
E. coli Biomolecules Increase Glycolysis and Invasive Potential in Lung Adenocarcinoma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Sequencing
2.3. Measurement of Lactate Levels
2.4. Measurement of Glucose Levels
2.5. Deoxyglucose Measurement
2.6. Wound-Healing Assay
2.7. Migration and Invasion Assay
2.8. Immunoblotting
2.9. Real-Time qPCR
2.10. Combination KEGG Analysis
2.11. Statistical Analysis
3. Results
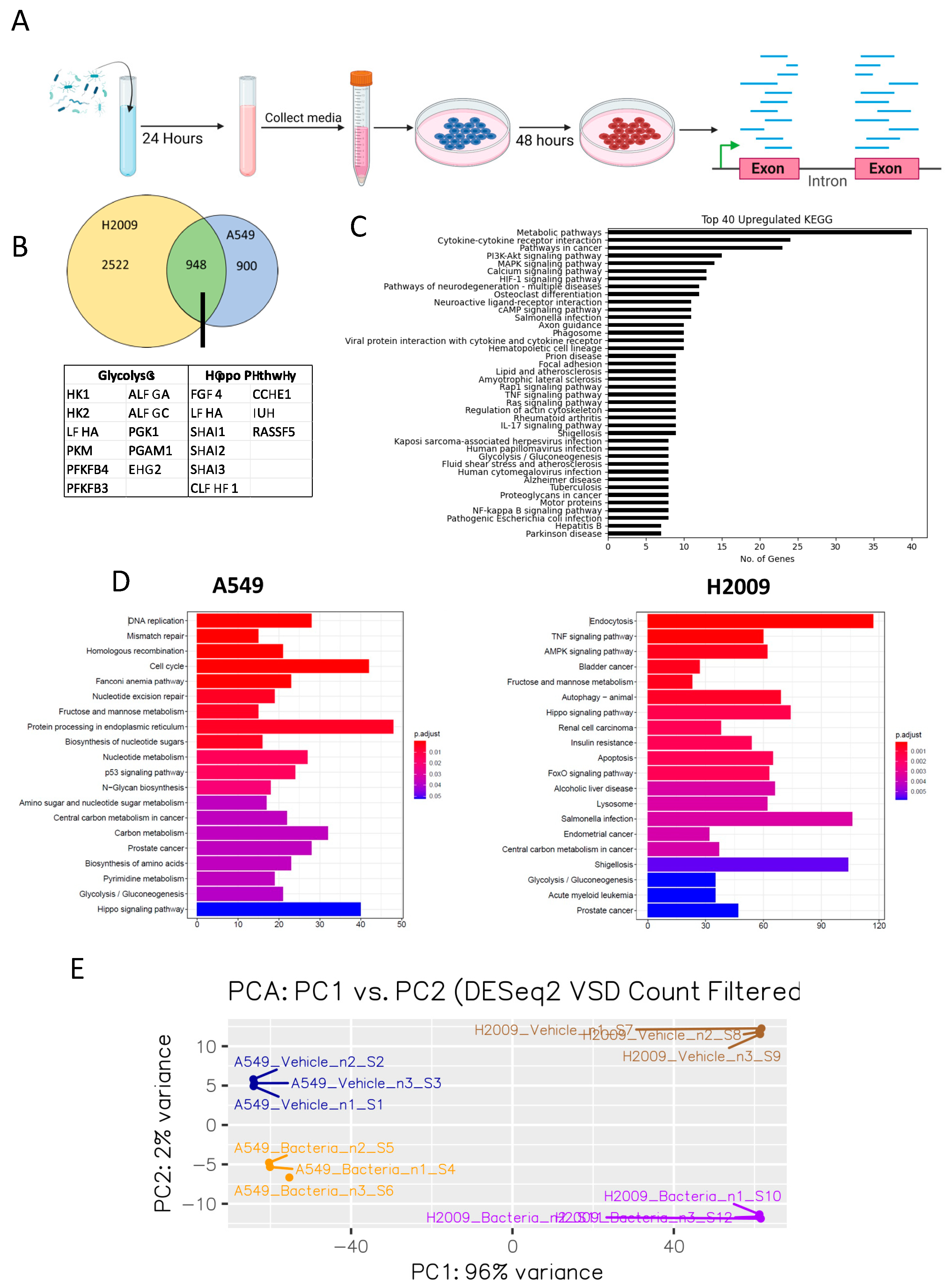
3.1. E. coli Biomolecules Elicit a Number of Transcriptomic Changes in LUAD Cell Lines
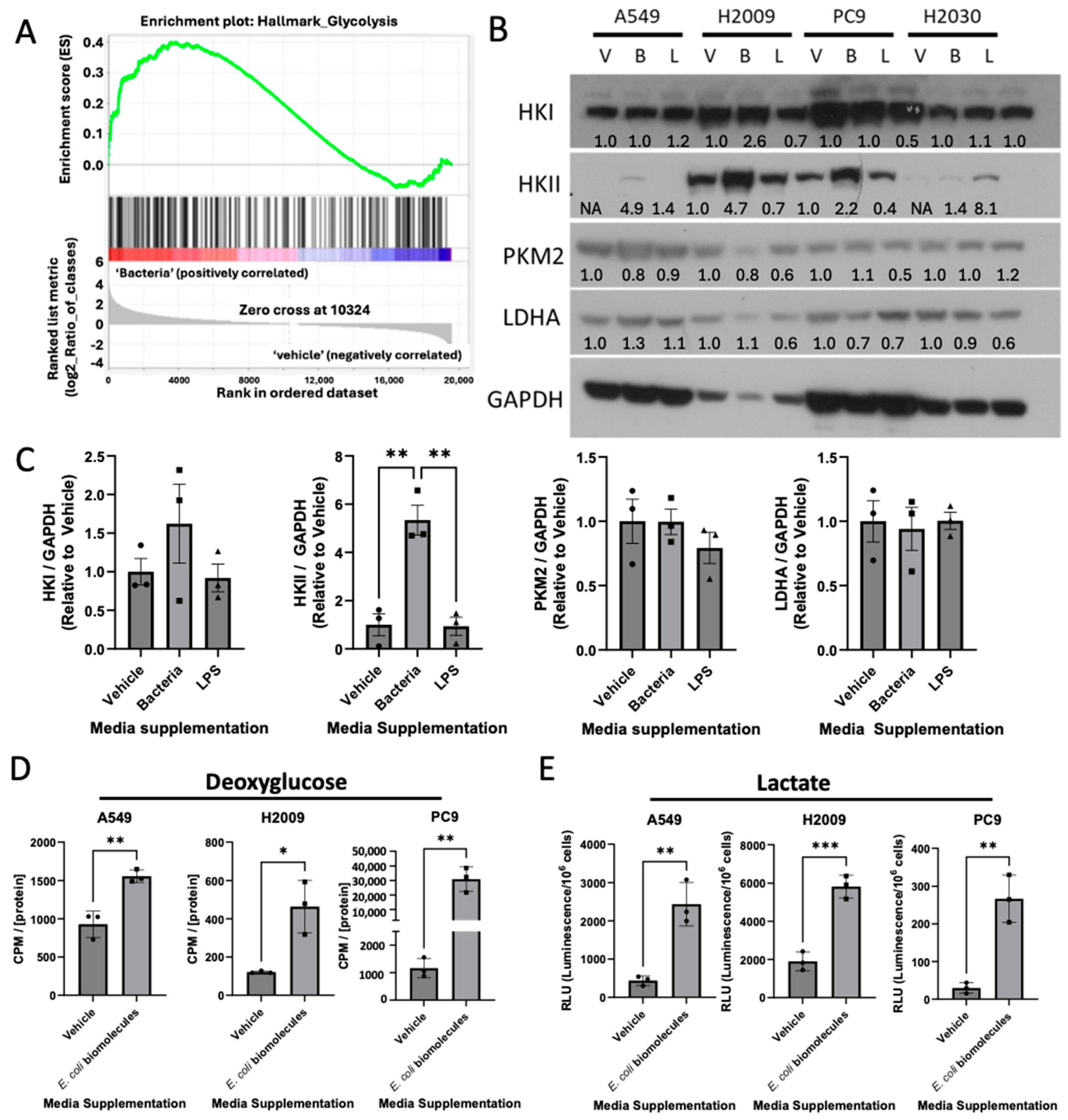
3.2. E. coli Biomolecules Increase Glycolysis in LUAD Cell Lines
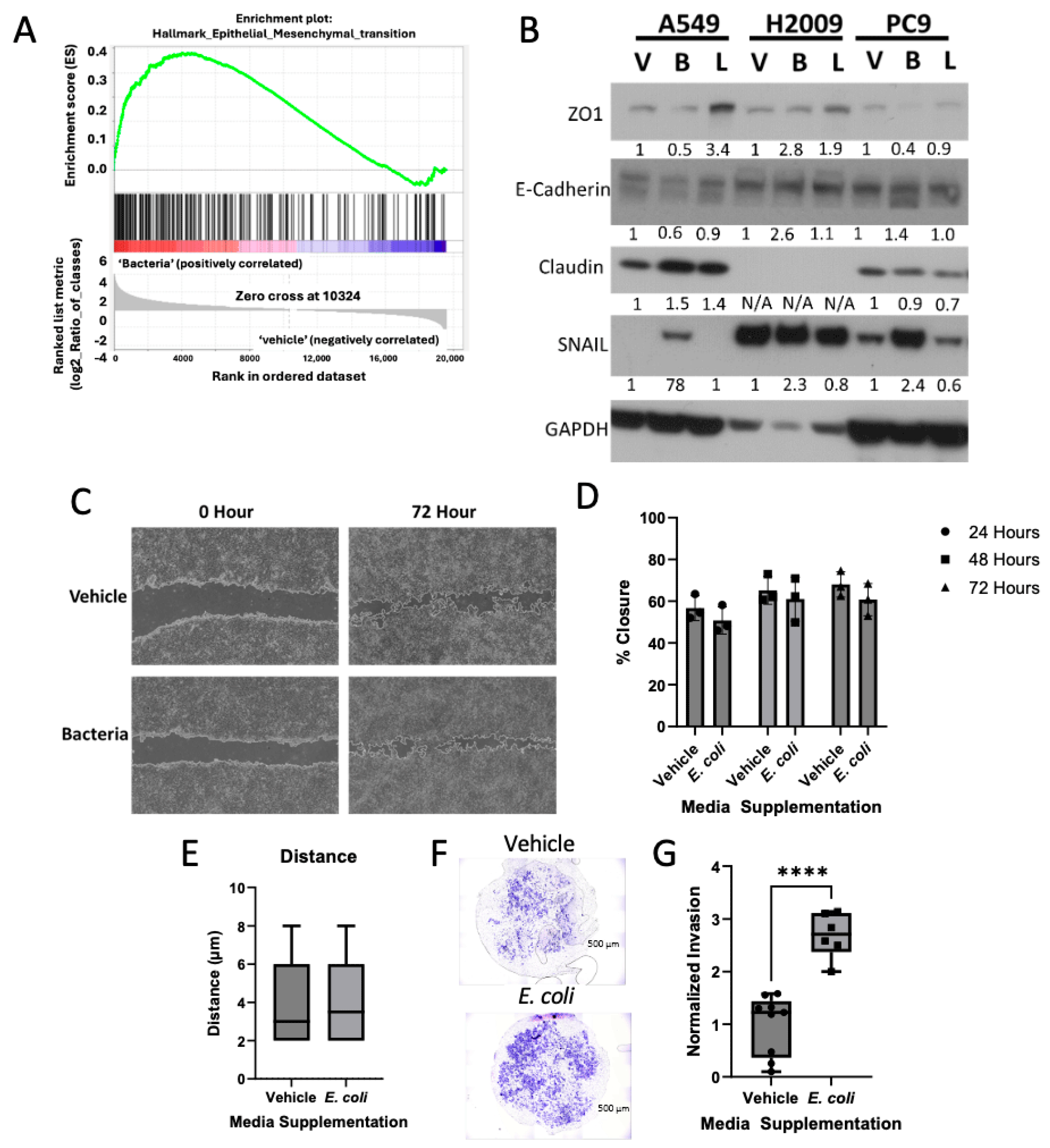
3.3. Bacterial Biomolecules Enhance Invasive Potential of LUAD Cell Lines
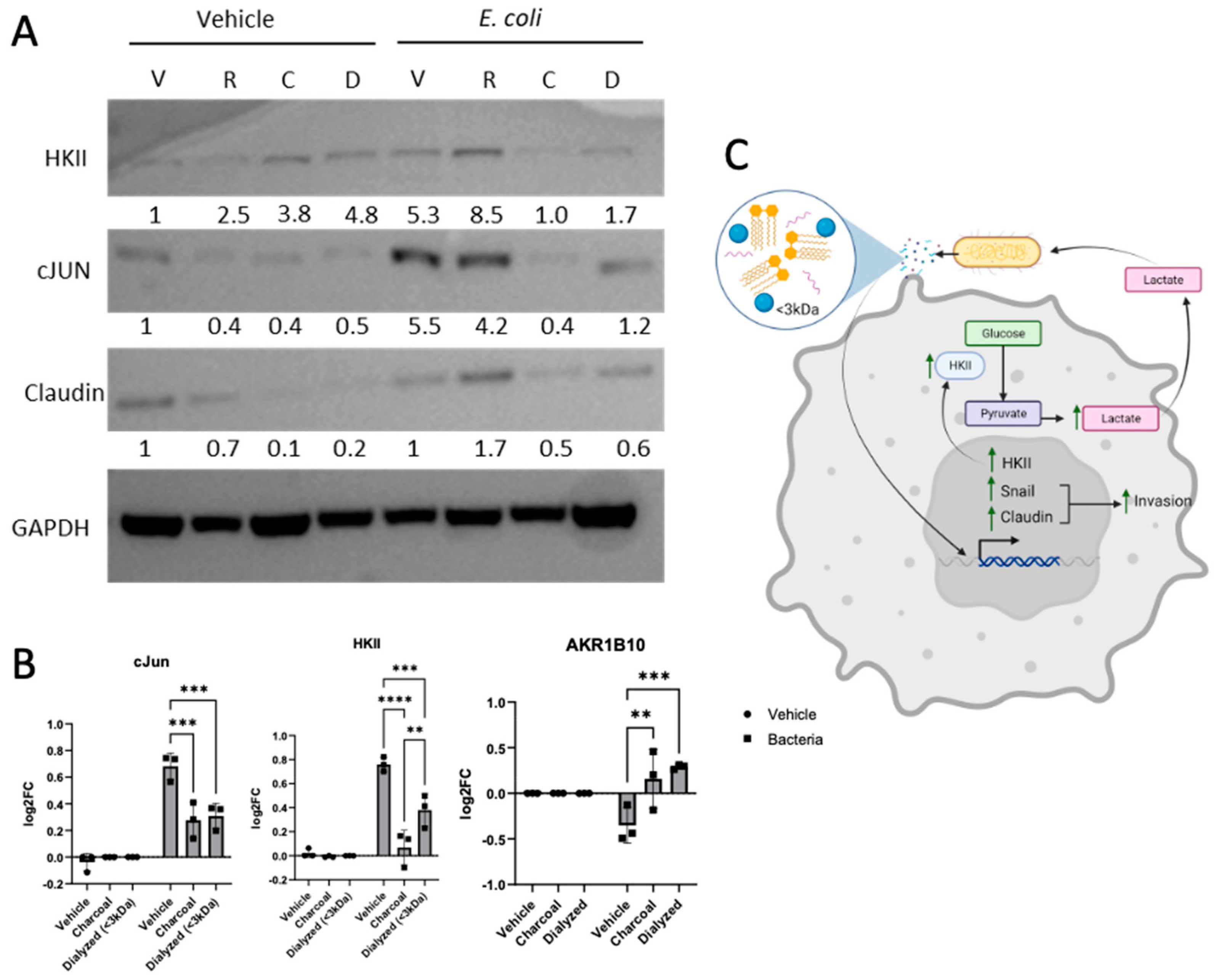
3.4. Bacterially Induced LUAD Phenotype Observed Is TLR4-Independent
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Kosibaty, Z.; Murata, Y.; Minami, Y.; Noguchi, M.; Sakamoto, N. ECT2 promotes lung adenocarcinoma progression through extracellular matrix dynamics and focal adhesion signaling. Cancer Sci. 2021, 112, 703–714. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef]
- Saforo, D.; Omer, L.; Smolenkov, A.; Barve, A.; Casson, L.; Boyd, N.; Clark, G.; Siskind, L.; Beverly, L. Primary lung cancer samples cultured under microenvironment-mimetic conditions enrich for mesenchymal stem-like cells that promote metastasis. Sci. Rep. 2019, 9, 4177. