1. Introduction
High-risk neuroblastoma is an aggressive pediatric malignancy of the sympathetic nervous tissue characterized by poor prognosis and few effective therapeutic options. The disialoganglioside GD2 is highly expressed by neuroblastoma cells with limited presence in normal tissues, making it an ideal target for immunotherapy. Monoclonal antibodies (mAbs) targeting GD2 have become an integral part of treatment protocols and significantly contributed to improving survival rates in patients with high-risk neuroblastoma [
1].
mAbs have emerged as a cornerstone in cancer therapy and represent one of the fastest-growing classes of biological therapeutics. Their Fc (fragment crystallizable region, Fc) part of the antibody facilitates effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [
2,
3]. Additionally, mAbs exhibit favorable biophysical properties such as high conformational stability and solubility, enabling formulation and extended storage with minimal aggregation [
4,
5].
To date, there are three anti-GD2 mAbs approved for the treatment of high-risk neuroblastoma including dinutuximab (Unituxin
®), dinutuximab beta (Qarziba
®, DB), and naxitamab (Danyelza
®, NAXI). Dinutuximab and DB share the protein sequence of ch14.18, and both are human/mouse chimeric mAbs but differ in the glycosylation pattern. NAXI is a humanized derivative of mAb 3F8. All three anti-GD2 antibodies are of human IgG1 isotype. However, one key difference between NAXI versus DB and dinutuximab is the binding affinity to GD2, with DB showing an intermediate and NAXI exhibiting an approximately 10-fold higher binding affinity to GD2 [
2,
6].
Recent research indicated that increasing antibody affinity via engineering strategies can enhance ADCC [
7]. High affinity also promotes improved antibody internalization, which is particularly beneficial in antibody–drug conjugates [
8]. However, other studies showed that high affinity can lead to the bivalent binding of antibodies to their target and therefore reduce the available number of antibodies per antigen [
9]. In contrast, moderate-affinity antibodies with faster off-rates may bind monovalently, increasing Fc density on target cells and thereby enhancing immune engagement [
9,
10]. Moreover, antibodies may be affected by the antigen sink effect, also known as target-mediated drug disposition (TMDD) [
11,
12,
13]. This phenomenon occurs when antibodies bind strongly to abundant target antigens, leading to rapid internalization and degradation of the antibody–antigen complex within cells. As a result, high-affinity antibodies can exhibit nonlinear pharmacokinetics characterized by rapid clearance from the systemic circulation, especially at lower doses [
12]. In this context, the role of the affinity difference between DB and NAXI has not been investigated yet.
Moreover, the clinical efficacy of DB and NAXI in patients with high-risk neuroblastoma was reported in separate clinical trials [
1,
14], and there is no trial so far testing both antibodies in a head-to-head comparison. However, the differences in the patient populations of the reported clinical studies make it difficult to compare the reported outcome data. In this context, we investigated a head-to-head comparison of the intermediate affinity anti-GD2 antibody DB and the higher affinity antibody NAXI using preclinical neuroblastoma spheroid models of varying GD2 expression levels. We determined the capability of both antibodies to mediate cellular and complement-dependent cytotoxicity and show the impact of binding affinity on efficacy and its connection to the TMDD effect.
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
Peripheral blood samples were provided by the Department of Transfusion Medicine, University Medicine Greifswald. All experimental protocols involving human participants were reviewed and approved by the Ethics Committee of the Medical Faculty, University Medicine Greifswald, Germany (approval code BB 014/14; approval date 24 January 2014). Written informed consent was obtained from every participant prior to sample collection. All methods were carried out in accordance with the Declaration of Helsinki (1964 and its later amendments) and with the relevant institutional and national regulations and guidelines.
2.2. Antibodies
NAXI was purchased from Y-mAbs Therapeutics, Inc. (New York, NY, USA). DB was obtained from EUSA Pharma (Hemel Hempstead, UK) and rituximab from Roche (Basel, Switzerland).
2.3. Cell Culture
The human neuroblastoma cells were cultured in respective media under standard cell culture conditions (37 °C/5% CO2). LAN-1 cells were cultured in RPMI (PAN BIOTECH, Aidenbach, Germany, P04–16520) supplemented with 10% FCS and 100 U/mL penicillin and 0.1 mg/mL streptomycin (1× P/S; PAN BIOTECH, P06–07100). CHLA-20, COG-N-440, and COG-N-519 cells were cultured in IMDM (PAN BIOTECH, P04–20250) supplemented with 4 mM stable glutamine, 20% FCS, 1× Insulin–Transferrin–Selenium (ITS) solution I (PAN-BIOTECH; P07-03110) and 1× P/S. SHEP-2, SK-N-SH, and COG-N-519 cells were cultured using MEM Eagle (PAN BIOTECH, P04-08056) supplemented with 10% FCS and 1× P/S. To detect potential mycoplasma contamination, all cell lines were regularly analyzed using a MYCOALERT detection Kit (Lonza Cologne GmbH, Cologne, Germany, LT07–318); only mycoplasma-negative cells were utilized for experiments. For ADCC experiments, human peripheral blood mononuclear cells (PBMCs) were separated from whole blood concentrates without serum of healthy donors using the Pancoll separating method (human, density 1.077 g/mL, BIOTECH, P04-60500) and cultured for 72 h in RPMI supplemented with 10% FCS, 50 µM, 100 IU/mL IL-2, β-mercaptoethanol, and 1× P/S. LAN-1 and SK-N-SH were commercially obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and the other cell lines were provided by the childhood cancer repository (Texas Tech University Health Sciences Center, Lubbock, TX, USA).
