
Scaffold-Free Functional Deconvolution Identifies Clinically Relevant Metastatic Melanoma EV Biomarkers

 , ,
, ,  , ,
, ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Exosome Isolation
2.3. Sample Preparation for LC-MS Analysis
2.4. Nano LC and High-Field Orbitrap MS
2.5. Protein Identification and Quantification
2.6. Functional Analysis
2.7. Exosome Capture Using Magnetic Beads
2.8. Western Blot Analysis of Melanoma-Specific Surface Proteins
2.9. Flow Cytometry
2.10. Survival Analysis
2.11. Statistical Analysis
2.12. Network Analysis
3. Results
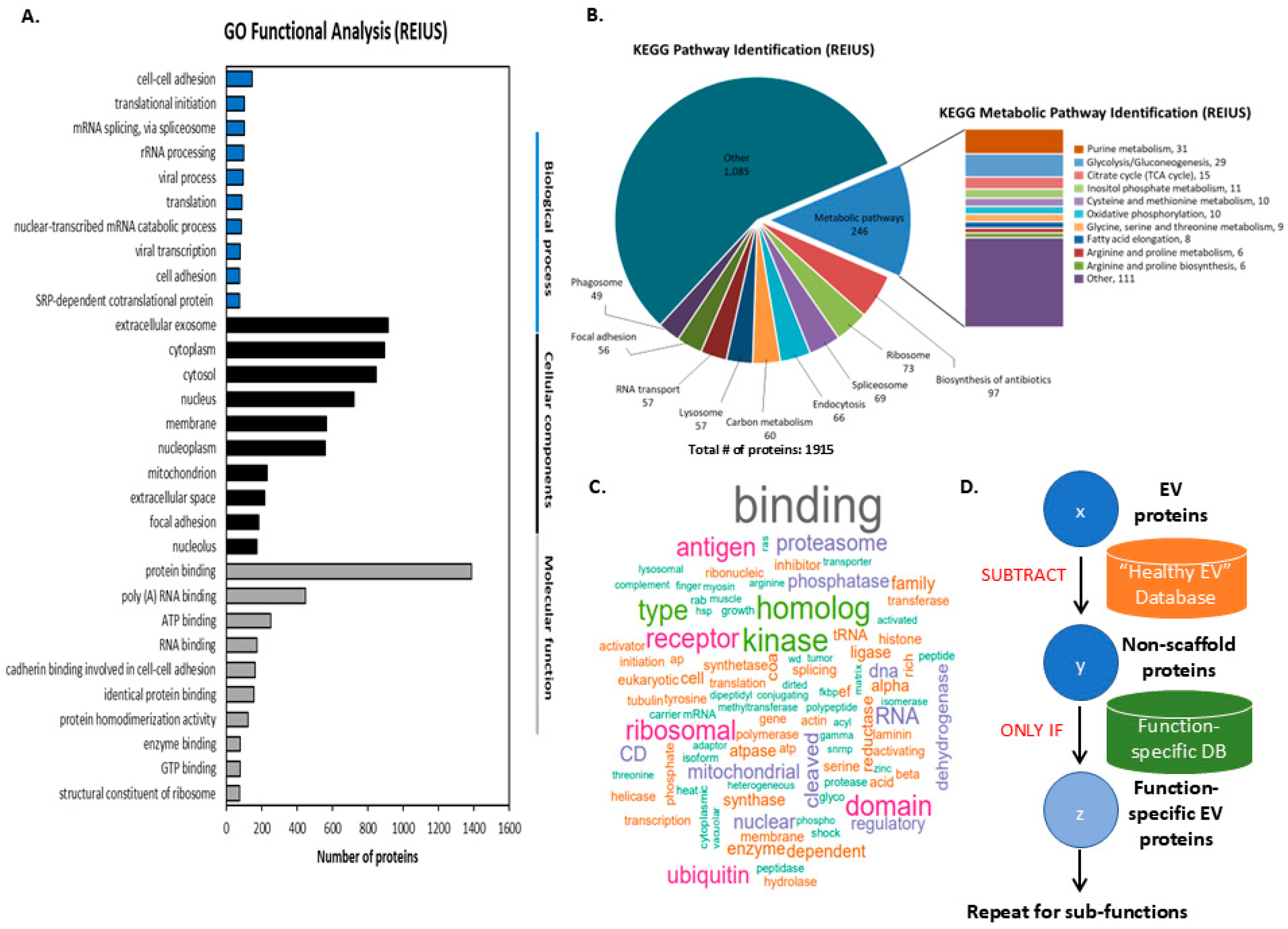
3.1. Metastatic Proteins from REIUS-Derived Exosomes

3.2. Classification of Proteomic Data Derived from REIUS Isolated Exosomes
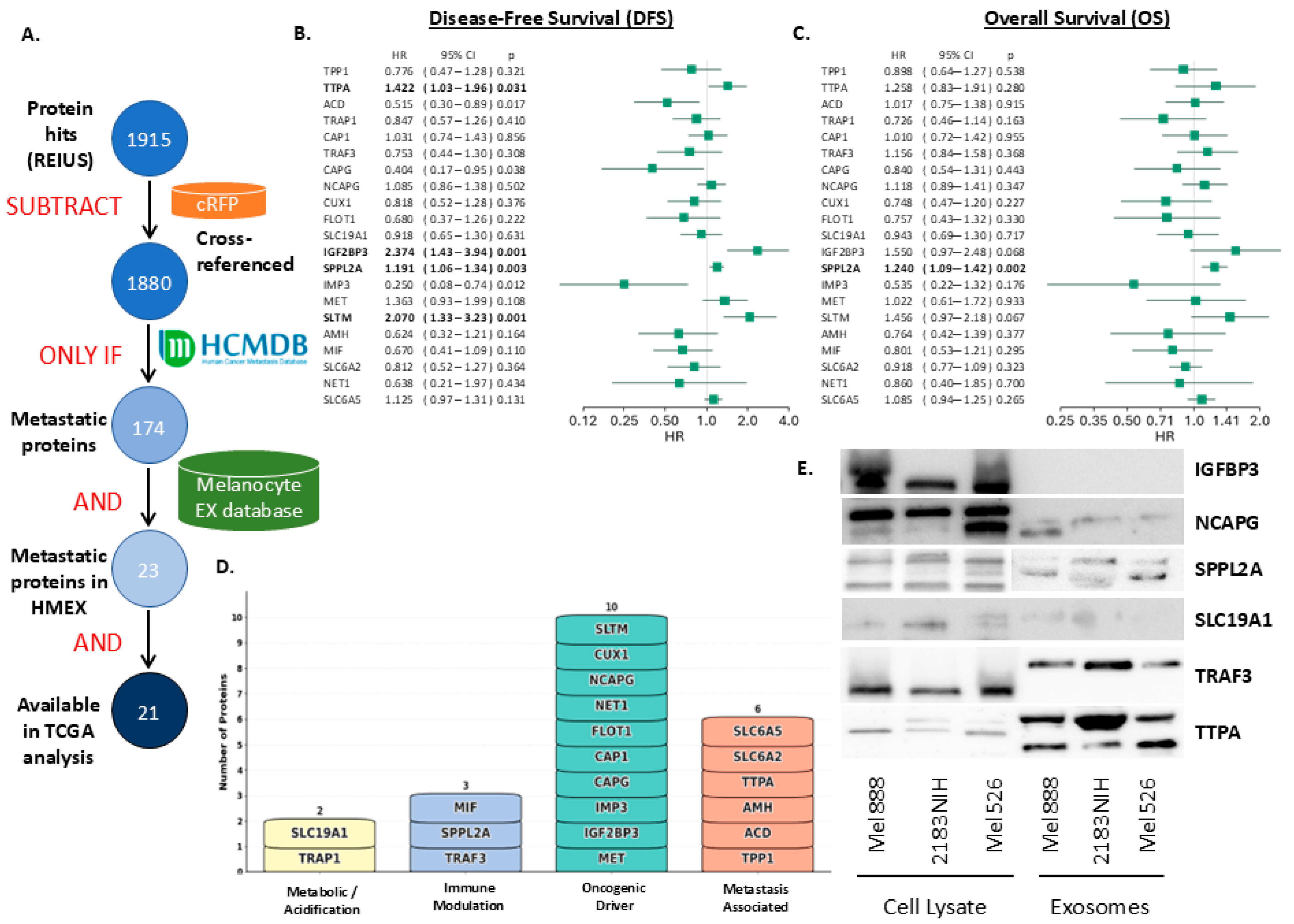
3.3. Finding Clinically Relevant Biomarkers on Melanoma Exosomes Using SFD
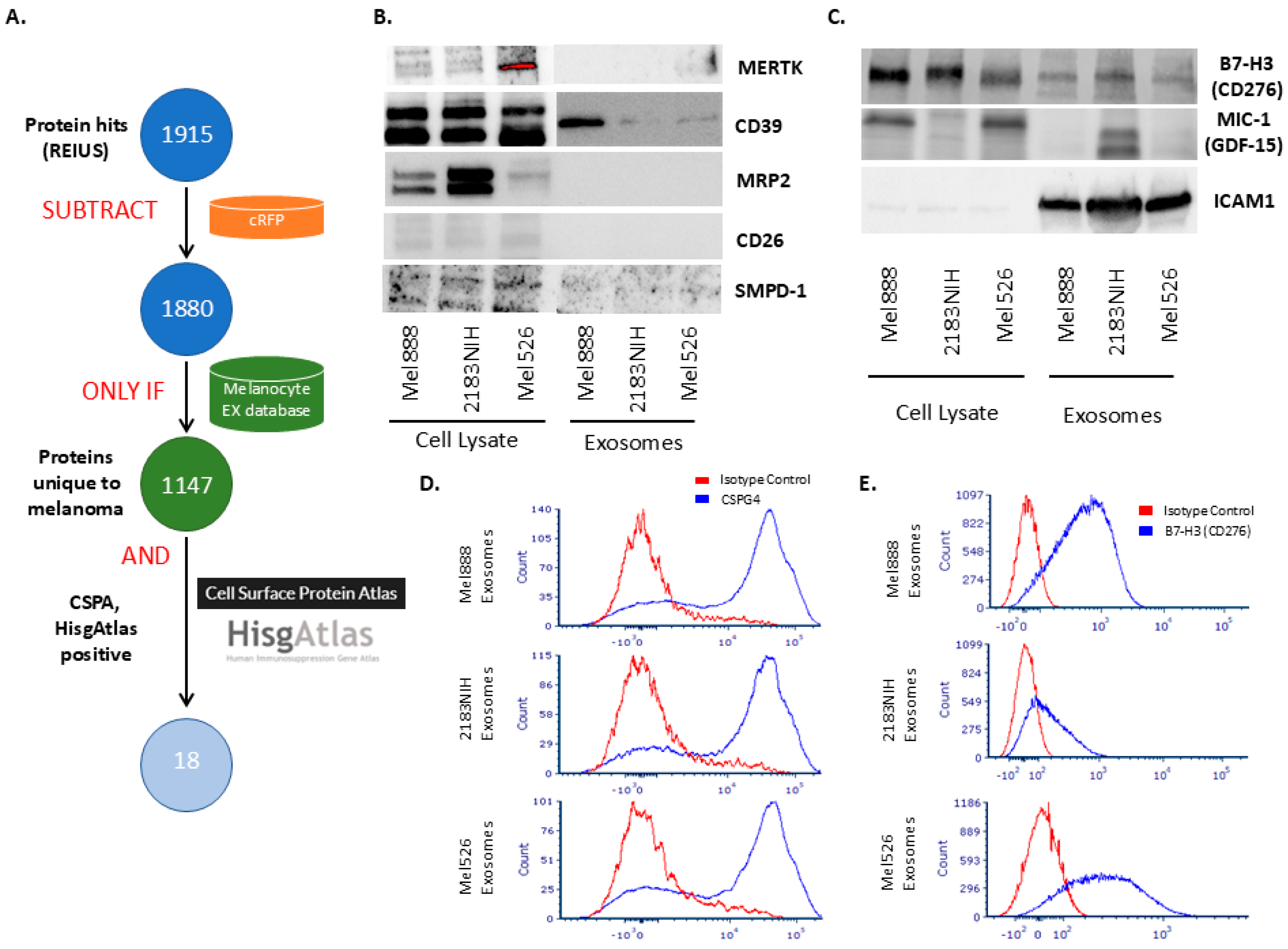
3.4. Using SFD to Identify Immunosuppressive Surface Biomarkers
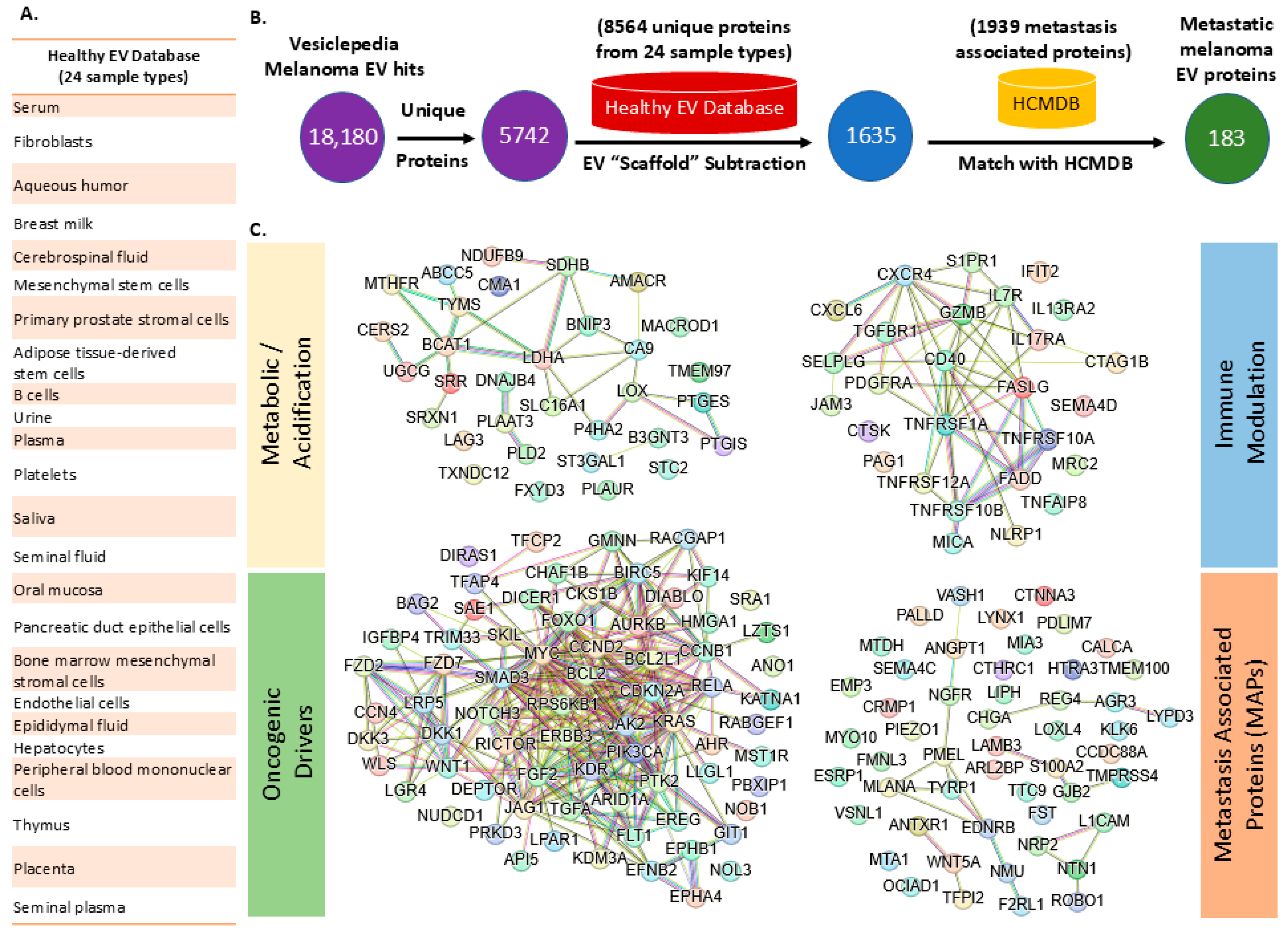
3.5. Meta-Analysis of Vesiclepedia Using SFD Approach
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braeuer, R.R.; Watson, I.R.; Wu, C.J.; Mobley, A.K.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Why is melanoma so metastatic? Pigment. Cell Melanoma Res. 2014, 27, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.C., Jr.; Slominski, A. Current concepts of metastasis in melanoma. Expert. Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Li, K.; Chen, Y.; Li, A.; Tan, C.; Liu, X. Exosomes play roles in sequential processes of tumor metastasis. Int. J. Cancer 2019, 144, 1486–1495. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudas, J.; Riechelmann, H.; Skvortsova, I.I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Wang, S.E. Extracellular Vesicles and Metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a037275. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Fridman, E.S.; Ginini, L.; Gil, Z. The Role of Extracellular Vesicles in Metabolic Reprogramming of the Tumor Microenvironment. Cells 2022, 11, 1433. [Google Scholar] [CrossRef]
- Schey, K.L.; Luther, J.M.; Rose, K.L. Proteomics characterization of exosome cargo. Methods 2015, 87, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lasser, C.; Szabo, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzas, E.I.; Lotvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2, 20677. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Rider, M.A.; Bundy, J.L.; Liu, X.; Singh, R.K.; Meckes, D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef]
- L’Imperio, V.; Smith, A.; Chinello, C.; Pagni, F.; Magni, F. Proteomics and glomerulonephritis: A complementary approach in renal pathology for the identification of chronic kidney disease related markers. Proteom. Clin. Appl. 2016, 10, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Allen, C.L.; Benjamin-Davalos, S.; Koroleva, M.; MacFarland, D.; Minderman, H.; Ernstoff, M.S. A Rapid Exosome Isolation Using Ultrafiltration and Size Exclusion Chromatography (REIUS) Method for Exosome Isolation from Melanoma Cell Lines. Methods Mol. Biol. 2021, 2265, 289–304. [Google Scholar] [CrossRef]
