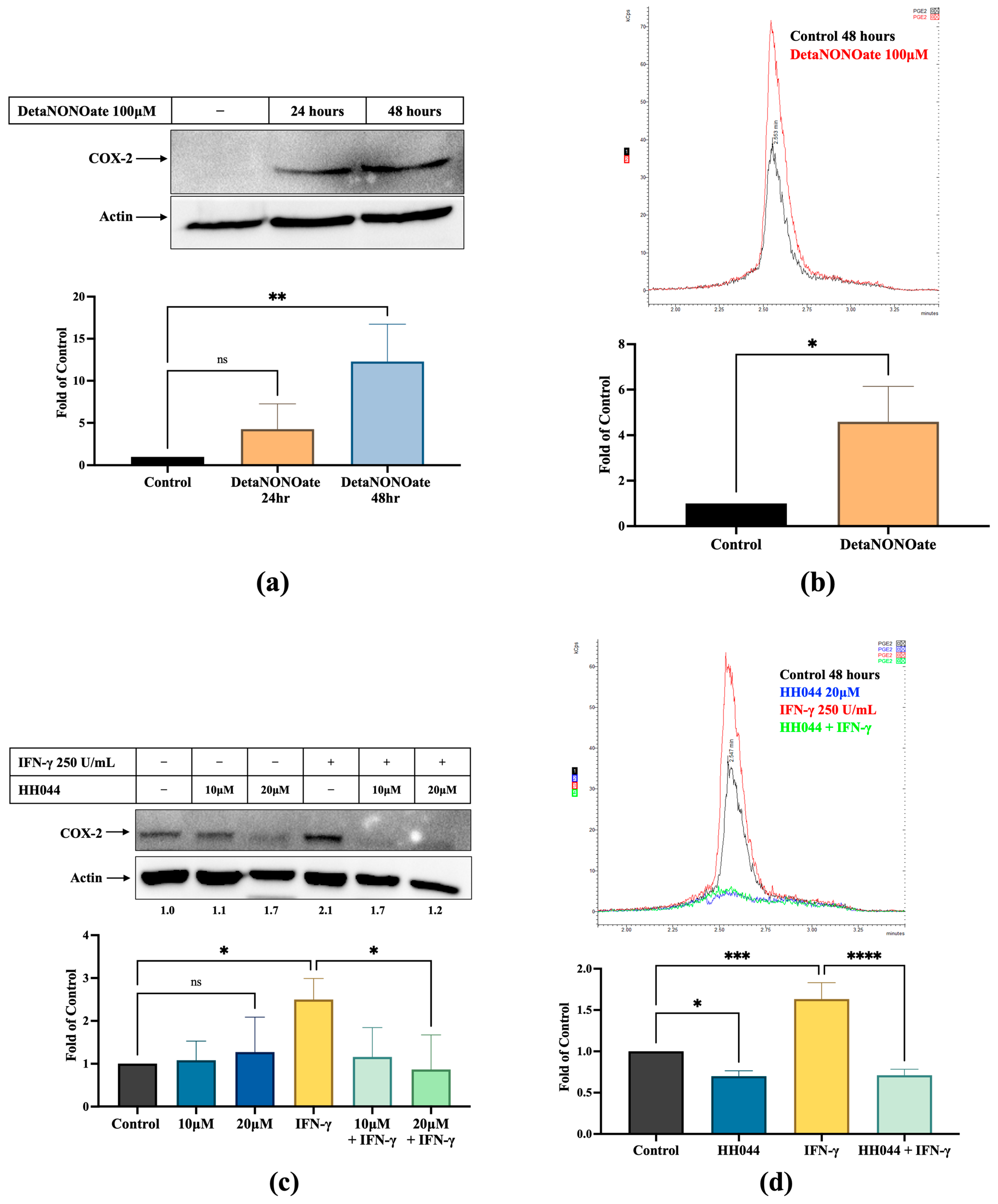
3.4. NO Increases COX-2 Expression and PGE2 Levels in Melanoma Cells, Which Is Inhibited by nNOS Inhibitor HH044
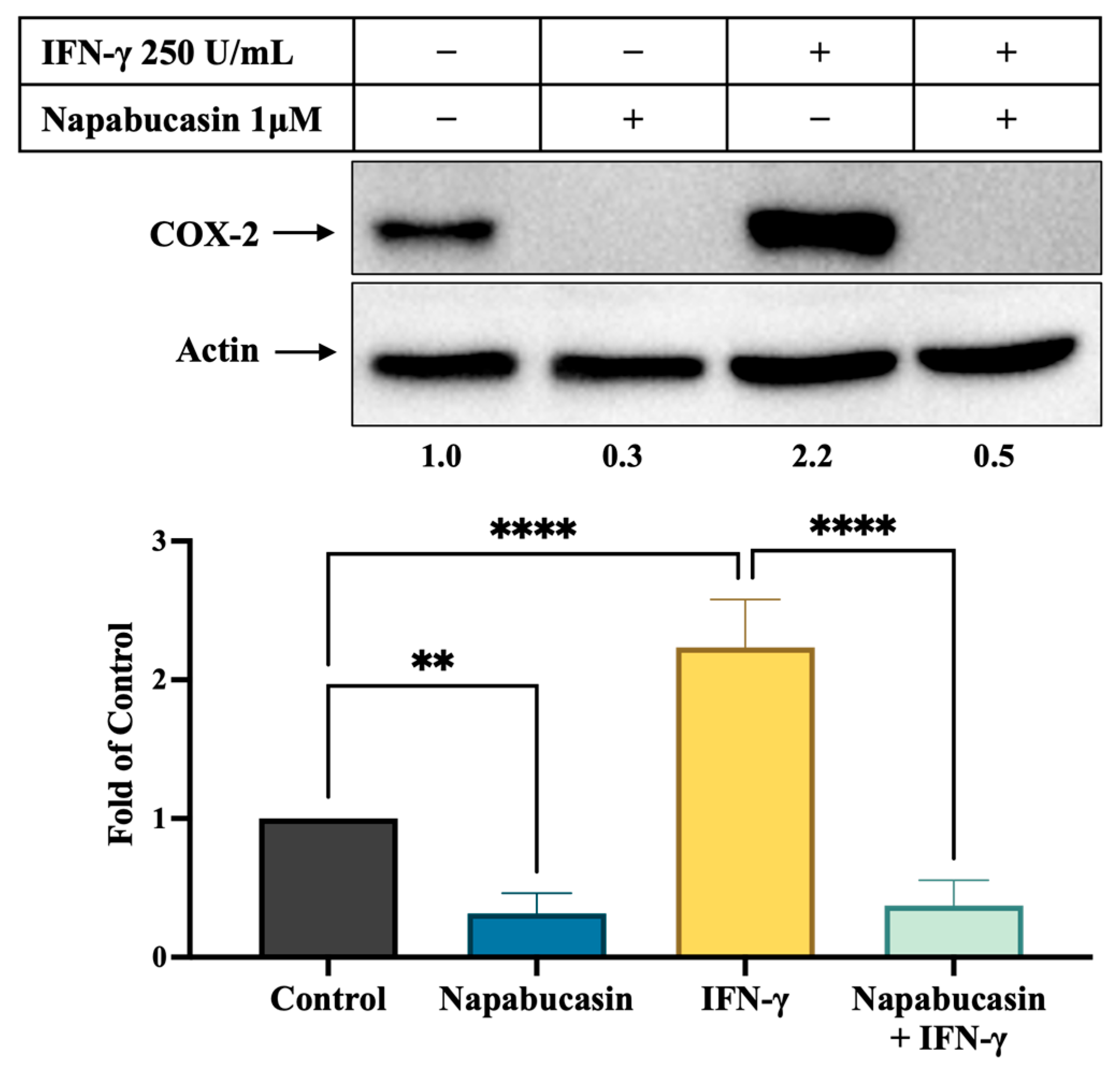
3.5. STAT3 Inhibitor Napabucasin Decreases IFN-γ-Stimulated COX-2 Expression
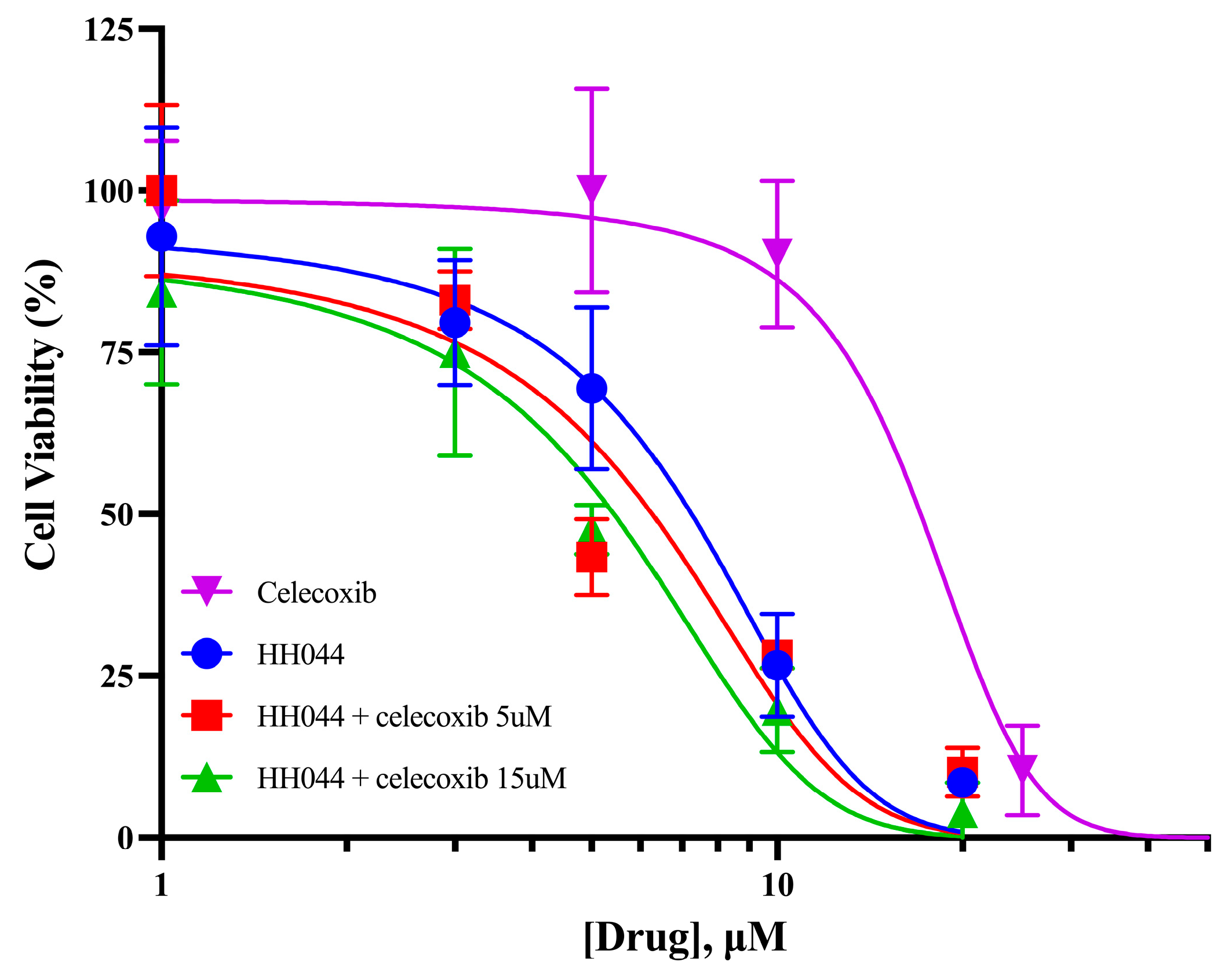
3.6. Celecoxib Increases the Cytotoxicity of nNOS Inhibition in Human Melanoma Cells
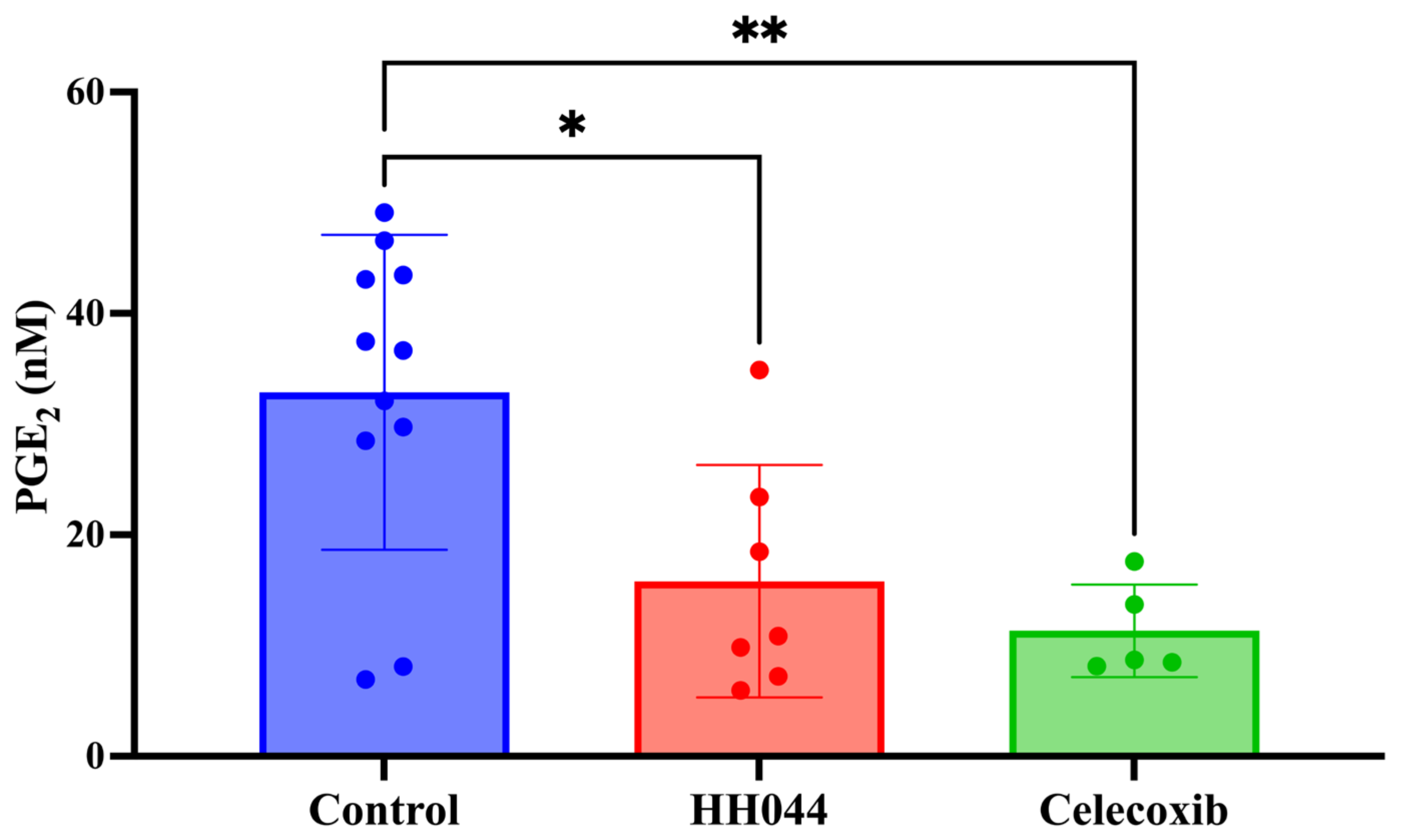
3.7. In Vivo Tumor PGE2 Levels Using an Immunocompetent Syngeneic Melanoma Mouse Model
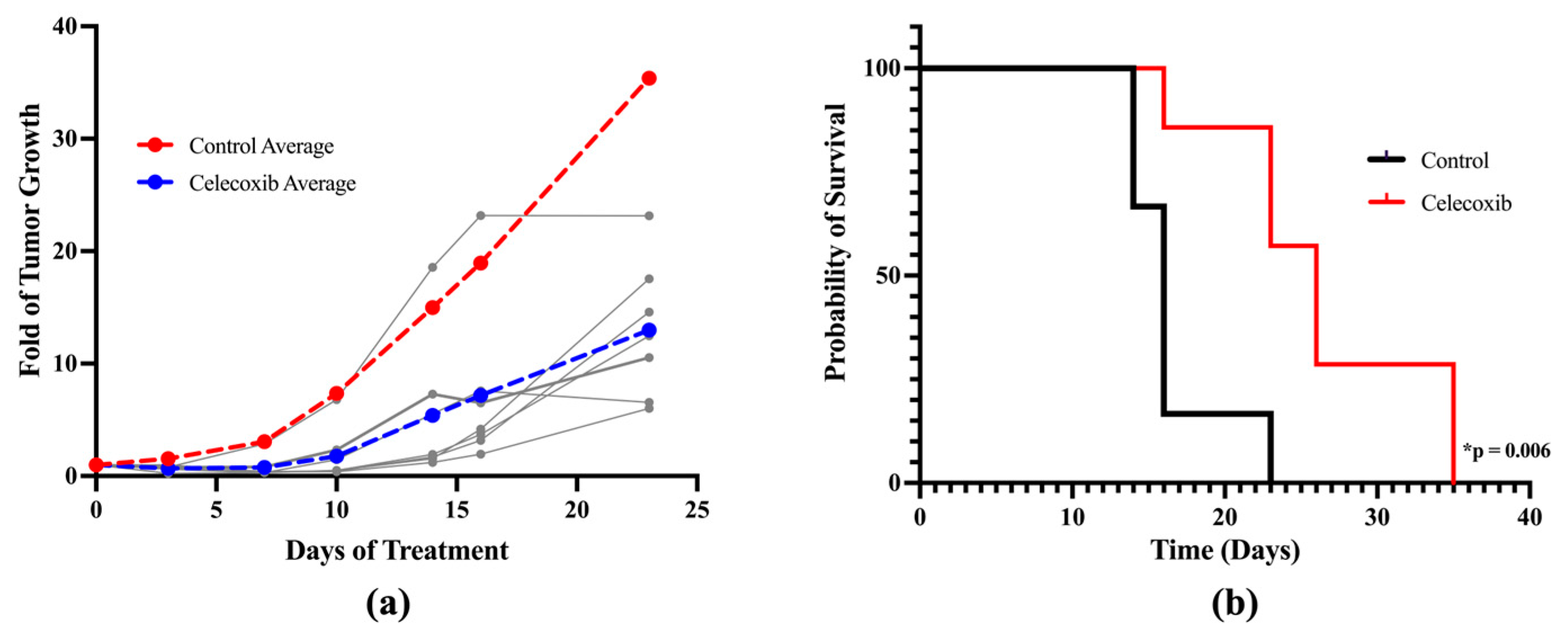
3.8. In Vivo Anti-Melanoma Activity of Celecoxib Using a Xenograft Mouse Model