2.4. Lentiviral Transduction
For the generation of neuroblastoma tumor cells expressing near-infrared reporter protein (iRFP-680) used as a viability marker, a second-generation lentiviral vector system was utilized. To produce recombinant lentivirus, CalPhos Mammalian Transfection Kit (Takara Bio Europe, Saint-Germain-en-Laye, France) was used according to the manufacturer’s protocol for co-transfection of Lenti-X™ 293T cells with purified pVSV-G envelope expressing plasmid (Addgene, Watertown, MA, USA), psPAX2 (Addgene, USA) vector encoding virus polymerase and packaging genes, and lentiviral vector pWPXL (Addgene, USA) encoding for iRFP-680. Tumor cells (target cells) were transduced using 8 mg/mL Polybrene (Merck, Burlington, MA, USA) and virus-containing supernatants. After 24 h, medium was replaced by a fresh standard culture medium, and after 72 h, target cells were checked for iRFP-680 fluorescence using the IncuCyte® SX5 live-cell analysis system (Sartorius, Göttingen, Germany).
2.5. Long-Term Live-Cell Spheroid Viability Assay
Tumor spheroids were established by seeding 3000 iRFP-680-expressing tumor cells per well into a 384 ultra-low attachment plate (S-BIO, Hudson, NH, USA, PrimeSurface®, MS-90384UZ), centrifuged at 150× g for 10 min and cultured under cell culture conditions. After 72 h, tumor spheroids were deployed for experiments using the IncuCyte® SX5 live-cell analysis system (Sartorius, Germany) with image acquisitions every 8 h. Viability was determined as the ratio of integrated spheroid fluorescence intensity of every time point and fluorescence at baseline (0 h). The area under the curve for each time period was calculated using the following formula: ((viability-time point 1 (t1) (%) + viability t2)/2) × (t2 − t1). Experiments were performed in six technical replicates, and viability is given in % ± SEM.
2.6. ADCC and CDC Live-Cell Viability Assay
To determine antibody-dependent cytotoxicity against neuroblastoma spheroids in a long-term live-cell spheroid viability assay, 75.000 effector cells (PBMCs) per well, as well as respective therapeutic antibody in a concentration range of 10−5–1 µg/mL, were added to the tumor spheroids (ADCC) and cultured in a total volume of 200 µL for 168 h (7 days) under cell culture conditions. PBMCs were taken from at least five different donors to ensure accuracy and reliability of the results.
In complement-dependent cytotoxicity experiments, 12.5% active human serum was used instead of PBMCs. To assess reproducibility and account for donor-specific variability in complement activity, independent experiments were performed using sera from multiple donors.
Tumor spheroids were only treated with PBMCs alone (antibody-independent cellular cytotoxicity, AICC) or heat-inactivated serum alone without therapeutic antibodies, and untreated tumor spheroids (medium) served as controls. To determine the impact of soluble target antigen, we added 100 nM and 1000 nM sGD2 (Merck, Burlington, MA, USA, 345743-500UG) in addition to the ADCC setting.
2.7. CDC Calcein–AM Assay
CDC was assessed using a calcein–acetoxymethyl ester (AM)-based cytotoxicity assay as published [
15]. In brief, 5000 calcein–AM-labeled, GD2-positive neuroblastoma cells were incubated with patient serum at a final concentration of 12.5% for 4 h. Heat-inactivated serum alone (i.e., without leukocytes) served as the negative control for CDC. After the incubation period, cytotoxicity was quantified by measuring the calcein fluorescence in the supernatant at excitation and emission wavelengths of 495 nm and 515 nm, respectively.
2.8. Analysis of GD2 Expression by Neuroblastoma Cells by Flow Cytometry
To determine GD2 expression by flow cytometry, iRFP-680-expressing neuroblastoma cells were harvested at 80–100% confluency. A total of 1 × 106 cells per sample were washed in washing buffer (1× PBS without Ca and Mg (CAPRICORN SCIENTIFIC, Ebsdorfergrund, Germany, PBS-1A) supplemented with 2% BSA and 1 mM EDTA; 300× g, 5 min) and incubated with PE-conjugated anti-GD2 antibody (BioLegend, San Diego, CA, USA, #357304, mouse IgG2a, κ, clone 14G2a, 1:80) for 20 min at 4 °C in the dark. After removing unbound antibodies by centrifugation (300× g, 5 min), cells were resuspended in washing buffer and analyzed using a BD CANTO II flow cytometer and FACS Diva software V10.10.0 (BD Biosciences, Heidelberg, Germany). A total of 20,000 live cells were analyzed by default. Samples stained with PE-labeled mouse IgG2a, κ isotype control antibody (BioLegend, #400214) were used to detect unspecific binding; unstained samples served as control.
2.9. Assessing Loss of Anti-GD2 Antibody Binding Activity by Pre-Incubation with GD2-Positive Dead Neuroblastoma Cells Using Flow Cytometry
To compare the off-rate characteristics of NAXI and DB, we exposed both antibodies to GD2-positive dead neuroblastoma cells before staining viable LAN-1 or CHLA-20 neuroblastoma cells. For this, neuroblastoma cells were cultured until 80–100% confluency, harvested, and washed using washing buffer (300× g, 5 min). To kill the neuroblastoma cells, 0.5 × 106/mL cells were exposed to 65 °C for 5 min followed by 4 °C for 5 min (dead cells were identified with DAPI staining). Dead cells were then incubated with either NAXI or DB (10–0.001 µg/mL) for 60 min at 4 °C in a reaction volume of 1000 µL. After centrifugation (300× g, 5 min), the supernatants were used for the staining of live cells (0.5 × 106, 20 min at 4 °C) (samples 1). In parallel, 0.5 × 106 live cells were stained with NAXI or DB (10–0.001 µg/mL) for 20 min at 4 °C (sample 2). After washing cells (washing buffer, 300× g, 5 min), the secondary antibody was incubated (Alexa Fluor 488-conjugated anti-human IgG1, BioLegend, #410706, rat IgG2a, κ, clone M1310G05, application 1:20 in 100 µL incubation volume, 20 min, 4 °C). Cells were washed again (washing buffer, 300× g, 5 min) and resuspended in washing buffer for the detection of the mean fluorescence intensity (MFI) of GD2 by flow cytometry, as described above. Tumor cells stained with the secondary antibody only, as well as unstained cells, served as negative controls. The loss of fluorescence intensity is given as fold decrease calculated as the ratio of the gMFI GD2 of live cells (sample 2) and live cells stained with supernatant obtained following pre-incubation with dead cells (sample 1). Experiments were performed in at least four biological replicates; values are given in fold change ± SEM.
