
Hallmarks of Cancer Expression in Oral Leukoplakia: A Scoping Review of Systematic Reviews and Meta-Analyses

 and
and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Eligibility Criteria
2.3. Study Selection Process
2.4. Data Extraction
2.5. Evaluation of Risk of Bias and Quality of Evidence
2.6. Rationale, Critical Analysis, and Evidence Synthesis
3. Results
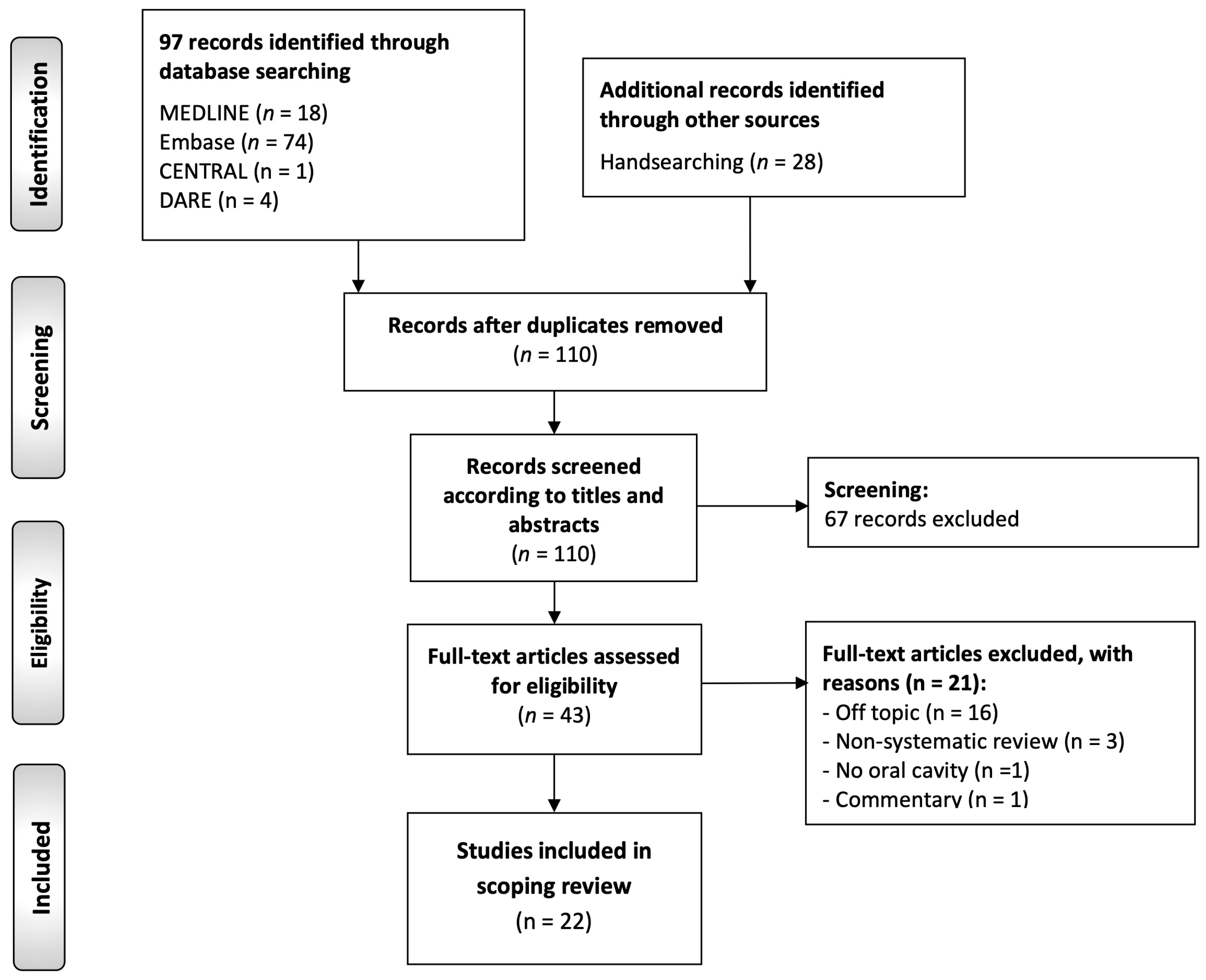
3.1. Results of the Literature Search
3.2. Study Characteristics
3.3. Risk of Bias and Quality of Evidence
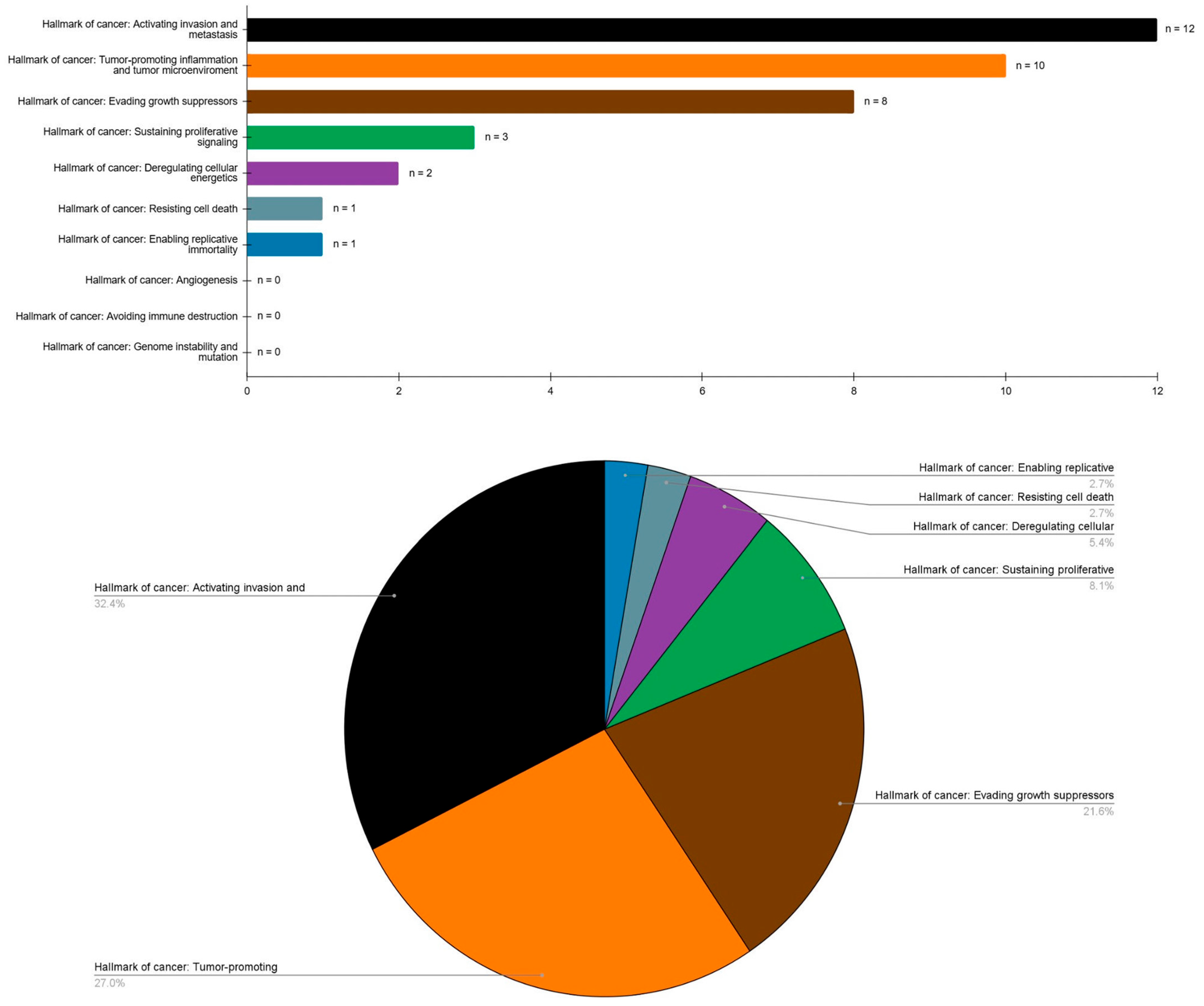
3.4. Critical Analysis and Evidence Synthesis
4. Discussion
4.1. Maintenance of Proliferative Signaling
4.2. Evasion of Growth-Suppressive Signals and Development of Resistance to Cell Death
4.3. Enabling Replicative Immortality
4.4. Induction of Angiogenesis
4.5. Activation of Invasion and Metastasis
4.6. Enabling Characteristics
4.7. Reprogramming of Energy Metabolism
4.8. Evading the Antitumor Immune Response
4.9. Study Limitations and Potential Strengths
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Kujan, O.; Aguirre-Urizar, J.M.; Bagan, J.V.; González-Moles, M.Á.; Kerr, A.R.; Lodi, G.; Mello, F.W.; Monteiro, L.; Ogden, G.R.; et al. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by the WHO Collaborating Centre for Oral Cancer. Oral Dis. 2021, 27, 1862–1880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, B.; Zeng, X.; Hu, X.S.; Hua, H. The global prevalence of oral leukoplakia: A systematic review and meta-analysis from 1996 to 2022. BMC Oral Health 2023, 23, 645. [Google Scholar] [CrossRef] [PubMed]
- Mello, F.W.; Miguel, A.F.P.; Dutra, K.L.; Porporatti, A.L.; Warnakulasuriya, S.; Guerra, E.N.S.; Rivero, E.R.C. Prevalence of oral potentially malignant disorders: A systematic review and meta-analysis. J. Oral Pathol. Med. 2018, 47, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Petti, S. Pooled estimate of world leukoplakia prevalence: A systematic review. Oral Oncol. 2003, 39, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Pimenta-Barros, L.A.; Ramos-García, P.; González-Moles, M.Á.; Aguirre-Urizar, J.M.; Warnakulasuriya, S. Malignant transformation of oral leukoplakia: Systematic review and comprehensive meta-analysis. Oral Dis. 2025, 31, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Mithani, S.; Mydlarz, W.; Grumbine, F.; Smith, I.; Califano, J. Molecular genetics of premalignant oral lesions. Oral Dis. 2007, 13, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA extension for scoping reviews (PRISMA-ScR): Checklist and explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.; Sampson, M.; Salzwedel, D.M.; Cogo, E.; Foerster, V.; Lefebvre, C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J. Clin. Epidemiol. 