

A Sequencing Overview of Malignant Peripheral Nerve Sheath Tumors: Findings and Implications for Treatment



Simple Summary
Abstract

1. Overview of MPNSTs
2. Sequencing Technologies and Its Application in MPNST
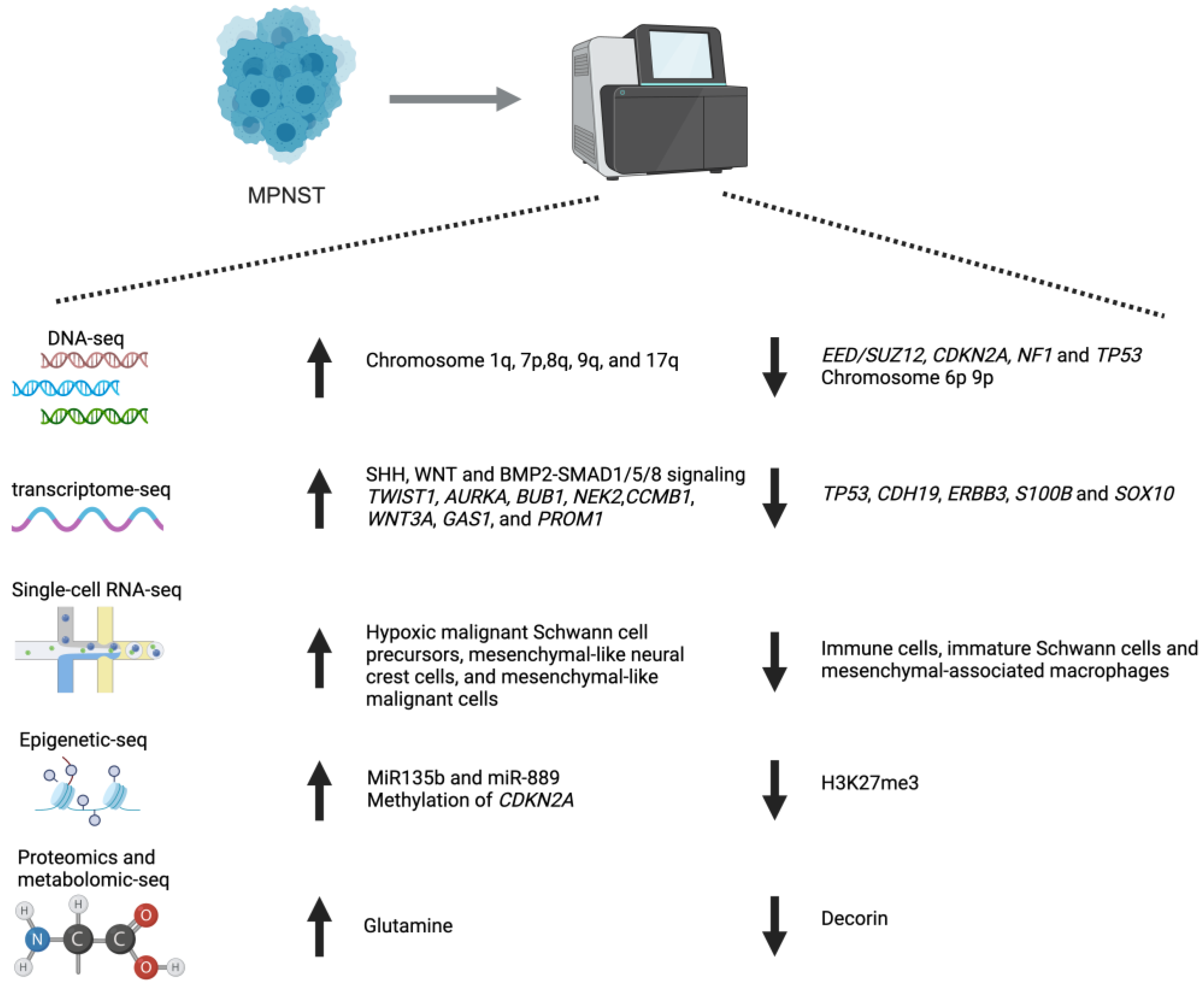
2.1. Microarray and RNA-Seq Analysis

{kind=link}
{kind=link}
| Data Type | Organism | Sample Settings | Reference | ||||
|---|---|---|---|---|---|---|---|
| MPNST | PN | NF | ANNUBP | ||||
| Microarray | Human | Tissue | 64 | NA | 15 | NA | GSE241224 (Høland et al., 2023) [29] |
| Microarray | Human | Tissue | 6 | 10 | 9 | 10 | GSE239561 (Rhodes, 2023) [30] |
| Microarray | Human | Tissue | 10 | NA | NA | NA | GSE52390, GSE52391 (Wolf et al., 2013) [31] |
| DNA methylation | Human | Tissue | 10 | NA | NA | NA | |
| Microarray | Human | Tissue | 6 | NA | 26 | NA | GSE41747 (Jessen et al., 2012) [32] |
| Microarray | Mouse | Tissue | 18 | NA | 15 | NA | |
| Microarray | Human | Tissue | 16 | NA | NA | NA | GSE17118 (Lafferty-Whyte et al., 2010) [33] |
| Microarray | Human | Tissue | 3 | NA | 3 | NA | GSE52252 (Wang et al., 2014) [34] |
| Microarray | Human | Tissue | 30 | NA | 8 | NA | GSE66743 (Kolberg et al., 2015) [35] |
| Microarray | Human | Tissue | 4 | NA | NA | NA | GSE77203 (Yasuhiro et al., 2019) [36] |
| Microarray | Human | Cell | 21 | NA | NA | NA | GSE39764 (Sun et al., 2013) [37] |
| Microarray | Human | Tissue | 3 | NA | NA | NA | GSE35852, GSE35851 (Kelly et al., 2012) [38] |
| micro-RNA | Human | Tissue | 3 | NA | NA | NA | |
| Microarray | Human | Cell | 22 | NA | NA | NA | GSE8717 (Mahller et al., 2007) [39] |
| Microarray | Human | Cell | 20 | NA | NA | NA | GSE47476 and GSE47477 (Zhang et al., 2013) [40] |
| micro-RNA | Human | Cell | 12 | NA | NA | NA | |
| Microarray | Human | Cell | 9 | NA | NA | NA | GSE62500 (De Raedt et al., 2014) [41] |
| Microarray | Human | Cell | 12 | NA | NA | NA | GSE84205 (Malone et al., 2017) [42] |
| Microarray | Human | PDX/Tissue | 11 | NA | NA | NA | GSE60082 (Castellsagué et al., 2015) [43] |
| Microarray | Mouse | Tissue | 8 | NA | NA | NA | GSE57141 (Malone et al., 2014) [44] |
| Bulk RNA-seq | Human | PDX | 13 | NA | NA | NA | syn11638893 (Hirbe et al., 2022) [45] |
| WES/WGS | Human | PDX | 16 | NA | NA | NA | |
| scRNA-seq | Human | PDX | 7 MPNST | ||||
| Bulk RNA-seq | Human | Tissue | 73 | NA | 1 | 2 | EGAD00001008608 (Genomics of Malignant Peripheral Nerve Sheath Tumor (GeM) Consortium, 2020) [46] |
| WGS | Human | Tissue | 72 | NA | 2 | 3 | |
| Bulk RNA-seq | Human | Tissue | 41 | NA | NA | NA | GSE206527 and GSE179699 (Chi et al., 2022) [47] |
| ATAC-seq | Human | Cell | 12 | NA | NA | NA | |
| Bulk RNA-seq | Human | Tissue | 9 | NA | 8 | NA | GSE178989, GSE179033 and GSE179041 (Wu et al., 2022) [48] |
| scRNA-seq | Human | Tissue | PN vs. MPNST | ||||
| scRNA-seq | Mouse | Tissue | 1.5-month MPNST vs. 4-months MPNST | ||||
| DNA methylation | Human | Cell | 63 | NA | NA | NA | GSE141438, GSE141435 and GSE141437 (Kochat et al., 2021) [49] |
| Bulk RNA-seq | Human | Tissue | 7 | NA | 3 | NA | |
| Bulk RNA-seq | Human | Cell | 36 | NA | NA | NA | |
| Bulk RNA-seq | Human | Tissue | 25 | 21 | NA | NA | GSE145064 (Kohlmeyer et al., 2020) [50] |
| Bulk RNA-seq | Human | Tissue | 12 | 12 | NA | NA | GSE212964 (Vasudevan et al., 2023) [51] |
| scRNA-seq | Human | Tissue | 3 MPNST vs. 3 PN | ||||
| Bulk RNA-seq | Human | Tissue | 14 | NA | 34 | NA | GSE207400, PRJNA854920 and GSe207399 (Suppiah et al., 2023) [52] |
| WES | Human | Tissue | 18 | NA | 16 | NA | |
| scRNA-seq | Human | Tissue | ANNUBP vs. MPNST | ||||
| Bulk RNA-seq | Human | Tissue | 10 | NA | NA | NA | TCGA-SARC (https://www.cancer.gov/tcga, accesed on 28 August 2024) |
| Bulk RNA-seq | Human | Tissue | 6 | NA | NA | NA | GSE120685 (Woodhoo et al., 2021) [53] |
| Bulk RNA-seq | Human | Cell | 28 | NA | NA | NA | GSE183308, GSE183307 (Zhang et al., 2022) [54] |
| scRNA-seq | Human | Tissue | primary vs. metastasis | ||||
| Bulk RNA-seq | Human | Tissue | 3 | NA | 3 | NA | GSE270880 (Zhang et al., 2024) [55] |
| Bulk RNA-seq | Human | Cell | 16 | NA | NA | NA | GSE179585, GSe179586 (Patel et al., 2022) [56] |
| DNA methylation | Human | Cell | 16 | NA | NA | NA | |
| Bulk RNA-seq | Human | Cell | 6 | NA | 10 | NA | GSE118185 (Wassef et al., 2019) [57] |