- Shu, S.; Yang, Y.; Allen, C.L.; Hurley, E.; Tung, K.H.; Minderman, H.; Wu, Y.; Ernstoff, M.S. Purity and yield of melanoma exosomes are dependent on isolation method. J. Extracell. Vesicles 2020, 9, 1692401. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; An, B.; Wang, X.; Hilchey, S.P.; Li, J.; Cao, J.; Tian, Y.; Hu, C.; Jin, L.; Ng, A.; et al. Surfactant Cocktail-Aided Extraction/Precipitation/On-Pellet Digestion Strategy Enables Efficient and Reproducible Sample Preparation for Large-Scale Quantitative Proteomics. Anal. Chem. 2018, 90, 10350–10359. [Google Scholar] [CrossRef]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Poulsen, D.J.; Orsburn, B.C.; Wang, J.; et al. IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. USA 2018, 115, E4767–E4776. [Google Scholar] [CrossRef]
- Shen, X.; Hu, Q.; Li, J.; Wang, J.; Qu, J. Experimental Null Method to Guide the Development of Technical Procedures and to Control False-Positive Discovery in Quantitative Proteomics. J. Proteome Res. 2015, 14, 4147–4157. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Benito-Martin, A.; Jasiulionis, M.G.; Garcia-Silva, S. Extracellular vesicles and melanoma: New perspectives on tumor microenvironment and metastasis. Front. Cell Dev. Biol. 2022, 10, 1061982. [Google Scholar] [CrossRef]