4. Discussion
4.1. COX-2, PGE2, and Melanoma
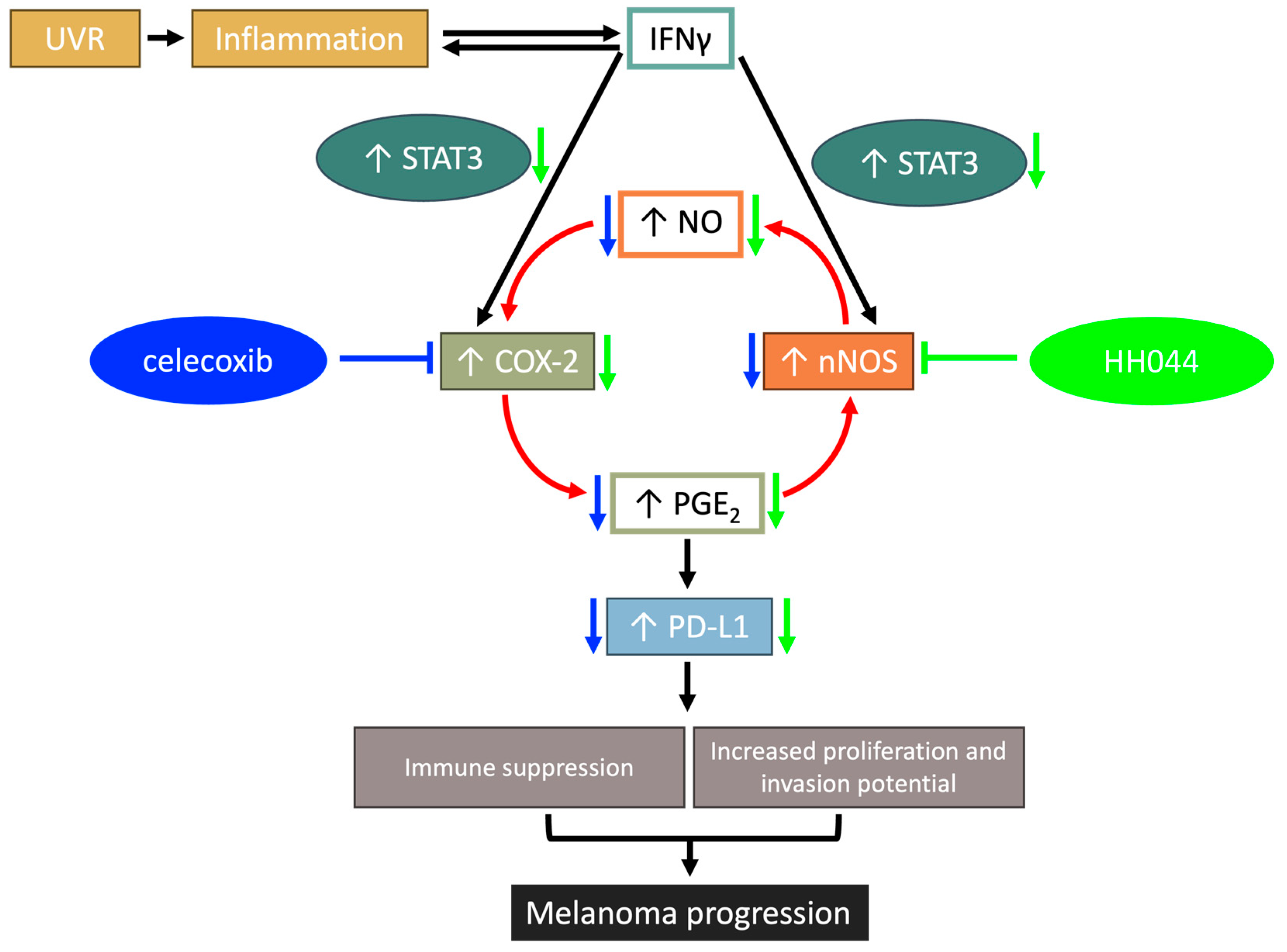
4.2. Crosstalk Between nNOS and COX-2 Plays an Important Role in IFN-γ-Stimulated Melanoma Progression
4.3. The Role of STAT3 Induced by IFN-γ in the Tumor Microenvironment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Stat Facts: Melanoma of the Skin. 2022. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 27 January 2025).
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Nagler, A.; Wargo, J.A.; Samuels, Y. Mechanisms of immune activation and regulation: Lessons from melanoma. Nat. Rev. Cancer 2022, 22, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wu, L.; Han, L.; Zheng, X.; Tong, R.; Li, L.; Bai, L.; Bian, Y. Immune-related adverse events of immune checkpoint inhibitors: A review. Front. Immunol. 2023, 14, 1167975. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Abu-Sbeih, H.; Ascierto, P.A.; Brufsky, J.; Cappelli, L.C.; Cortazar, F.B.; Gerber, D.E.; Hamad, L.; Hansen, E.; Johnson, D.B.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 2021, 9, e002435. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to Molecularly Targeted Therapies in Melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2021, 39, 122. [Google Scholar] [CrossRef]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2018, 19, 89. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Kopecky, K.; Samson, M.; Hersh, E.; Macdonald, J.; Jaffe, H.; Crowley, J.; Coltman, C. Recombinant human interferon gamma: Adverse effects in high-risk stage I and II cutaneous malignant melanoma. J. Natl. Cancer Inst. 1990, 82, 1071. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2019, 29, 3766. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef]
- Ueda, Y.; Richmond, A. NF-kappaB activation in melanoma. Pigment. Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef]
- Yang, Z.; Misner, B.; Ji, H.; Poulos, T.L.; Silverman, R.B.; Meyskens, F.L.; Yang, S. Targeting nitric oxide signaling with nNOS inhibitors as a novel strategy for the therapy and prevention of human melanoma. Antioxid. Redox Signal. 2013, 19, 433–447. [Google Scholar] [CrossRef]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef]
- Tong, S.; Cinelli, M.A.; El-Sayed, N.S.; Huang, H.; Patel, A.; Silverman, R.B.; Yang, S. Inhibition of interferon-gamma-stimulated melanoma progression by targeting neuronal nitric oxide synthase (nNOS). Sci. Rep. 2022, 12, 1701. [Google Scholar] [CrossRef]
- Vasu, D.; Reidl, C.T.; Wang, E.; Yang, S.; Silverman, R.B. Improved synthesis and anticancer activity of a potent neuronal nitric oxide synthase inhibitor. Bioorg Med. Chem. Lett. 2023, 90, 129329. [Google Scholar] [CrossRef]
- Davila-Gonzalez, D.; Chang, J.C.; Billiar, T.R. NO and COX2: Dual targeting for aggressive cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13591–13593. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Salvemini, D. Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney Int. 2007, 71, 290–297. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Storey, J.D.; Zhang, Y.; Torres, L.C. sva: Surrogate Variable Analysis. R Package Version 3.34.0. 2019. Available online: https://code.bioconductor.org/browse/sva/RELEASE_3_20/ (accessed on 2 May 2020).