| Bulk RNA-seq | Human | Cell | 6 | NA | NA | NA | GSE213988 (Chung et al., 2022) [58] |
| Bulk RNA-seq | Human | Cell | 4 | NA | NA | NA | GSE216792 |
| WGS | Human | Tissue (blood) | 46 | 23 | NA | NA | syn23651229 (Shern et al., 2020) [59] |
| WES | Human | Tissue | 51 | NA | NA | NA | EGAS0000100452 (Lyskjær et al., 2020) [60] |
| WES | Human | Tissue | 2 | 3 | NA | NA | Hirbe et al., 2015 [61] |
| WES | Human | Tissue | 15 | NA | NA | NA | Lee et al., 2014 [19] |
| WES | Human | Tissue | 6 | NA | NA | NA | Godec et al., 2020 [62] |
| WES/WGS | Human | Tissue | 11 | NA | 2 | NA | Kinoshita et al., 2020 [63] |
| DNA methylation | Human | Tissue | 1 | NA | 1 | NA | GSE21714 (Feber et al., 2011) [64] |
| DNA methylation | Human | Cell | 16 | NA | NA | NA | GSE263127 (Bhunia et al., 2024) [65] |
| DNA methylation | Human | Tissue | 102 | NA | NA | NA | E-MTAB-8864 www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-6961, accesed on 12 December 2024) |
| DNA methylation | Human | Tissue | 8 | NA | NA | NA | GSE36982 (Renner et al., 2012) [66] |
| micro-RNA | Human | Tissue | 19 | 9 | NA | NA | GSE140987 (Wiemer, 2019) [67] |
| ATAC-seq | Human | Cell | 6 | NA | NA | NA | GSE275047 |
2.2. Single-Cell RNA-Seq
2.3. Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS)
2.4. Epigenetics Sequencing
2.5. Emerging Sequencing Technologies: Proteomics and Metabolomic Sequencing
3. From Sequencing to Implications: Targeted Therapy
3.1. MEK Inhibitor
3.2. Histone Deacetylase (HDAC) Inhibitors
3.3. Other Emerging Targeted Therapies
3.4. Implications for Immunotherapy
4. Conclusions and Implications for Clinical Practice
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cuneo, H.M.; Rand, C.W. Tumors of the Gasserian Ganglion: Tumor of the Left Gasserian Ganglion Associated with Enlargement of the Mandibular Nerve a Review of the Literature and Case Report. J. Neurosurg. 1952, 9, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Ducatman, B.; Scheithauer, B.; Piepgras, D.; Reiman, H. Malignant Peripheral Nerve Sheath Tumors in Childhood. J. Neuro-Oncology 1984, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant Peripheral Nerve Sheath Tumors. A Clinicopathologic Study of 120 Cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef]
- Anghileri, M.; Miceli, R.; Fiore, M.; Mariani, L.; Ferrari, A.; Mussi, C.; Lozza, L.; Collini, P.; Olmi, P.; Casali, P.G.; et al. Malignant Peripheral Nerve Sheath Tumors: Prognostic Factors and Survival in a Series of Patients Treated at a Single Institution. Cancer 2006, 107, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.-H.G.; Gross, A.M.; Romo, C.G.; Dehner, C.A.; Lazar, A.J.; Miettinen, M.; Pekmezci, M.; Quezado, M.; Rodriguez, F.J.; Stemmer-Rachamimov, A.; et al. Consensus Recommendations for an Integrated Diagnostic Approach to Peripheral Nerve Sheath Tumors Arising in the Setting of Neurofibromatosis Type 1 (NF1). Neuro-Oncology 2024, noae235. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Dehner, C.A.; Dombi, E.; Eulo, V.; Gross, A.M.; Sundby, T.; Lazar, A.J.; Widemann, B.C. Contemporary Approach to Neurofibromatosis Type 1–Associated Malignant Peripheral Nerve Sheath Tumors. Am. Soc. Clin. Oncol. Educ. Book. 2024, 44, e432242. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Clark, M.A.; Fisher, C.; Judson, I.; Thomas, J.M. Soft-Tissue Sarcomas in Adults. N. Engl. J. Med. 2005, 353, 701–711. [Google Scholar] [CrossRef]
- Dunn, G.P.; Spiliopoulos, K.; Plotkin, S.R.; Hornicek, F.J.; Harmon, D.C.; Delaney, T.F.; Williams, Z. Role of Resection of Malignant Peripheral Nerve Sheath Tumors in Patients with Neurofibromatosis Type 1: Clinical Article. J. Neurosurg. 2013, 118, 142–148. [Google Scholar] [CrossRef]
- Lucas, C.-H.G.; Vasudevan, H.N.; Chen, W.C.; Magill, S.T.; Braunstein, S.E.; Jacques, L.; Dahiya, S.; Rodriguez, F.J.; Horvai, A.E.; Perry, A.; et al. Histopathologic Findings in Malignant Peripheral Nerve Sheath Tumor Predict Response to Radiotherapy and Overall Survival. Neuro-Oncol. Adv. 2020, 2, vdaa131. [Google Scholar] [CrossRef] [PubMed]