2.10. Assessing Loss of Anti-GD2 Antibody Binding Activity in Presence of Soluble GD2 Using Flow Cytometry
To assess the off-rate characteristics of NAXI and DB in presence of sGD2, NAXI and DB were used in a competitive binding assay to CHLA-20 tumor cells in addition of 10, 100, and 1000 nM sGD2. For this, NAXI or DB was diluted in washing buffer in a concentration range from 0.001 to 1 µg/mL. GD2 (Merck, CAS# 65988-71-8) was diluted in methanol (Carl Roth GmbH + Co. KG, Karlsruhe, Germany, # 4627.5) at 10, 100, and 1000 nM. First, 0.001–1 µg/mL antibody and 10, 100, and 1000 nM GD2 were incubated for 30 min in a reaction volume of 1000 µL. Antibody solution added with methanol only (without GD2) as well as washing buffer, and methanol (without antibody and GD2) served as control. Second, CHLA-20 neuroblastoma cells were harvested at a confluency of 80–100% and washed using washing buffer (300× g, 5 min). A total of 0.5 × 106 live neuroblastoma cells resuspended in 50 µL of washing buffer were added to the GD2 antibody solution and controls. After incubation for 20 min at 4 °C, samples were centrifuged (300× g, 5 min), and the supernatant was removed, followed by another washing step (washing buffer, 300× g, 5 min). Then, samples were incubated with the secondary antibody (Alexa Fluor 488-conjugated anti-human IgG1, BioLegend, #410706, rat IgG2a, κ, clone M1310G05, application 1:20 in 100 µL incubation volume, 20 min, 4 °C). After washing (washing buffer, 300× g, 5 min), cells were resuspended in washing buffer for cytometric analysis of GD2 gMFI, as described above. Tumor cells stained with the secondary antibody only, as well as unstained cells, served as negative controls. The loss of fluorescence intensity is given as fold decrease calculated as the ratio of the gMFI GD2 of live cells and live cells stained with GD2 antibody solution. Experiments were performed in at least four biological replicates; values are given in fold change ± SEM.
2.11. Antibody-Internalization Assay Using Live-Cell Imaging
To measure antibody internalization in relation to binding affinity and concentration of the therapeutic antibodies, we established an internalization assay using NAXI or DB tagged with a pH-sensitive dye and the live-cell imaging method (IncuCyte
®). First, a desalting and buffer exchange procedure of both antibodies was performed to remove low molecular weight components and primary amines, which may inhibit protein labeling. For this, Zeba Spin Desalting Columns (ThermoFisher Scientific, Waltham, MA, USA, #89889) were used according to the manufacturer’s instructions. Secondly, antibody labeling was conducted using a pHrodo™ iFL Red Antibody Labeling Kit (ThermoFisher Scientific, # P36021) followed by the measurement of protein contraction and degree of labeling (DOL) according to the manufacturer’s protocol to obtain a covalently attached fluorogenic pH probe. DOL was calculated according to the following formula:
The absorbance was measured using a ThermoScientific Varioskan™ LUX plate reader at 280 nm for protein concentration and 560 nm for the label dye as per the manufacturer’s instructions. The degree of labeling of DB and NAXI was similar with 2.27 and 2.23, respectively, rendering corrections for DOL unnecessary (correction factor = 1.02).
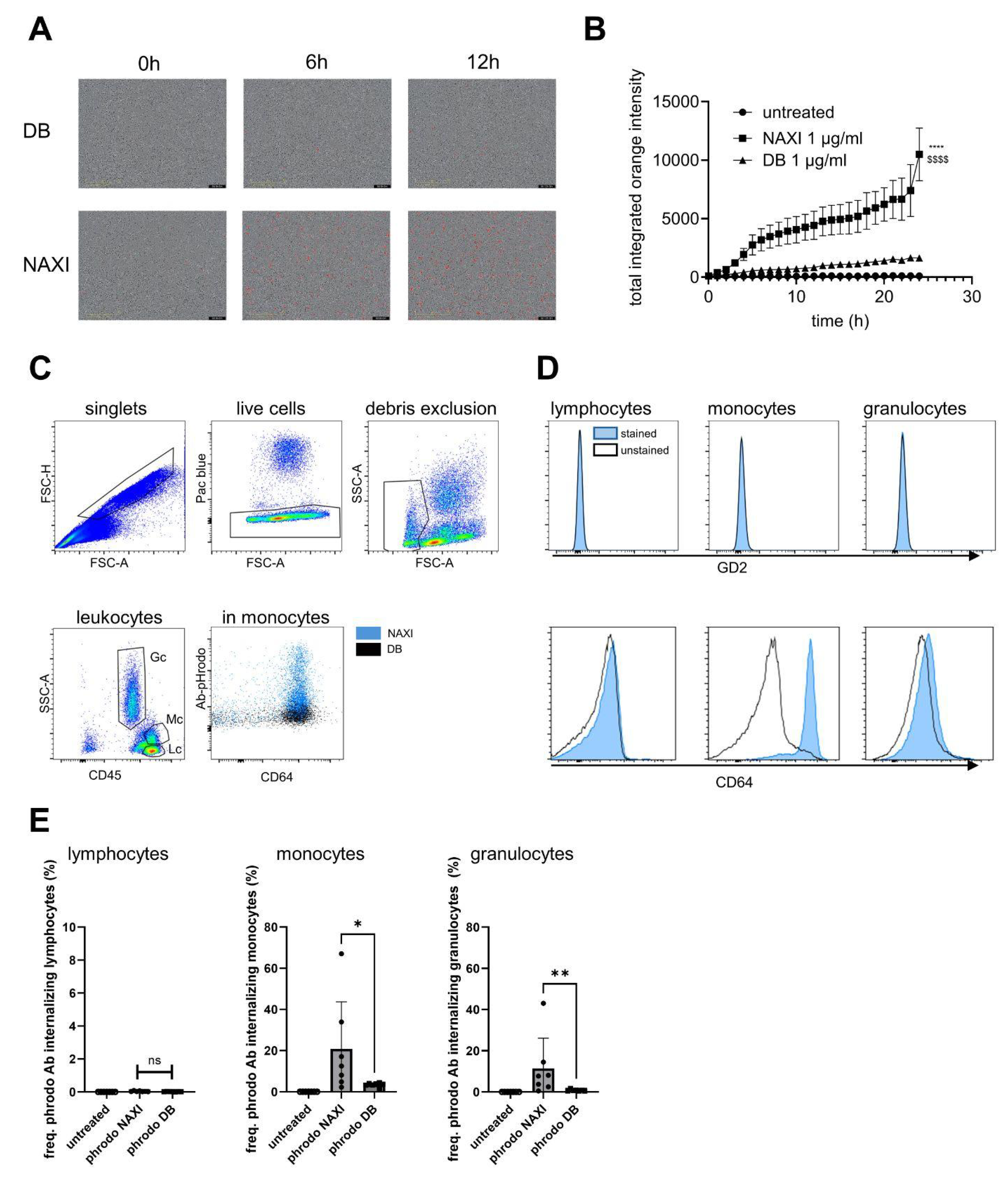