2016, 75, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Centre for Reviews and Dissemination Group. Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care; York Publishing Services Ltd.: York, UK, 2009. [Google Scholar]
- Lee, E.; Dobbins, M.; Decorby, K.; McRae, L.; Tirilis, D.; Husson, H. An optimal search filter for retrieving systematic reviews and meta-analyses. BMC Med. Res. Methodol. 2012, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Stang, A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur. J. Epidemiol. 2010, 25, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Hayden, J.A.; van der Windt, D.A.; Cartwright, J.L.; Côté, P.; Bombardier, C. Assessing Bias in Studies of Prognostic Factors. Ann. Intern. Med. 2013, 158, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Aromataris, E.; Lockwood, C.; Porritt, K.; Pilla, B.; Jordan, Z. (Eds.) JBI Manual for Evidence Synthesis; JBI: Adelaide, Australia, 2024; ISBN 9780648848820. [Google Scholar]
- Higgins, J.P.T.; Thomas, J.; Chandler, J.; Cumpston, M.; Li, T.; Page, M.J. Cochrane Handbook for Systematic Reviews of Interventions; Welch, V.A., Ed.; Wiley: New York, NY, USA, 2019; ISBN 9781119536628. [Google Scholar]
- Guyatt, G.; Oxman, A.D.; Akl, E.A.; Kunz, R.; Vist, G.; Brozek, J.; Norris, S.; Falck-Ytter, Y.; Glasziou, P.; DeBeer, H. GRADE guidelines: 1. Introduction—GRADE evidence profiles and summary of findings tables. J. Clin. Epidemiol. 2011, 64, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Sucharew, H. Methods for Research Evidence Synthesis: The Scoping Review Approach. J. Hosp. Med. 2019, 14, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Munn, Z.; Peters, M.D.J.; Stern, C.; Tufanaru, C.; McArthur, A.; Aromataris, E. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach. BMC Med. Res. Methodol. 2018, 18, 143. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, E.; Donís, S.P.; Petronacci, C.M.C.; Alves, M.G.O.; Mendía, X.M.; Fernandes, D.; Pouso, A.I.L.; Bufalino, A.; Bravo López, S.; Sayáns, M.P. Usefulness of protein-based salivary markers in the diagnosis of oral potentially malignant disorders: A systematic review and meta-analysis. Cancer Biomark. 