- Sloan, L.; Terezakis, S.A.; Blakeley, J.O.; Slobogean, B.; Kleinberg, L.R. Long-Term Outcomes of Radiation Therapy (RT) in the Management of Malignant Peripheral Nerve Sheath Tumors (MPNST) in Patients with Neurofibromatosis Type 1 (NF1). Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, e474–e475. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Orbach, D.; Brennan, B.; Kelsey, A.; Zanetti, I.; De Salvo, G.L.; Gaze, M.N.; Craigie, R.J.; McHugh, K.; Francotte, N.; et al. Outcome and Prognostic Factors in Pediatric Malignant Peripheral Nerve Sheath Tumors: An Analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 Prospective Study. Pediatr. Blood Cancer 2019, 66, e27833. [Google Scholar] [CrossRef]
- Hruban, R.H.; Shiu, M.H.; Senie, R.T.; Woodruff, J.M. Malignant Peripheral Nerve Sheath Tumors of the Buttock and Lower Extremity. A Study of 43 Cases. Cancer 1990, 66, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Puj, K.; Salunke, A.; Pandya, S.; Gandhi, J.; Parikh, A. Malignant Peripheral Nerve Sheath Tumor with Analysis of Various Prognostic Factors: A Single-Institutional Experience. J. Can. Res. Ther. 2021, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.-J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients With Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open 2021, 4, e210945. [Google Scholar] [CrossRef] [PubMed]
- Stucky, C.-C.H.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann. Surg. Oncol. 2012, 19, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of Human Malignant Peripheral Nerve Sheath Tumors. Neuro-Oncol. Adv. 2020, 2, i50–i61. [Google Scholar] [CrossRef]
- Spurlock, G.; Knight, S.J.L.; Thomas, N.; Kiehl, T.-R.; Guha, A.; Upadhyaya, M. Molecular Evolution of a Neurofibroma to Malignant Peripheral Nerve Sheath Tumor (MPNST) in an NF1 Patient: Correlation between Histopathological, Clinical and Molecular Findings. J. Cancer Res. Clin. Oncol. 2010, 136, 1869–1880. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 Is Recurrently Inactivated through EED or SUZ12 Loss in Malignant Peripheral Nerve Sheath Tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Yao, C.; Zhou, H.; Dong, Y.; Alhaskawi, A.; Hasan Abdullah Ezzi, S.; Wang, Z.; Lai, J.; Goutham Kota, V.; Hasan Abdulla Hasan Abdulla, M.; Lu, H. Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management. Cancers 2023, 15, 1077. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Gutmann, D.H.; Rubenstein, A.; Viskochil, D.; Zimmerman, R.A.; Vezina, G.; Small, J.; Korf, B. Plexiform Neurofibromas in NF1: Toward Biologic-Based Therapy. Neurology 2002, 58, 1461–1470. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic Evaluation of Atypical Neurofibromatous Tumors and Their Transformation into Malignant Peripheral Nerve Sheath Tumor in Patients with Neurofibromatosis 1—A Consensus Overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) Loss Drives NF1-Associated Atypical Neurofibroma and Malignant Transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef] [PubMed]
- Brockman, Q.R.; Scherer, A.; McGivney, G.R.; Gutierrez, W.R.; Voigt, A.P.; Isaacson, A.L.; Laverty, E.A.; Roughton, G.; Knepper-Adrian, V.; Darbro, B.; et al. PRC2 Loss Drives MPNST Metastasis and Matrix Remodeling. JCI Insight 2022, 7, e157502. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Lingo, J.J.; Voigt, E.; Leidinger, M.R.; McGivney, G.R.; Scherer, A.; Koppenhafer, S.L.; Gordon, D.J.; Breheny, P.; et al. Oncogenic RABL6A Promotes NF1-Associated MPNST Progression in Vivo. Neuro-Oncol. Adv. 2022, 4, vdac047. [Google Scholar] [CrossRef]
- Rahrmann, E.P.; Watson, A.L.; Keng, V.W.; Choi, K.; Moriarity, B.S.; Beckmann, D.A.; Wolf, N.K.; Sarver, A.; Collins, M.H.; Moertel, C.L.; et al. Forward Genetic Screen for Malignant Peripheral Nerve Sheath Tumor Formation Identifies New Genes and Pathways Driving Tumorigenesis. Nat. Genet. 2013, 45, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.J. DNA Microarray Technology: Devices, Systems, and Applications. Annu. Rev. Biomed. Eng. 2002, 4, 129–153. [Google Scholar] [CrossRef]
- Reilly, C.; Raghavan, A.; Bohjanen, P. Global Assessment of Cross-Hybridization for Oligonucleotide Arrays. J. Biomol. Tech. 2006, 17, 163–172. [Google Scholar]