- Shen, Y.; Rahman, M.; Piccolo, S.R.; Gusenleitner, D.; El-Chaar, N.N.; Cheng, L.; Monti, S.; Bild, A.H.; Johnson, W.E. ASSIGN: Context-specific genomic profiling of multiple heterogeneous biological pathways. Bioinformatics 2015, 31, 1745–1753. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xiao, L.; Park, G.; Wang, X.; Azim, A.C.; Christman, J.W.; van Breemen, R.B. An improved LC-MS/MS method for the quantification of prostaglandins E2 and D2 production in biological fluids. Anal. Biochem. 2008, 372, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Irani, K.; Heffron, S.E.; Jurnak, F.; Meyskens, F.L., Jr. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol. Cancer Ther. 2005, 4, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Nemeth, J.; Furstenberger, G.; Muller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Becker, M.R.; Siegelin, M.D.; Rompel, R.; Enk, A.H.; Gaiser, T. COX-2 expression in malignant melanoma: A novel prognostic marker? Melanoma Res. 2009, 19, 8–16. [Google Scholar] [CrossRef]
- Tudor, D.V.; Baldea, I.; Lupu, M.; Kacso, T.; Kutasi, E.; Hopartean, A.; Stretea, R.; Gabriela Filip, A. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20–31. [Google Scholar] [CrossRef]
- Minisini, A.M.; Pascoletti, G.; Intersimone, D.; Poletto, E.; Driol, P.; Spizzo, R.; Scott, C.A.; Puglisi, F.; Fasola, G.; Di Loreto, C. Expression of thymidine phosphorylase and cyclooxygenase-2 in melanoma. Melanoma Res. 2013, 23, 96–101. [Google Scholar] [CrossRef]
- Pinto-Proenca, R.; Santos, M.; Fonseca, C.; Fernandes, J.; Gaspar, M.F.; Proenca, R. Conjunctival melanoma: Association of cyclooxygenase-2 tumor expression to prognosis. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 989–995. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, M.H.; Kim, H.T.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E. Prognostic significance of CD163 expression and its correlation with cyclooxygenase-2 and vascular endothelial growth factor expression in cutaneous melanoma. Melanoma Res. 2019, 29, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Kuzbicki, L.; Lange, D.; Stanek-Widera, A.; Chwirot, B.W. Intratumoral expression of cyclooxygenase-2 (COX-2) is a negative prognostic marker for patients with cutaneous melanoma. Melanoma Res. 2016, 26, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Cahlin, C.; Gelin, J.; Delbro, D.; Lonnroth, C.; Doi, C.; Lundholm, K. Effect of cyclooxygenase and nitric oxide synthase inhibitors on tumor growth in mouse tumor models with and without cancer cachexia related to prostanoids. Cancer Res. 2000, 60, 1742–1749. [Google Scholar] [PubMed]
- Denkert, C.; Kobel, M.; Berger, S.; Siegert, A.; Leclere, A.; Trefzer, U.; Hauptmann, S. Expression of cyclooxygenase 2 in human malignant melanoma. Cancer Res. 2001, 61, 303–308. [Google Scholar]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- NIH. Polarized Dendritic Cell (aDC1) Based Treatment, Interferon Alpha-2, Rintatolimod, and Celecoxib for the Treatment of HLA-A2+ Refractory Melanoma. 2024. Available online: https://clinicaltrials.gov/study/NCT04093323 (accessed on 18 September 2024).