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Zella, D.; Gallo, R.C. Viruses and Bacteria Associated with Cancer: An Overview. Viruses 2021, 13, 1039. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.A.; Filho, F.S.L.; Sin, D.D.; Lam, S.; Leung, J.M.; Lam, W.L. Distinct bronchial microbiome precedes clinical diagnosis of lung cancer. Mol. Cancer 2022, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Naffouje, S.A.; Goto, M.; Wang, J.; Christov, K.; Rademacher, D.J.; Green, A.; Stecenko, A.A.; Chakrabarty, A.M.; Das Gupta, T.K.; et al. Cross-talk between cancer and Pseudomonas aeruginosa mediates tumor suppression. Commun. Biol. 2023, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Narunsky-Haziza, L.; Sepich-Poore, G.D.; Livyatan, I.; Asraf, O.; Martino, C.; Nejman, D.; Gavert, N.; Stajich, J.E.; Amit, G.; Gonzalez, A.; et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 2022, 185, 3789–3806.e17. [Google Scholar] [CrossRef] [PubMed]
- Packey, C.D.; Sartor, R.B. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr. Opin. Infect. Dis. 2009, 22, 292–301. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Varon, C.; Azzi-Martin, L.; Khalid, S.; Seeneevassen, L.; Menard, A.; Spuul, P. Helicobacters and cancer, not only gastric cancer? Semin. Cancer Biol. 2022, 86, 1138–1154. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Vega, A.A.; Marshall, E.A.; Noonan, A.J.C.; Filho, F.S.L.; Yang, J.; Stewart, G.L.; Johnson, F.D.; Vucic, E.A.; Pewarchuk, M.E.; Shah, P.P.; et al. Methionine-producing tumor micro(be) environment fuels growth of solid tumors. Cell. Oncol. 