- Høland, M.; Berg, K.C.G.; Eilertsen, I.A.; Bjerkehagen, B.; Kolberg, M.; Boye, K.; Lingjærde, O.C.; Guren, T.K.; Mandahl, N.; Van Den Berg, E.; et al. Transcriptomic Subtyping of Malignant Peripheral Nerve Sheath Tumours Highlights Immune Signatures, Genomic Profiles, Patient Survival and Therapeutic Targets. eBioMedicine 2023, 97, 104829. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.K.; Burgess, B.; White, E.E.; Smith, A.E.; Sierra Potchanant, E.A.; Mang, H.; Hickey, B.E.; Lu, Q.; Qian, S.; Bessler, W.; et al. Spatial Gene-Expression Profiling Unveils Immuno-Oncogenic Programs of NF1-Associated Peripheral Nerve Sheath Tumor Progression. Clin. Cancer Res. 2024, OF1–OF16. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Renner, M.; Straub, M.; Mughal, S.S.; Hutter, B.; Kosaloglu, Z.; Schweßinger, R.; Scheffler, M.; Alldinger, I.; Schimmack, S.; et al. Targeting Fibroblast Growth Factor Receptor 1 for Treatment of Soft-Tissue Sarcoma. Clin. Cancer Res. 2017, 23, 962–973. [Google Scholar] [CrossRef]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.-O.; Dombi, E.; et al. MEK Inhibition Exhibits Efficacy in Human and Mouse Neurofibromatosis Tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Lafferty-Whyte, K.; Bilsland, A.; Hoare, S.F.; Burns, S.; Zaffaroni, N.; Cairney, C.J.; Keith, W.N. TCEAL7 Inhibition of C-Myc Activity in Alternative Lengthening of Telomeres Regulates hTERT Expression. Neoplasia 2010, 12, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bledsoe, K.L.; Graham, R.P.; Asmann, Y.W.; Viswanatha, D.S.; Lewis, J.E.; Lewis, J.T.; Chou, M.M.; Yaszemski, M.J.; Jen, J.; et al. Recurrent PAX3-MAML3 Fusion in Biphenotypic Sinonasal Sarcoma. Nat. Genet. 2014, 46, 666–668. [Google Scholar] [CrossRef]
- Kolberg, M.; Høland, M.; Lind, G.E.; Ågesen, T.H.; Skotheim, R.I.; Sundby Hall, K.; Mandahl, N.; Smeland, S.; Mertens, F.; Davidson, B.; et al. Protein Expression of BIRC5, TK1, and TOP2A in Malignant Peripheral Nerve Sheath Tumours—A Prognostic Test after Surgical Resection. Mol. Oncol. 2015, 9, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Komura, S.; Ito, K.; Ohta, S.; Ukai, T.; Kabata, M.; Itakura, F.; Semi, K.; Matsuda, Y.; Hashimoto, K.; Shibata, H.; et al. Cell-Type Dependent Enhancer Binding of the EWS/ATF1 Fusion Gene in Clear Cell Sarcomas. Nat. Commun. 2019, 10, 3999. [Google Scholar] [CrossRef]
- Sun, D.; Haddad, R.; Kraniak, J.M.; Horne, S.D.; Tainsky, M.A. RAS/MEK–Independent Gene Expression Reveals BMP2-Related Malignant Phenotypes in the Nf1 -Deficient MPNST. Mol. Cancer Res. 2013, 11, 616–627. [Google Scholar] [CrossRef]
- Fountzilas, E.; Kelly, A.D.; Perez-Atayde, A.R.; Goldsmith, J.; Konstantinopoulos, P.A.; Francoeur, N.; Correll, M.; Rubio, R.; Hu, L.; Gebhardt, M.C.; et al. A microRNA Activity Map of Human Mesenchymal Tumors: Connections to Oncogenic Pathways; an Integrative Transcriptomic Study. BMC Genom. 2012, 13, 332. [Google Scholar] [CrossRef]
- Mahller, Y.Y.; Sakthivel, B.; Baird, W.H.; Aronow, B.J.; Hsu, Y.-H.; Cripe, T.P.; Mehrian-Shai, R. Molecular Analysis of Human Cancer Cells Infected by an Oncolytic HSV-1 Reveals Multiple Upregulated Cellular Genes and a Role for SOCS1 in Virus Replication. Cancer Gene Ther. 2008, 15, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Garnett, J.; Creighton, C.J.; Al Sannaa, G.A.; Igram, D.R.; Lazar, A.; Liu, X.; Liu, C.; Pollock, R.E. EZH2-miR-30d-KPNB1 Pathway Regulates Malignant Peripheral Nerve Sheath Tumour Cell Survival and Tumourigenesis. J. Pathol. 2014, 232, 308–318. [Google Scholar] [CrossRef]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 Loss Amplifies Ras-Driven Transcription and Confers Sensitivity to BRD4-Based Therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed]
- Castellsagué, J.; Gel, B.; Fernández-Rodríguez, J.; Llatjós, R.; Blanco, I.; Benavente, Y.; Pérez-Sidelnikova, D.; García-Del Muro, J.; Viñals, J.M.; Vidal, A.; et al. Comprehensive Establishment and Characterization of Orthoxenograft Mouse Models of Malignant Peripheral Nerve Sheath Tumors for Personalized Medicine. EMBO Mol. Med. 2015, 7, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.F.; Fromm, J.A.; Maertens, O.; DeRaedt, T.; Ingraham, R.; Cichowski, K. Defining Key Signaling Nodes and Therapeutic Biomarkers in NF1-Mutant Cancers. Cancer Discov. 2014, 4, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Dehner, C.; Moon, C.I.; Zhang, X.; Zhou, Z.; Miller, C.; Xu, H.; Wan, X.; Yang, K.; Mashl, J.; Gosline, S.J.C.; et al. Chromosome 8 Gain Is Associated with High-Grade Transformation in MPNST. JCI Insight 2021, 6, e146351. [Google Scholar] [CrossRef]