For the antibody internalization assay, 10,000 tumor cells or 100,000 PBMCs per well were seeded into a 96-well plate (Corning, New York, NY, USA, 3595) and cultured for 24 h. Then, pHrodo™ iFL Red-labeled NAXI or DB in a concentration from 1 to 0.001 µg/mL diluted in standard culture medium were added to the wells. A total of 1 µg/mL rituximab and non-treated cells served as controls. Antibody internalization was assessed using the IncuCyte® SX5 live-cell analysis system with image acquisitions every 2 h. Antibody internalization was calculated as a ratio of integrated fluorescence intensity of every time point and fluorescence at baseline (0 h). Values are shown as areas under the total integrated red fluorescence intensity curve ± SEM.
2.12. Measurement of Antibody Internalization by Leukocytes Using Flow Cytometry
To measure antibody internalization by leukocytes, 100 µL of fresh whole blood collected in heparin tubes were incubated with 1 µg of pHrodo-labeled NAXI or DB (detection equivalent to PE) for 4 h under cell culture conditions. For immunostaining, 20 µL of anti-human FcR blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany, #130-059-901) was added per sample and incubated for 10 min at 4 °C, followed by adding of APC/Vio770-labeled anti-human CD45 antibody (Miltenyi Biotec #130-110-635, human IgG1, REA 747, 1:50) and APC-conjugated anti-human CD64 antibody (BioLegend #399510, mouse IgG1κ, clone S18012C, 1:40). After incubation for 30 min at 4 °C in the dark, samples were washed (4 mL of washing buffer) and centrifuged at 300× g for 5 min, and the supernatant was aspirated. To remove red blood cells, 2 mL of erythrocyte lysis buffer was added, gently mixed, and incubated for 10 min at room temperature. After adding 2 mL of washing buffer, samples were again centrifuged (300× g for 5 min), and the supernatant was discarded. For flow cytometry measurement, the cell pellet was resuspended using 500 µL of washing buffer. A total of 0.1 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) was added 5 min before acquisition to exclude dead cells from analysis. Flow cytometry analysis was performed as described above.
To investigate the expression of GD2 by leukocytes, whole blood samples without addition of pHrodo-labeled antibody were stained with anti-human CD45-APC/Vio770, anti-human CD64-APC (see above), and PE-conjugated anti-GD2 antibody (BioLegend, #357304, mouse IgG2aκ, clone 14G2a, 1:80) in the manner described above.
2.13. Statistics
Statistical analyses were conducted using GraphPad version 10.4.1. Normality was assessed before further analysis. For comparisons between two groups, a two-sided t-test was used, while one-way ANOVA was used for comparisons involving more than two groups, followed by the appropriate post hoc test. Data are presented as mean ± SEM or as fold change compared to untreated control ± SEM.
4. Discussion
We evaluated the functional potency of two anti-GD2 antibodies approved for the treatment of high-risk neuroblastoma, NAXI and DB. Although the constant regions of both antibodies were engineered to be of human IgG1 isotype, there are substantial structural differences between both molecules. NAXI is a humanized variant of the murine anti-GD2 antibody 3F8 created by CDR grafting, whereas DB is a human–mouse chimeric variant of the murine anti-GD2 antibody 14.G2A. Although both antibodies are produced in Chinese Hamster Ovary (CHO) cells, suggesting similar post-translational glycosylation patterns, the protein sequences of both molecules are completely different, reflected by a ten-fold higher binding affinity of NAXI to GD2 compared to DB, which influences their function [
2].