2023, 46, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Canale, F.P.; Basso, C.; Antonini, G.; Perotti, M.; Li, N.; Sokolovska, A.; Neumann, J.; James, M.J.; Geiger, S.; Jin, W.; et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature 2021, 598, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Jain, S.; Dash, P.; Minz, A.P.; Satpathi, S.; Samal, A.G.; Behera, P.K.; Satpathi, P.S.; Senapati, S. Lipopolysaccharide (LPS) enhances prostate cancer metastasis potentially through NF-kappaB activation and recurrent dexamethasone administration fails to suppress it in vivo. Prostate 2019, 79, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tiruthani, K.; Wang, Y.; Shen, L.; Hu, M.; Dorosheva, O.; Qiu, K.; Kinghorn, K.A.; Liu, R.; Huang, L. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv. Mater. 2018, 30, e1805007. [Google Scholar] [CrossRef]
- Melkamu, T.; Qian, X.; Upadhyaya, P.; O’Sullivan, M.G.; Kassie, F. Lipopolysaccharide enhances mouse lung tumorigenesis: A model for inflammation-driven lung cancer. Vet. Pathol. 2013, 50, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ehmer, U.; Sage, J. Control of Proliferation and Cancer Growth by the Hippo Signaling Pathway. Mol. Cancer Res. 2016, 14, 127–140. [Google Scholar] [CrossRef]
- Akrida, I.; Bravou, V.; Papadaki, H. The deadly cross-talk between Hippo pathway and epithelial-mesenchymal transition (EMT) in cancer. Mol. Biol. Rep. 2022, 49, 10065–10076. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Wu, J.; Yu, S.; Zhou, Y.; Zhou, C. AKR1B10 inhibits the proliferation and migration of gastric cancer via regulating epithelial-mesenchymal transition. Aging 2021, 13, 22298–22314. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Liu, P.; Wang, Z.C.; Zou, L.; Santiago, S.; Garbitt, V.; Gjoerup, O.V.; Iglehart, J.D.; Miron, A.; Richardson, A.L.; et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci. Signal 2009, 2, ra35. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qian, S.; Zhang, J.; Feng, J.; Luo, K.; Sun, L.; Zhao, L.; Ran, Y.; Sun, L.; Wang, J.; et al. Lipopolysaccharide promotes metastasis via acceleration of glycolysis by the nuclear factor-kappaB/snail/hexokinase3 signaling axis in colorectal cancer. Cancer Metab. 2021, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Yao, B.; Dong, T.; Chen, Y.; Yao, J.; Liu, Y.; Li, H.; Bai, H.; Liu, X.; Zhang, Y.; et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 2022, 185, 1356–1372.e26. [Google Scholar] [CrossRef]
- Szajnik, M.; Szczepanski, M.J.; Czystowska, M.; Elishaev, E.; Mandapathil, M.; Nowak-Markwitz, E.; Spaczynski, M.; Whiteside, T.L. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene 2009, 28, 4353–4363. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.H.; Mi, M.; Jiang, F.L.; Yue, Z.G.; Sun, Y.; Fan, L.; Meng, J.; Zhang, X.; Liu, L.; et al. Modified apple polysaccharides suppress the migration and invasion of colorectal cancer cells induced by lipopolysaccharide. Nutr. Res. 2013, 33, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pei, C.; Yan, S.; Liu, G.; Liu, G.; Chen, W.; Cui, Y.; Liu, Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 2015, 36, 1493–1502. [Google Scholar] [CrossRef]