- Miller, D.T.; Cortés-Ciriano, I.; Pillay, N.; Hirbe, A.C.; Snuderl, M.; Bui, M.M.; Piculell, K.; Al-Ibraheemi, A.; Dickson, B.C.; Hart, J.; et al. Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs. Genes 2020, 11, 387. [Google Scholar] [CrossRef]
- Yan, J.; Chen, Y.; Patel, A.J.; Warda, S.; Lee, C.J.; Nixon, B.G.; Wong, E.W.P.; Miranda-Román, M.A.; Yang, N.; Wang, Y.; et al. Tumor-Intrinsic PRC2 Inactivation Drives a Context-Dependent Immune-Desert Microenvironment and Is Sensitized by Immunogenic Viruses. J. Clin. Investig. 2022, 132, e153437. [Google Scholar] [CrossRef]
- Wu, L.M.N.; Zhang, F.; Rao, R.; Adam, M.; Pollard, K.; Szabo, S.; Liu, X.; Belcher, K.A.; Luo, Z.; Ogurek, S.; et al. Single-Cell Multiomics Identifies Clinically Relevant Mesenchymal Stem-like Cells and Key Regulators for MPNST Malignancy. Sci. Adv. 2022, 8, eabo5442. [Google Scholar] [CrossRef] [PubMed]
- Kochat, V.; Raman, A.T.; Landers, S.M.; Tang, M.; Schulz, J.; Terranova, C.; Landry, J.P.; Bhalla, A.D.; Beird, H.C.; Wu, C.-C.; et al. Enhancer Reprogramming in PRC2-Deficient Malignant Peripheral Nerve Sheath Tumors Induces a Targetable de-Differentiated State. Acta Neuropathol. 2021, 142, 565–590. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs That Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997–3011. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Payne, E.; Delley, C.L.; John Liu, S.; Mirchia, K.; Sale, M.J.; Lastella, S.; Nunez, M.S.; Lucas, C.-H.G.; Eaton, C.D.; et al. Functional Interactions between Neurofibromatosis Tumor Suppressors Underlie Schwann Cell Tumor De-Differentiation and Treatment Resistance. Nat. Commun. 2024, 15, 477. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, S.; Mansouri, S.; Mamatjan, Y.; Liu, J.C.; Bhunia, M.M.; Patil, V.; Rath, P.; Mehani, B.; Heir, P.; Bunda, S.; et al. Multiplatform Molecular Profiling Uncovers Two Subgroups of Malignant Peripheral Nerve Sheath Tumors with Distinct Therapeutic Vulnerabilities. Nat. Commun. 2023, 14, 2696. [Google Scholar] [CrossRef] [PubMed]
- Palomo-Irigoyen, M.; Pérez-Andrés, E.; Iruarrizaga-Lejarreta, M.; Barreira-Manrique, A.; Tamayo-Caro, M.; Vila-Vecilla, L.; Moreno-Cugnon, L.; Beitia, N.; Medrano, D.; Fernández-Ramos, D.; et al. HuR/ELAVL1 Drives Malignant Peripheral Nerve Sheath Tumor Growth and Metastasis. J. Clin. Investig. 2020, 130, 3848–3864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lou, H.E.; Gopalan, V.; Liu, Z.; Jafarah, H.M.; Lei, H.; Jones, P.; Sayers, C.M.; Yohe, M.E.; Chittiboina, P.; et al. Single-Cell Sequencing Reveals Activation of Core Transcription Factors in PRC2-Deficient Malignant Peripheral Nerve Sheath Tumor. Cell Rep. 2022, 40, 111363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, C.; Sun, S.; Guo, C.; Bu, Y.; Wang, Z.; Liu, Z.; Zhang, X.; Li, D.; Liu, S. TSPO Deficiency Promotes the Progression of Malignant Peripheral Sheath Tumors by Regulating the G2/M Phase of the Cell Cycle via CDK1. Sci. Rep. 2024, 14, 26235. [Google Scholar] [CrossRef]
- Patel, A.J.; Warda, S.; Maag, J.L.V.; Misra, R.; Miranda-Román, M.A.; Pachai, M.R.; Lee, C.J.; Li, D.; Wang, N.; Bayshtok, G.; et al. PRC2-Inactivating Mutations in Cancer Enhance Cytotoxic Response to DNMT1-Targeted Therapy via Enhanced Viral Mimicry. Cancer Discov. 2022, 12, 2120–2139. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Luscan, A.; Aflaki, S.; Zielinski, D.; Jansen, P.W.T.C.; Baymaz, H.I.; Battistella, A.; Kersouani, C.; Servant, N.; Wallace, M.R.; et al. EZH1/2 Function Mostly within Canonical PRC2 and Exhibit Proliferation-Dependent Redundancy That Shapes Mutational Signatures in Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6075–6080. [Google Scholar] [CrossRef]
- Chung, M.-H.; Aimaier, R.; Yu, Q.; Li, H.; Li, Y.; Wei, C.; Gu, Y.; Wang, W.; Guo, Z.; Long, M.; et al. RRM2 as a Novel Prognostic and Therapeutic Target of NF1-Associated MPNST. Cell Oncol. 2023, 46, 1399–1413. [Google Scholar] [CrossRef]
- Szymanski, J.J.; Sundby, R.T.; Jones, P.A.; Srihari, D.; Earland, N.; Harris, P.K.; Feng, W.; Qaium, F.; Lei, H.; Roberts, D.; et al. Cell-Free DNA Ultra-Low-Pass Whole Genome Sequencing to Distinguish Malignant Peripheral Nerve Sheath Tumor (MPNST) from Its Benign Precursor Lesion: A Cross-Sectional Study. PLoS Med. 2021, 18, e1003734. [Google Scholar] [CrossRef]