- Waster, P.; Eriksson, I.; Vainikka, L.; Rosdahl, I.; Ollinger, K. Extracellular vesicles are transferred from melanocytes to keratinocytes after UVA irradiation. Sci. Rep. 2016, 6, 27890. [Google Scholar] [CrossRef]
- Lee, K.H.; Panelli, M.C.; Kim, C.J.; Riker, A.I.; Bettinotti, M.P.; Roden, M.M.; Fetsch, P.; Abati, A.; Rosenberg, S.A.; Marincola, F.M. Functional dissociation between local and systemic immune response during anti-melanoma peptide vaccination. J. Immunol. 1998, 161, 4183–4194. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Ferrone, S.; Whiteside, T.L. Targeting CSPG4 for isolation of melanoma cell-derived exosomes from body fluids. HNO 2020, 68, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef]
- Welton, J.L.; Khanna, S.; Giles, P.J.; Brennan, P.; Brewis, I.A.; Staffurth, J.; Mason, M.D.; Clayton, A. Proteomics analysis of bladder cancer exosomes. Mol. Cell Proteom. 2010, 9, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ou, X.; Wu, X. Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis. Int. J. Oncol. 2019, 54, 1719–1733. [Google Scholar] [CrossRef]
- Tanase, C.P.; Codrici, E.; Popescu, I.D.; Mihai, S.; Enciu, A.M.; Necula, L.G.; Preda, A.; Ismail, G.; Albulescu, R. Prostate cancer proteomics: Current trends and future perspectives for biomarker discovery. Oncotarget 2017, 8, 18497–18512. [Google Scholar] [CrossRef]