- Liu, W.T.; Jing, Y.Y.; Yu, G.F.; Han, Z.P.; Yu, D.D.; Fan, Q.M.; Ye, F.; Li, R.; Gao, L.; Zhao, Q.D.; et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015, 358, 136–143. [Google Scholar] [CrossRef]
- Li, J.; Yin, J.; Shen, W.; Gao, R.; Liu, Y.; Chen, Y.; Li, X.; Liu, C.; Xiang, R.; Luo, N. TLR4 Promotes Breast Cancer Metastasis via Akt/GSK3beta/beta-Catenin Pathway upon LPS Stimulation. Anat. Rec. 2017, 300, 1219–1229. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Aublin-Gex, A.; Diaz, O.; Ramiere, C.; Peri, F.; Andre, P.; Lotteau, V. Toll-like Receptor 4-Induced Glycolytic Burst in Human Monocyte-Derived Dendritic Cells Results from p38-Dependent Stabilization of HIF-1alpha and Increased Hexokinase II Expression. J. Immunol. 2018, 201, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, G.T.; Wang, Y.Q.; Xian, S.; Zhang, L.; Zhu, S.M.; Pan, F.; Cheng, Y.X. TLR4 promotes the expression of HIF-1alpha by triggering reactive oxygen species in cervical cancer cells in vitro-implications for therapeutic intervention. Mol. Med. Rep. 2018, 17, 2229–2238. [Google Scholar] [CrossRef]
- Vallance, T.M.; Zeuner, M.T.; Williams, H.F.; Widera, D.; Vaiyapuri, S. Toll-Like Receptor 4 Signalling and Its Impact on Platelet Function, Thrombosis, and Haemostasis. Mediators Inflamm. 2017, 2017, 9605894. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed]