- Lyskjær, I.; Lindsay, D.; Tirabosco, R.; Steele, C.D.; Lombard, P.; Strobl, A.; Rocha, A.M.; Davies, C.; Ye, H.; Bekers, E.; et al. H3K27me3 Expression and Methylation Status in Histological Variants of Malignant Peripheral Nerve Sheath Tumours. J. Pathol. 2020, 252, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Dahiya, S.; Miller, C.A.; Li, T.; Fulton, R.S.; Zhang, X.; McDonald, S.; DeSchryver, K.; Duncavage, E.J.; Walrath, J.; et al. Whole Exome Sequencing Reveals the Order of Genetic Changes during Malignant Transformation and Metastasis in a Single Patient with NF1-Plexiform Neurofibroma. Clin. Cancer Res. 2015, 21, 4201–4211. [Google Scholar] [CrossRef] [PubMed]
- Godec, A.; Jayasinghe, R.; Chrisinger, J.S.A.; Prudner, B.; Ball, T.; Wang, Y.; Srihari, D.; Kaushal, M.; Dietz, H.; Zhang, X.; et al. Whole Exome Sequencing Reveals the Maintained Polyclonal Nature from Primary to Metastatic Malignant Peripheral Nerve Sheath Tumor in Two Patients with NF1. Neuro-Oncol. Adv. 2020, 2, i75–i84. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, I.; Yamada, Y.; Kohashi, K.; Yamamoto, H.; Iwasaki, T.; Ishihara, S.; Toda, Y.; Ito, Y.; Susuki, Y.; Kawaguchi, K.; et al. Frequent MN1 Gene Mutations in Malignant Peripheral Nerve Sheath Tumor. Anticancer Res. 2020, 40, 6221–6228. [Google Scholar] [CrossRef]
- Feber, A.; Wilson, G.A.; Zhang, L.; Presneau, N.; Idowu, B.; Down, T.A.; Rakyan, V.K.; Noon, L.A.; Lloyd, A.C.; Stupka, E.; et al. Comparative Methylome Analysis of Benign and Malignant Peripheral Nerve Sheath Tumors. Genome Res. 2011, 21, 515–524. [Google Scholar] [CrossRef]
- Bhunia, M.M.; Stehn, C.M.; Jubenville, T.A.; Novacek, E.L.; Larsson, A.T.; Madala, M.; Suppiah, S.; Velez-Reyes, G.L.; Williams, K.B.; Sokolowski, M.; et al. Multiomic Analyses Reveal New Targets of Polycomb Repressor Complex 2 in Schwann Lineage Cells and Malignant Peripheral Nerve Sheath Tumors. Neuro-Oncol. Adv. 2024, 6, vdae188. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Czwan, E.; Hartmann, W.; Penzel, R.; Brors, B.; Eils, R.; Wardelmann, E.; Büttner, R.; Lichter, P.; Schirmacher, P.; et al. MicroRNA Profiling of Primary High-grade Soft Tissue Sarcomas. Genes Chromosomes Cancer 2012, 51, 982–996. [Google Scholar] [CrossRef]
- Amirnasr, A.; Verdijk, R.M.; Van Kuijk, P.F.; Kartal, P.; Vriends, A.L.M.; French, P.J.; Van Royen, M.E.; Taal, W.; Sleijfer, S.; Wiemer, E.A.C. Deregulated microRNAs in Neurofibromatosis Type 1 Derived Malignant Peripheral Nerve Sheath Tumors. Sci. Rep. 2020, 10, 2927. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.F.; Tycksen, E.D.; Sandell, L.J.; Brophy, R.H. Advantages of RNA-seq Compared to RNA Microarrays for Transcriptome Profiling of Anterior Cruciate Ligament Tears. J. Orthop. Res. 2018, 36, 484–497. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Bagchi, A.; Dhanda, S.K.; Dunphy, P.; Sioson, E.; Robinson, G.W. Molecular Classification Improves Therapeutic Options for Infants and Young Children With Medulloblastoma. J. Natl. Compr. Cancer Netw. 2023, 21, 1097–1105. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Steele, C.D.; Piculell, K.; Al-Ibraheemi, A.; Eulo, V.; Bui, M.M.; Chatzipli, A.; Dickson, B.C.; Borcherding, D.C.; Feber, A.; et al. Genomic Patterns of Malignant Peripheral Nerve Sheath Tumor (MPNST) Evolution Correlate with Clinical Outcome and Are Detectable in Cell-Free DNA. Cancer Discov. 2023, 13, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, D.C.; Amin, N.V.; He, K.; Zhang, X.; Lyu, Y.; Dehner, C.; Bhatia, H.; Gothra, A.; Daud, L.; Ruminski, P.; et al. MEK Inhibition Synergizes with TYK2 Inhibitors in NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2023, 29, 1592–1604. [Google Scholar] [CrossRef]
- Bottillo, I.; Ahlquist, T.; Brekke, H.; Danielsen, S.A.; Van Den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and Somatic NF1 Mutations in Sporadic and NF1-associated Malignant Peripheral Nerve Sheath Tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Magallón-Lorenz, M.; Fernández-Rodríguez, J.; Terribas, E.; Creus-Batchiller, E.; Romagosa, C.; Estival, A.; Perez Sidelnikova, D.; Salvador, H.; Villanueva, A.; Blanco, I.; et al. Chromosomal Translocations Inactivating CDKN2A Support a Single Path for Malignant Peripheral Nerve Sheath Tumor Initiation. Hum. Genet. 2021, 140, 1241–1252. [Google Scholar] [CrossRef]