- Stefanius, K.; Servage, K.; de Souza Santos, M.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. Elife 2019, 8, 40226. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Xue, L.; Hsu, C.C.; Paez, J.S.; Pan, L.; Andaluz, H.; Wendt, M.K.; Iliuk, A.B.; Zhu, J.K.; Tao, W.A. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3175–3180. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Zoller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Zhang, W.; Zhong, W.; Wang, B.; Yang, J.; Yang, J.; Yu, Z.; Qin, Z.; Shi, A.; Xu, W.; Zheng, C.; et al. ICAM-1-mediated adhesion is a prerequisite for exosome-induced T cell suppression. Dev. Cell 2022, 57, 329–343.e327. [Google Scholar] [CrossRef]
- Zhou, W.T.; Jin, W.L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Y.; Zhao, Y.; Kim, J.J.; Li, H.; Meng, C.; Chen, F.; Zhang, J.; Mak, D.H.; Van, V.; et al. Immune checkpoint B7-H3 is a therapeutic vulnerability in prostate cancer harboring PTEN and TP53 deficiencies. Sci. Transl. Med. 2023, 15, eadf6724. [Google Scholar] [CrossRef]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tille, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, 3311. [Google Scholar] [CrossRef] [PubMed]
- Shirley, C.A.; Chhabra, G.; Amiri, D.; Chang, H.; Ahmad, N. Immune escape and metastasis mechanisms in melanoma: Breaking down the dichotomy. Front. Immunol. 2024, 15, 1336023. [Google Scholar] [CrossRef]
- Wischhusen, J.; Melero, I.; Fridman, W.H. Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front. Immunol. 2020, 11, 951. [Google Scholar] [CrossRef]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef]