2021, 32, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Benito-Ramal, E.; Egido-Moreno, S.; González-Navarro, B.; Jané-Salas, E.; Roselló-Llabrés, X.; López-López, J. Role of selected salivary inflammatory cytokines in the diagnosis and prognosis of oral squamous cell carcinoma. A Systematic Review and Meta-analysis. Med. Oral Patol. Oral Y Cir. Bucal 2023, 28, e474–e486. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.; Mello, F.W.; Warnakulasuriya, S. Tissue biomarkers for predicting the risk of oral cancer in patients diagnosed with oral leukoplakia: A systematic review. Oral Dis. 2021, 27, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.; Mariano, L.C.; Warnakulasuriya, S. Podoplanin could be a predictive biomarker of the risk of patients with oral leukoplakia to develop oral cancer: A systematic review and meta-analysis. Oral Dis. 2024, 30, 207–215. [Google Scholar] [CrossRef] [PubMed]
- de Morais, E.F.; Pinheiro, J.C.; Lira, J.A.S.; Mafra, R.P.; Barboza, C.A.G.; de Souza, L.B.; Freitas, R. de A. Prognostic value of the immunohistochemical detection of epithelial-mesenchymal transition biomarkers in oral epithelial dysplasia: A systematic review. Med. Oral Patol. Oral Cir. Bucal 2020, 25, e205–e216. [Google Scholar]
- Normando, A.G.C.; dos Santos, E.S.; de Oliveira Sá, J.; Busso-Lopes, A.F.; De Rossi, T.; de Sá Patroni, F.M.; Granato, D.C.; Guerra, E.N.S.; Santos-Silva, A.R.; Lopes, M.A.; et al. A meta-analysis reveals the protein profile associated with malignant transformation of oral leukoplakia. Front. Oral Health 2023, 4, 1088022. [Google Scholar] [CrossRef] [PubMed]
- Piyarathne, N.S.; Rasnayake, R.M.S.G.K.; Angammana, R.; Chandrasekera, P.; Ramachandra, S.; Weerasekera, M.; Yasawardene, S.; Abu-Eid, R.; Jayasinghe, J.A.P.; Gupta, E. Diagnostic salivary biomarkers in oral cancer and oral potentially malignant disorders and their relationships to risk factors—A systematic review. Expert Rev. Mol. Diagn. 2021, 21, 789–807. [Google Scholar] [CrossRef] [PubMed]
- Ramos-García, P.; González-Moles, M.Á.; Warnakulasuriya, S. Significance of p53 overexpression in the prediction of the malignant transformation risk of oral potentially malignant disorders: A systematic review and meta-analysis. Oral Oncol. 2022, 126, 105734. [Google Scholar] [CrossRef] [PubMed]