- Banerjee, J.; Allaway, R.J.; Taroni, J.N.; Baker, A.; Zhang, X.; Moon, C.I.; Pratilas, C.A.; Blakeley, J.O.; Guinney, J.; Hirbe, A.; et al. Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways That Define Tumor Heterogeneity in NF1. Genes 2020, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Brockman, Q.R.; Rytlewski, J.D.; Milhem, M.; Monga, V.; Dodd, R.D. Integrated Epigenetic and Transcriptomic Analysis Identifies Interleukin 17 DNA Methylation Signature of Malignant Peripheral Nerve Sheath Tumor Progression and Metastasis. JCO Precis. Oncol. 2024, 8, e2300325. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, S.A.; Lind, G.E.; Kolberg, M.; Høland, M.; Bjerkehagen, B.; Sundby Hall, K.; Van Den Berg, E.; Mertens, F.; Smeland, S.; Picci, P.; et al. Methylated RASSF1A in Malignant Peripheral Nerve Sheath Tumors Identifies Neurofibromatosis Type 1 Patients with Inferior Prognosis. Neuro-Oncology 2015, 17, 63–69. [Google Scholar] [CrossRef]
- Röhrich, M.; Koelsche, C.; Schrimpf, D.; Capper, D.; Sahm, F.; Kratz, A.; Reuss, J.; Hovestadt, V.; Jones, D.T.W.; Bewerunge-Hudler, M.; et al. Methylation-Based Classification of Benign and Malignant Peripheral Nerve Sheath Tumors. Acta Neuropathol. 2016, 131, 877–887. [Google Scholar] [CrossRef]
- Tsuchiya, R.; Yoshimatsu, Y.; Noguchi, R.; Sin, Y.; Ono, T.; Akiyama, T.; Kosako, H.; Yoshida, A.; Ohtori, S.; Kawai, A.; et al. Integrating Analysis of Proteome Profile and Drug Screening Identifies Therapeutic Potential of MET Pathway for the Treatment of Malignant Peripheral Nerve Sheath Tumor. Expert Rev. Proteom. 2023, 20, 109–119. [Google Scholar] [CrossRef]
- Jia, X.; Chen, C.; Chen, L.; Yu, C.; Kondo, T. Decorin as a Prognostic Biomarker in Patients with Malignant Peripheral Nerve Sheath Tumors. Oncol. Lett. 2019. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Zhao, L.; Wu, Y.; Veeravalli, V.; Alt, J.; Aguilar, J.M.H.; Dash, R.P.; Lam, J.; Tenora, L.; Rodriguez, C.; et al. The Novel Glutamine Antagonist Prodrug JHU395 Has Antitumor Activity in Malignant Peripheral Nerve Sheath Tumor. Mol. Cancer Ther. 2020, 19, 397–408. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Ali, E.S.; Krecmerova, M.; Aguilar, J.M.H.; Alt, J.; Peters, D.E.; Zhao, L.; Wu, Y.; Nuha, N.; Asara, J.M.; et al. Pro-905, a Novel Purine Antimetabolite, Combines with Glutamine Amidotransferase Inhibition to Suppress Growth of Malignant Peripheral Nerve Sheath Tumor. Mol. Cancer Ther. 2023, 22, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The Genomic Landscape of Malignant Peripheral Nerve Sheath Tumors: Diverse Drivers of Ras Pathway Activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, W.; Li, Y.; Li, H.; Guo, Z.; Wei, C.; Long, M.; Chung, M.; Aimaier, R.; Li, Q.; et al. Preclinical Assessment of MEK Inhibitors for Malignant Peripheral Nerve Sheath Tumors Reveals Differences in Efficacy and Adaptive Response. Front. Oncol. 2022, 12, 903177. [Google Scholar] [CrossRef]
- Wang, J.; Pollard, K.; Calizo, A.; Pratilas, C.A. Activation of Receptor Tyrosine Kinases Mediates Acquired Resistance to MEK Inhibition in Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2021, 81, 747–762. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Lingo, J.J.; Kaemmer, C.A.; Scherer, A.; Warrier, A.; Voigt, E.; Raygoza Garay, J.A.; McGivney, G.R.; Brockman, Q.R.; Tang, A.; et al. CDK4/6-MEK Inhibition in MPNSTs Causes Plasma Cell Infiltration, Sensitization to PD-L1 Blockade, and Tumor Regression. Clin. Cancer Res. 2023, 29, 3484–3497. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pollard, K.; Allen, A.N.; Tomar, T.; Pijnenburg, D.; Yao, Z.; Rodriguez, F.J.; Pratilas, C.A. Combined Inhibition of SHP2 and MEK Is Effective in Models of NF1-Deficient Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2020, 80, 5367–5379. [Google Scholar] [CrossRef]