Our study demonstrates a higher ADCC potency of DB compared to NAXI (
Figure 1) attributed to a stronger TMDD effect for NAXI compared to DB (
Figure 2,
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8), a phenomenon that has been described for high- versus intermediate-affinity antibodies. The enhancement of the binding affinity of a mAb can lead to trade-offs with other interdependent properties [
4], including an increased susceptibility for the antigen sink effect, also known as TMDD [
11]. This phenomenon occurs when antibodies bind strongly to abundant target antigens, leading to rapid internalization and degradation of the antibody–antigen complex within cells [
11,
12,
18]. TMDD is also driven by additional mechanisms linked to elevated affinity, such as the binding of soluble target antigens or an increased tendency for the antibody to remain bound, thereby reducing its potential for re-engagement with target cells. As a result, high-affinity antibodies can exhibit nonlinear pharmacokinetics characterized by rapid clearance from the systemic circulation, especially at lower doses [
19]. The TMDD effect is more pronounced in conditions with high antigen expression, such as neuroblastoma tumors overexpressing GD2, and shedding of the tumor antigen increases the TMDD effect of a high-affinity antibody [
19]. A high sGD2 serum concentration of up to 1000 nM was reported for neuroblastoma patients [
17]. In general, intermediate-affinity antibodies are less affected by internalization and degradation, maintain a more stable concentration, and are therefore available to engage immune effector cells more efficiently compared to high-affinity antibodies [
11,
13,
20]. Furthermore, intermediate-affinity antibodies exhibit faster off-rates and may predominantly bind antigens monovalently, thus increasing Fc density on target cells and enhancing immune engagement [
9,
10,
13,
21]. In contrast, high-affinity antibodies, which typically bind bivalently (i.e., using both Fab arms), can reduce Fc accessibility and consequently attenuate ADCC responses when compared to intermediate-affinity counterparts [
9,
10,
13].
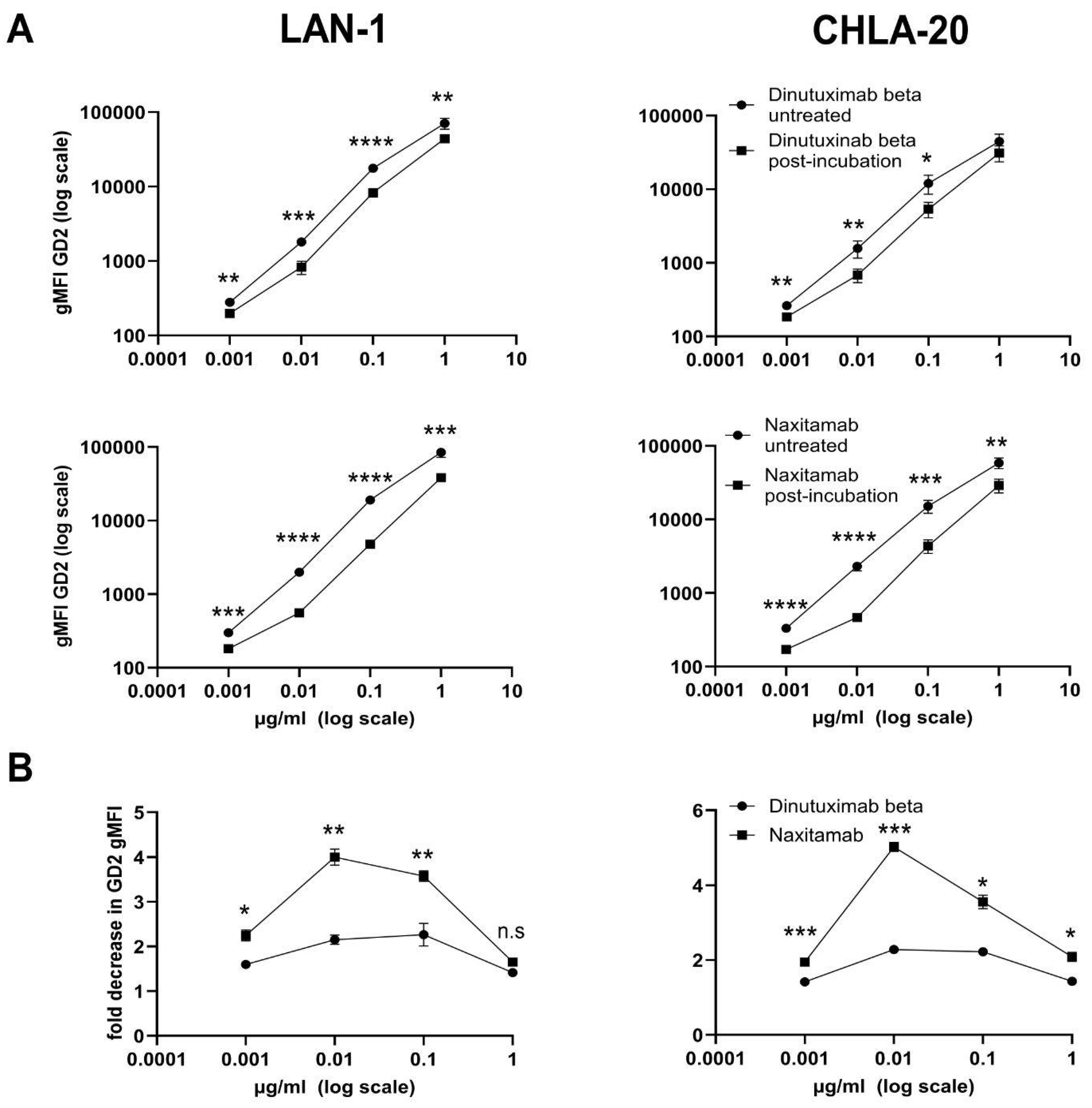
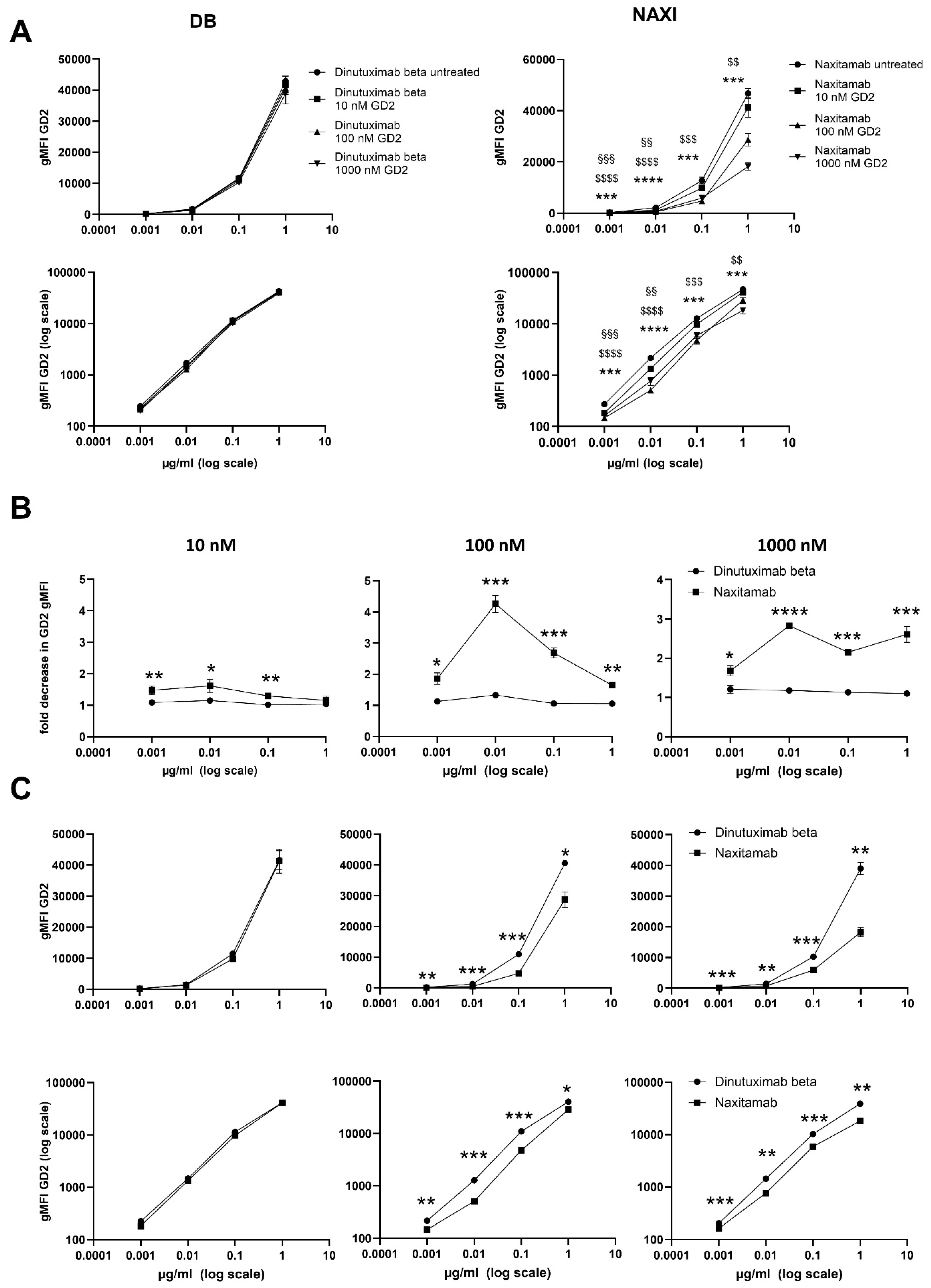
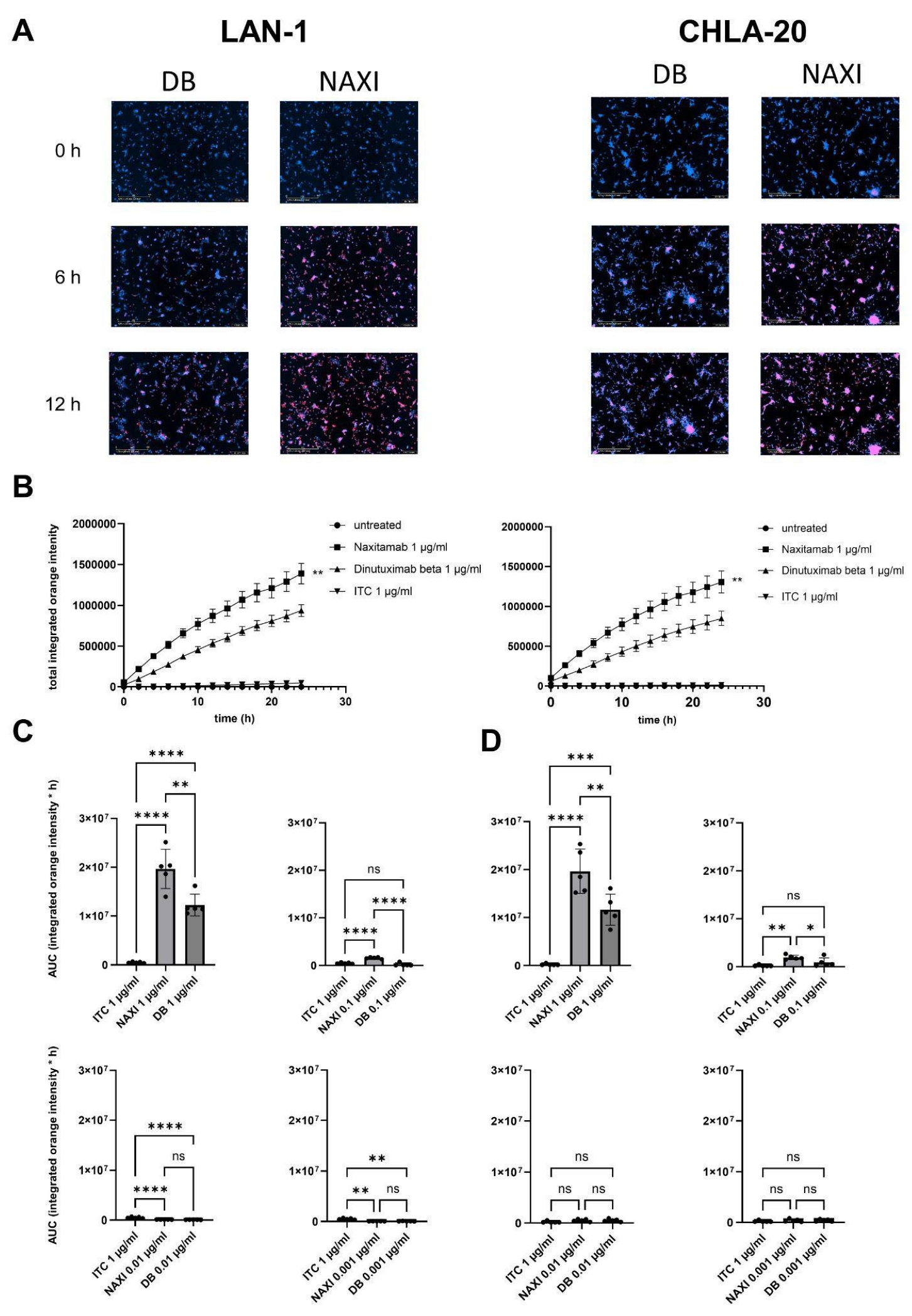