- Dransfield, I.; Zagorska, A.; Lew, E.D.; Michail, K.; Lemke, G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 2015, 6, e1646. [Google Scholar] [CrossRef] [PubMed]
- Maric, G.; Rose, A.A.; Annis, M.G.; Siegel, P.M. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco Targets Ther. 2013, 6, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ding, Y.; Wu, N.; Jiang, J.; Huang, Y.; Zhang, F.; Wang, H.; Zhou, Q.; Yang, Y.; Zhuo, W.; et al. FSTL1 promotes growth and metastasis in gastric cancer by activating AKT related pathway and predicts poor survival. Am. J. Cancer Res. 2021, 11, 712–728. [Google Scholar]
- Wu, R.; Zhang, Y.; Xu, X.; You, Q.; Yu, C.; Wang, W.; Mao, Y. Exosomal B7-H3 facilitates colorectal cancer angiogenesis and metastasis through AKT1/mTOR/VEGFA pathway. Cell Signal 2023, 109, 110737. [Google Scholar] [CrossRef] [PubMed]
- Herbold, K.W.; Zhou, J.; Haggerty, J.G.; Milstone, L.M. CD44 expression on epidermal melanocytes. J. Invest. Dermatol. 1996, 106, 1230–1235. [Google Scholar] [CrossRef]
- Angi, M.; Kalirai, H.; Prendergast, S.; Simpson, D.; Hammond, D.E.; Madigan, M.C.; Beynon, R.J.; Coupland, S.E. In-depth proteomic profiling of the uveal melanoma secretome. Oncotarget 2016, 7, 49623–49635. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Li, Y.; Zeng, Y.; Zhang, Y.; Jiang, Q.; Zhang, Q.; Zhu, J.; Gong, J. Advancements in melanoma immunotherapy: The emergence of Extracellular Vesicle Vaccines. Cell Death Discov. 