- Huang, P.-Y.; Shih, I.-A.; Liao, Y.-C.; You, H.-L.; Lee, M.-J. A Novel HDAC11 Inhibitor Potentiates the Tumoricidal Effects of Cordycepin against Malignant Peripheral Nerve Sheath Tumor through the Hippo Signaling Pathway. Am. J. Cancer Res. 2022, 12, 873–892. [Google Scholar]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic Survival in Resistance to Histone Deacetylase Inhibitors: Novel Strategies to Treat Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Bill, K.L.J.; Bid, H.K.; Braggio, D.; Constantino, D.; Prudner, B.; Zewdu, A.; Batte, K.; Lev, D.; Pollock, R.E. HDAC8, A Potential Therapeutic Target for the Treatment of Malignant Peripheral Nerve Sheath Tumors (MPNST). PLoS ONE 2015, 10, e0133302. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Pharmacoepigenomics. In Medical Epigenetics; Elsevier: Amsterdam, The Netherlands, 2016; pp. 585–617. ISBN 978-0-12-803239-8. [Google Scholar]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic Significance of AKT/mTOR and MAPK Pathways and Antitumor Effect of mTOR Inhibitor in NF1-Related and Sporadic Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.-O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in Vivo Targeting of the Mammalian Target of Rapamycin Pathway in Malignant Peripheral Nerve Sheath Tumors. Mol. Cancer Ther. 2008, 7, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Stanton, L.J.; Ge, L.; Sta Ana, S.; Chrisinger, J.; Titus, S.; Labadie, B.W.; May, M.S.; Lyu, Y.; Sender, N.; et al. A Phase II Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Malignant Peripheral Nerve Sheath Tumors. JCO 2024, 42, 11565. [Google Scholar] [CrossRef]
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef]
- Larsson, A.T.; Bhatia, H.; Calizo, A.; Pollard, K.; Zhang, X.; Conniff, E.; Tibbitts, J.F.; Rono, E.; Cummins, K.; Osum, S.H.; et al. Ex Vivo to in Vivo Model of Malignant Peripheral Nerve Sheath Tumors for Precision Oncology. Neuro-Oncology 2023, 25, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Kivlin, C.M.; Watson, K.L.; Al Sannaa, G.A.; Belousov, R.; Ingram, D.R.; Huang, K.-L.; May, C.D.; Bolshakov, S.; Landers, S.M.; Kalam, A.A.; et al. Poly (ADP) Ribose Polymerase Inhibition: A Potential Treatment of Malignant Peripheral Nerve Sheath Tumor. Cancer Biol. Ther. 2016, 17, 129–138. [Google Scholar] [CrossRef]
- Patwardhan, P.P.; Surriga, O.; Beckman, M.J.; De Stanchina, E.; Dematteo, R.P.; Tap, W.D.; Schwartz, G.K. Sustained Inhibition of Receptor Tyrosine Kinases and Macrophage Depletion by PLX3397 and Rapamycin as a Potential New Approach for the Treatment of MPNSTs. Clin. Cancer Res. 2014, 20, 3146–3158. [Google Scholar] [CrossRef]
- Somatilaka, B.N.; Madana, L.; Sadek, A.; Chen, Z.; Chandrasekaran, S.; McKay, R.M.; Le, L.Q. STING Activation Reprograms the Microenvironment to Sensitize NF1-Related Malignant Peripheral Nerve Sheath Tumors for Immunotherapy. J. Clin. Investig. 2024, 134, e176748. [Google Scholar] [CrossRef]
- Borcherding, D.; Zhang, X.; Zhang, M.; Bhatia, H.; Lyu, Y.; He, K.; Yang, L.; Yang, K.; DiBenedetto, H.; Tsai, A.; et al. EXTH-63. MTA-COOPERATIVE PRMT5 INHIBITORS ARE EFFICACIOUS IN MTAP-DELETED MALIGNANT PERIPHERAL NERVE SHEATH TUMOR MODELS. Neuro-Oncology 2023, 25, v238. [Google Scholar] [CrossRef]
- Somaiah, N.; Van Tine, B.A.; Chmielowski, B.; Drabick, J.J.; Chawla, S.P.; McKean, M.; Spira, A.I.; O’Byrne, K.J.; Foresto, S.A.; Movva, S.; et al. A Phase 2 Study of Alrizomadlin, a Novel MDM2/P53 Inhibitor, in Combination with Pembrolizumab for Treatment of Patients with Malignant Peripheral Nerve Sheath Tumor (MPNST). JCO 2023, 41, e14627. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, K.; Yang, K.; Hirbe, A.C. A Sequencing Overview of Malignant Peripheral Nerve Sheath Tumors: Findings and Implications for Treatment. Cancers 2025, 17, 180. https://doi.org/10.3390/cancers17020180
Xiao K, Yang K, Hirbe AC. A Sequencing Overview of Malignant Peripheral Nerve Sheath Tumors: Findings and Implications for Treatment. Cancers. 2025; 17(2):180. https://doi.org/10.3390/cancers17020180
Chicago/Turabian StyleXiao, Kangwen, Kuangying Yang, and Angela C. Hirbe. 2025. "A Sequencing Overview of Malignant Peripheral Nerve Sheath Tumors: Findings and Implications for Treatment" Cancers 17, no. 2: 180. https://doi.org/10.3390/cancers17020180
APA StyleXiao, K., Yang, K., & Hirbe, A. C. (2025). A Sequencing Overview of Malignant Peripheral Nerve Sheath Tumors: Findings and Implications for Treatment. Cancers, 17(2), 180. https://doi.org/10.3390/cancers17020180