We could demonstrate a clear role for TMDD likely attributed to the higher affinity of NAXI compared to DB to off-viable tumor GD2 (
Figure 4 and
Figure 5) as well as to a more rapid internalization of NAXI compared to DB (
Figure 6 and
Figure 7). This leads to a lower amount of antibody available for GD2 binding to on-viable tumor after exposure to dead GD2-expressing tumor cells (
Figure 4), soluble GD2 (
Figure 5), or following internalization into tumor- (
Figure 6) or immune cells (
Figure 7). The extent of this effect was dependent on the tumor spheroid model and most pronounced at lower antibody concentrations (
Figure 1 and
Figure 5). The internalization may also contribute to the rapid clearance of the antibody from the tumor microenvironment, further exacerbating the TMDD effect and diminishing its ability to mediate ADCC in vivo. Interestingly, even leukocytes, particularly monocytes, showed significantly greater internalization of NAXI than of DB mediated by CD64
+ cells (
Figure 7). This might be due to the Fc engineering of NAXI to improve Fc-FcR affinity, resulting in higher internalization rates [
2]. However, the binding signal of NAXI to neuroblastoma cells showed similar or higher gMFI values compared to DB across all concentrations, indicating similar or higher tumor cell surface coverage by NAXI (
Figure 3).
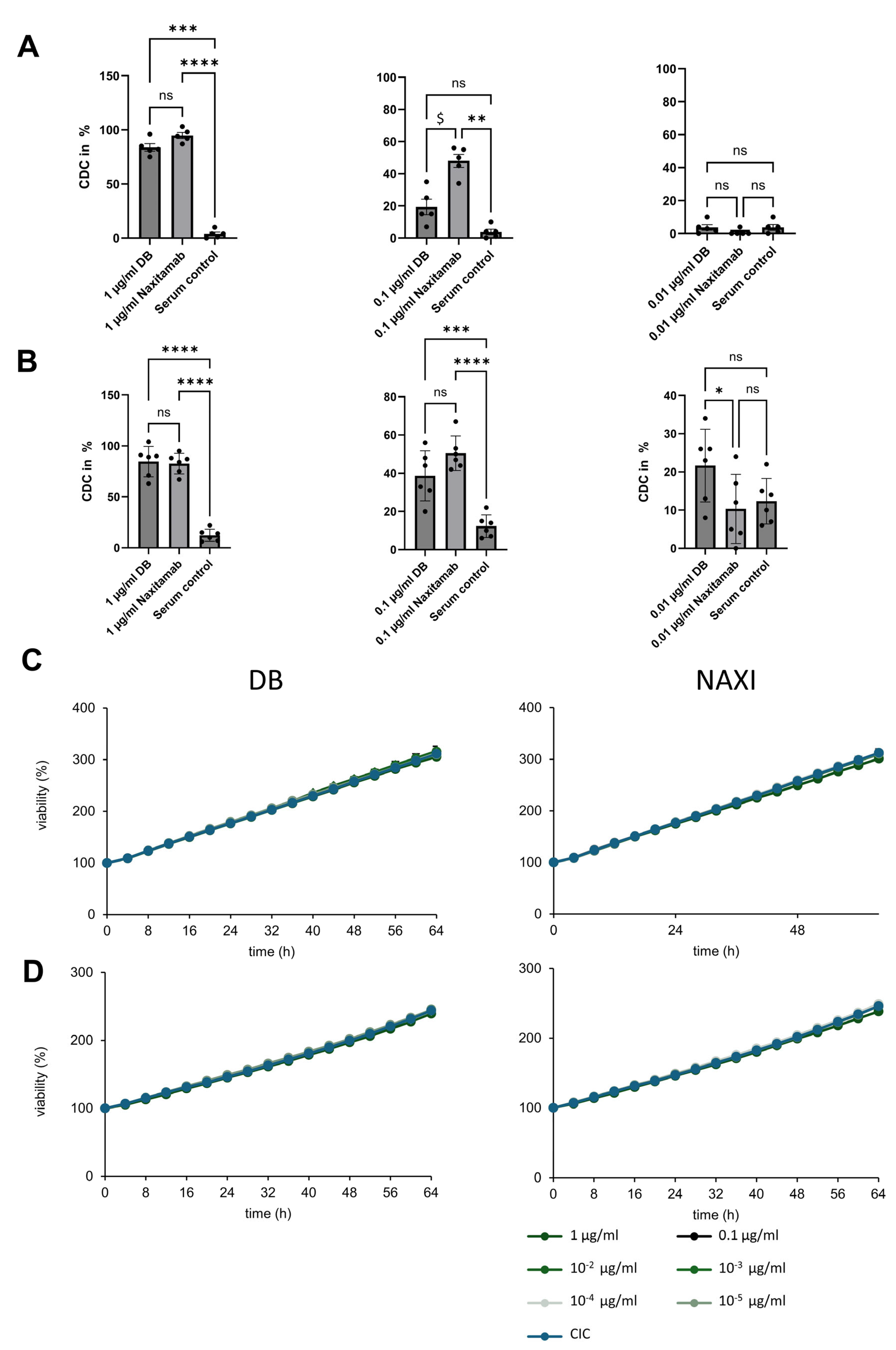
We could not show any CDC anti-tumor activity for both antibodies in the 3D spheroid models, which more closely mimic the 3D structure of tumors in vivo in contrast to 2D models (
Figure 2). This indicates that ADCC is the most effective mechanism of action, which has also been concluded from clinical trials with DB, where neuroblastoma patients with a high-affinity FcγR polymorphism showed a better outcome [
22]. Further evidence for a subordinate role of CDC compared to ADCC for antibody-mediated anti-neuroblastoma immune responses can be concluded from clinical trial data with hu14.18K322A, which is a humanized anti-GD2 antibody generated by CDR grafting using the CDR regions of the anti-GD2 antibody 14G2a. The constant human IgG1 regions were engineered introducing the K322A mutation to reduce complement activation. Despite decreased CDC activity, hu14.18K322A maintained its anti-tumor efficacy while significantly reducing neuropathic pain, a major on-target off-tumor side effect of anti-GD2 antibodies, attributed at least in part to CDC [