2024, 10, 374. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, S.-L.; Benjamin-Davalos, S.; Wang, X.; Katsuta, E.; Fitzgerald, M.; Koroleva, M.; Allen, C.L.; Qu, F.; Paragh, G.; Minderman, H.; et al. Scaffold-Free Functional Deconvolution Identifies Clinically Relevant Metastatic Melanoma EV Biomarkers. Cancers 2025, 17, 2509. https://doi.org/10.3390/cancers17152509
Shu S-L, Benjamin-Davalos S, Wang X, Katsuta E, Fitzgerald M, Koroleva M, Allen CL, Qu F, Paragh G, Minderman H, et al. Scaffold-Free Functional Deconvolution Identifies Clinically Relevant Metastatic Melanoma EV Biomarkers. Cancers. 2025; 17(15):2509. https://doi.org/10.3390/cancers17152509
Chicago/Turabian StyleShu, Shin-La, Shawna Benjamin-Davalos, Xue Wang, Eriko Katsuta, Megan Fitzgerald, Marina Koroleva, Cheryl L. Allen, Flora Qu, Gyorgy Paragh, Hans Minderman, and et al. 2025. "Scaffold-Free Functional Deconvolution Identifies Clinically Relevant Metastatic Melanoma EV Biomarkers" Cancers 17, no. 15: 2509. https://doi.org/10.3390/cancers17152509
APA StyleShu, S.-L., Benjamin-Davalos, S., Wang, X., Katsuta, E., Fitzgerald, M., Koroleva, M., Allen, C. L., Qu, F., Paragh, G., Minderman, H., Kalinski, P., Takabe, K., & Ernstoff, M. S. (2025). Scaffold-Free Functional Deconvolution Identifies Clinically Relevant Metastatic Melanoma EV Biomarkers. Cancers, 17(15), 2509. https://doi.org/10.3390/cancers17152509