- Russo, P.A.; Halliday, G.M. Inhibition of nitric oxide and reactive oxygen species production improves the ability of a sunscreen to protect from sunburn, immunosuppression and photocarcinogenesis. Br. J. Dermatol. 2006, 155, 408–415. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Fischer, S.M.; Pavone, A.; Mikulec, C.; Langenbach, R.; Rundhaug, J.E. Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol. Carcinog. 2007, 46, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef] [PubMed]
- Mandala, M.; Merelli, B.; Massi, D. PD-L1 in melanoma: Facts and myths. Melanoma Manag. 2016, 3, 187–194. [Google Scholar] [CrossRef]
- Ellebaek, E.; Khan, S.; Bastholt, L.; Schmidt, H.; Haslund, C.A.; Donia, M.; Svane, I.M. PD-L1 is a biomarker of real-world clinical outcomes for anti-CTLA-4 plus anti-PD-1 or anti-PD-1 monotherapy in metastatic melanoma. Eur. J. Cancer 2024, 198, 113476. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAK-STAT 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Kortylewski, M.; Jove, R.; Yu, H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005, 24, 315–327. [Google Scholar] [CrossRef]
- Cao, H.H.; Chu, J.H.; Kwan, H.Y.; Su, T.; Yu, H.; Cheng, C.Y.; Fu, X.Q.; Guo, H.; Li, T.; Tse, A.K.; et al. Inhibition of the STAT3 signaling pathway contributes to apigenin-mediated anti-metastatic effect in melanoma. Sci. Rep. 2016, 6, 21731. [Google Scholar] [CrossRef]
- Kulesza, D.W.; Ramji, K.; Maleszewska, M.; Mieczkowski, J.; Dabrowski, M.; Chouaib, S.; Kaminska, B. Search for novel STAT3-dependent genes reveals SERPINA3 as a new STAT3 target that regulates invasion of human melanoma cells. Lab. Investig. 2019, 99, 1607–1621. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Partridge, M.A.; Huang, S.X.; Hei, T.K. Suppression of the proinflammatory response of metastatic melanoma cells increases TRAIL-induced apoptosis. J. Cell. Biochem. 2011, 112, 463–475. [Google Scholar] [CrossRef]
- Wang, W.; Guo, C.; Zhu, P.; Lu, J.; Li, W.; Liu, C.; Xie, H.; Myatt, L.; Chen, Z.J.; Sun, K. Phosphorylation of STAT3 mediates the induction of cyclooxygenase-2 by cortisol in the human amnion at parturition. Sci. Signal. 2015, 8, ra106. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, A.; Tong, S.; Roosan, M.R.; Syed, B.; Awasthi, A.; Silverman, R.B.; Yang, S. Crosstalk Between nNOS/NO and COX-2 Enhances Interferon-Gamma-Stimulated Melanoma Progression. Cancers 2025, 17, 477. https://doi.org/10.3390/cancers17030477
Patel A, Tong S, Roosan MR, Syed B, Awasthi A, Silverman RB, Yang S. Crosstalk Between nNOS/NO and COX-2 Enhances Interferon-Gamma-Stimulated Melanoma Progression. Cancers. 2025; 17(3):477. https://doi.org/10.3390/cancers17030477
Chicago/Turabian StylePatel, Anika, Shirley Tong, Moom R. Roosan, Basir Syed, Amardeep Awasthi, Richard B. Silverman, and Sun Yang. 2025. "Crosstalk Between nNOS/NO and COX-2 Enhances Interferon-Gamma-Stimulated Melanoma Progression" Cancers 17, no. 3: 477. https://doi.org/10.3390/cancers17030477
APA StylePatel, A., Tong, S., Roosan, M. R., Syed, B., Awasthi, A., Silverman, R. B., & Yang, S. (2025). Crosstalk Between nNOS/NO and COX-2 Enhances Interferon-Gamma-Stimulated Melanoma Progression. Cancers, 17(3), 477. https://doi.org/10.3390/cancers17030477