23]. This supports the notion that ADCC, rather than CDC, is the primary driver of anti-tumor activity of anti-GD2 antibodies. In this context, a potential methodological consideration is the use of mixed PBMC populations instead of isolated NK cells for the ADCC assays. Although NK cells are the principal effectors of anti-GD2 antibody-mediated ADCC via FcγRIIIa (CD16), we opted to use unseparated peripheral blood mononuclear cells (PBMCs) rather than isolated NK cells or neutrophils in our cytotoxicity assays, in line with the established practice for ADCC evaluation [
24,
25,
26]. This decision was based on the intent to preserve a physiologically relevant immune cell context, which better reflects the in vivo situation. PBMCs include not only NK cells, but also monocytes and subsets of T cells that may contribute to or modulate antibody-dependent responses [
27]. While isolation of specific effector populations may provide more mechanistic insight, it may also lead to artificial activation or loss of functionality due to processing steps [
28,
29]. To minimize variability and control for non-specific effects, we included appropriate antibody-free controls in all experiments. Additionally, PBMCs from multiple donors were used in independent replicates, and consistent trends were observed across all datasets. We therefore consider the use of PBMCs to be a valid and informative approach for the assessment of ADCC in this study.
DB is approved for patients with high-risk neuroblastoma in the maintenance phase of newly diagnosed patients as well as for patients with relapsed and refractory disease, and it is given as a continuous infusion over 24 h on 10 consecutive days per cycle for 5 cycles (100 mg/m
2/cycle; 10 mg/m
2/day). The long-term infusion was developed because side effects are less intensive and less frequent [
30]. This is in contrast to NAXI, which is approved for patients with relapsed and refractory neuroblastoma with osteomedullary disease. It is given as short infusions of 30 min duration on days 1, 3, and 5 of each cycle (9 mg/kg/cycle = 270 mg/m
2/cycle; 3 mg/kg/day = 90 mg/m