- Kayahara, M.; Wang, X.; Tournier, C. Selective regulation of c-jun gene expression by mitogen-activated protein kinases via the 12-o-tetradecanoylphorbol-13-acetate- responsive element and myocyte enhancer factor 2 binding sites. Mol. Cell Biol. 2005, 25, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Eyer, B.R.; Yamaguchi, Y.; Zhang, F.; Neal, M.D.; Sodhi, C.P.; Good, M.; Branca, M.; Prindle, T., Jr.; Lu, P.; et al. Synthesis of anti-inflammatory alpha-and beta-linked acetamidopyranosides as inhibitors of toll-like receptor 4 (TLR4). Tetrahedron Lett. 2015, 56, 3097–3100. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Chen, L.; Hao, L.; Zhang, X.; Chen, Y.; Ruan, Z.; Liang, H. Stearic acid induces proinflammatory cytokine production partly through activation of lactate-HIF1alpha pathway in chondrocytes. Sci. Rep. 2015, 5, 13092. [Google Scholar] [CrossRef]
- Hu, X.; Fatima, S.; Chen, M.; Xu, K.; Huang, C.; Gong, R.H.; Su, T.; Wong, H.L.X.; Bian, Z.; Kwan, H.Y. Toll-like receptor 4 is a master regulator for colorectal cancer growth under high-fat diet by programming cancer metabolism. Cell Death Dis. 2021, 12, 791. [Google Scholar] [CrossRef]
- Flemer, B.; Lynch, D.B.; Brown, J.M.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Chen, X.; Deng, T.; Huo, T.; Dong, F.; Deng, J. MiR-140-5p/TLR4/NF-kappaB signaling pathway: Crucial role in inflammatory response in 16HBE cells induced by dust fall PM(2.5). Ecotoxicol. Environ. Saf. 2021, 208, 111414. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007, 44, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.S.; Burrichter, A.G.; Durai Raj, A.C.; von Strempel, A.; Meng, C.; Kleigrewe, K.; Munch, P.C.; Rossler, L.; Huber, C.; Eisenreich, W.; et al. In vitro interaction network of a synthetic gut bacterial community. ISME J. 2022, 16, 1095–1109. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vega, A.A.; Shah, P.P.; Rouchka, E.C.; Clem, B.F.; Dean, C.R.; Woodrum, N.; Tanwani, P.; Siskind, L.J.; Beverly, L.J. E. coli Biomolecules Increase Glycolysis and Invasive Potential in Lung Adenocarcinoma. Cancers 2025, 17, 380. https://doi.org/10.3390/cancers17030380
Vega AA, Shah PP, Rouchka EC, Clem BF, Dean CR, Woodrum N, Tanwani P, Siskind LJ, Beverly LJ. E. coli Biomolecules Increase Glycolysis and Invasive Potential in Lung Adenocarcinoma. Cancers. 2025; 17(3):380. https://doi.org/10.3390/cancers17030380
Chicago/Turabian StyleVega, Alexis A., Parag P. Shah, Eric C. Rouchka, Brian F. Clem, Calista R. Dean, Natassja Woodrum, Preeti Tanwani, Leah J. Siskind, and Levi J. Beverly. 2025. "E. coli Biomolecules Increase Glycolysis and Invasive Potential in Lung Adenocarcinoma" Cancers 17, no. 3: 380. https://doi.org/10.3390/cancers17030380
APA StyleVega, A. A., Shah, P. P., Rouchka, E. C., Clem, B. F., Dean, C. R., Woodrum, N., Tanwani, P., Siskind, L. J., & Beverly, L. J. (2025). E. coli Biomolecules Increase Glycolysis and Invasive Potential in Lung Adenocarcinoma. Cancers, 17(3), 380. https://doi.org/10.3390/cancers17030380