2/day). The treatment intervals between DB and NAXI cycles are 4–5 weeks for NAXI and 5 weeks for DB. For DB, pharmacokinetic data are available showing trough levels ranging from 0.1 to 1 µg/mL preceding the next cycle, which results in an immunologically active antibody concentration throughout the entire treatment period of approximately six months, as shown here for 3D targets (
Figure 1) but also in 2D models [
30,
31]. For NAXI, pharmacokinetic data are not reported; therefore, it is not clear if the 9-fold higher daily dosing or the 3-fold higher dose per cycle of NAXI compared to DB outweighs the TMDD disadvantage of NAXI compared to DB (
Figure 2,
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8). Furthermore, the current practice is to combine NAXI with GM-CSF, whereas DB is given without additional cytokine co-medication [
14]. The treatment of neuroblastoma patients with cytokines (IL-2 and GM-CSF) is associated with a strong increase of activated CD64
+ granulocytes and monocytes [
32]. This co-medication may lead to a further increase of the internalization rate and magnitude in particular of mAbs engineered to have a high affinity to the FcR, which may further aggravate the TMDD effect for NAXI.
While our data consistently demonstrate TMDD-driven effects across multiple models, the interpretation of these results should consider potential limitations. This proof-of-concept study relied on human in vitro systems and did not include in vivo mouse experiments. Thus, organism-level pharmacokinetics/biodistribution, continuous-infusion dosing and stromal components (e.g., cancer-associated fibroblasts) were not considered in the models used. Murine xenograft studies using 3F8 have shown potent PBMC/PMN-mediated ADCC and superior tumor control after humanization [
2]. Affinity maturation of hu3F8 (NAXI) improved binding (7-13-fold), ADCC (up to 12-fold), and tumor ablation in vivo, but also revealed an “affinity ceiling” beyond which additional gains were minimal [
33]. Our data demonstrate TMDD-driven functional loss at equimolar concentrations for clinical-grade NAXI compared to DB. However, the optimal affinity range for GD2 and clinical relevance require confirmation in further model systems and clinical trials.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}