- Ramos-García, P.; González-Moles, M.Á.; Ayén, Á.; González-Ruiz, L.; Gil-Montoya, J.A.; Ruiz-Ávila, I. Predictive value of CCND1/cyclin D1 alterations in the malignant transformation of potentially malignant head and neck disorders: Systematic review and meta-analysis. Head Neck 2019, 41, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Gallegos, R.; Figueroa, C. Biomarkers of progression to oral cancer in patients with dysplasia: A systematic review. Mol. Clin. Oncol. 2020, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Saluja, T.S.; Ali, M.; Mishra, P.; Kumar, V.; Singh, S.K. Prognostic value of cancer stem cell markers in potentially malignant disorders of oral mucosa: A meta-analysis. Cancer Epidemiol. Biomark. Prev. 2019, 28, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Rattay, T.; McConkey, C.; Helliwell, T.; Mehanna, H. Biomarkers in dysplasia of the oral cavity: A systematic review. Oral Oncol. 2009, 45, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Celentano, A.; Glurich, I.; Borgnakke, W.S.; Farah, C.S. World Workshop on Oral Medicine VII: Prognostic biomarkers in oral leukoplakia and proliferative verrucous leukoplakia—A systematic review of retrospective studies. Oral Dis. 2021, 27, 848–880. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, A.; Uma Maheswari, T. Expression of matrix metalloproteinase-9 in oral potentially malignant disorders: A systematic review. J. Oral Maxillofac. Pathol. 2016, 20, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Celentano, A.; Glurich, I.; Borgnakke, W.S.; Jensen, S.B.; Peterson, D.E.; Delli, K.; Ojeda, D.; Vissink, A.; Farah, C.S. World Workshop on Oral Medicine VII: Prognostic biomarkers in oral leukoplakia: A systematic review of longitudinal studies. Oral Dis. 2019, 25, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Cívico-Ortega, J.L.; Ramos-García, P.; González-Moles, M.Á. Significance of Epidermal Growth Factor Receptor (EGFR) upregulation in the prediction of the malignant transformation risk in oral potentially malignant disorders: A systematic review and meta-analysis. Front. Oral Health 2025, 6, 1578561. [Google Scholar] [CrossRef] [PubMed]
- Kasradze, D.; Juodzbalys, G.; Guobis, Z.; Albinas Gervickas, M.C. Genetic And Proteomic Biomarkers of Head-and-Neck Cancer: A Systematic Review. J. Pers. Med. 2022, 12, 1816. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Q.; Guo, Z.; Deng, G.; Chen, R.; Zheng, Y. Potential noninvasive biomarkers for the malignant transformation of oral leukoplakia: A systematic review and meta-analysis. Cancer Med. 2023, 12, 14718–14730. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Velázquez, Ó.; López-Pintor, R.M.; González-Serrano, J.; Casañas, E.; Torres, J.; Hernández, G. Salivary LDH in oral cancer and potentially malignant disorders: A systematic review and meta-analysis. Oral Dis. 2022, 28, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.L.; Naik, Z.; Lagali-Jirge, V.; Sridhar, M.; Arun Panwar, V.K. Salivary Lactate Dehydrogenase as a Potential Biomarker in Oral Potentially Malignant Disorderss and Head & Neck Cancer: A Systematic Review and Meta-analysis. Gulf. J. Oncol. 2023, 24, 941–949. [Google Scholar]
- López-Ansio, M.; Ramos-García, P.; González-Moles, M.Á. Predictive Value of the Loss of pRb Expression in the Malignant Transformation Risk of Oral Potentially Malignant Disorders: A Systematic Review and Meta-Analysis. Cancers 2025, 17, 329. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Pouso, A.I.; Caponio, V.C.A.; Vieira, E.; Silva, F.F.; Pérez-Jardón, A.; Álvarez-Calderón-Iglesias, Ó.; Gándara-Vila, P.; Pannone, G.; Pérez-Sayáns, M. Predictive value of CDKN2A/p16INK4a expression in the malignant transformation of oral potentially malignant disorders: Systematic review and meta-analysis. Pathol. Res. Pract. 2023, 248, 154656. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, K.A.A.S.; Robinson, D.; Evans, H. Multiple primary tumours following head and neck cancer in southern England during 1961-98. J. Oral Pathol. Med. 2003, 32, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.; Luca, A.; Caponigro, F.; Salomon, D. The ErbB Receptors and their Ligands in Cancer: An Overview. Curr. Drug Targets 2005, 6, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Rehim, D.M.; Pinder, S.E.; Paish, C.E.; Bell, J.A.; Rampaul, R.S.; Blamey, R.W.; Robertson, J.F.R.; Nicholson, R.I.; Ellis, I.O. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br. J. Cancer 2004, 91, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G. ErbB-4: Mechanism of action and biology. Exp. Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Skaria, K.B.; Yarden, Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. EGF Recept. Fam. Biol. Mech. Role Cancer 2003, 284, 57–68. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, M.P.; Lerman, M.A.; Coffey, R.J.; Muller, W.J.; Cardiff, R.D.; Stern, D.F. Active signaling by Neu in transgenic mice. Oncogene 1998, 17, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Munirajan, A.K.; Tsuchida, N. Ras oncogenes in oral cancer: The past 20 years. Oral Oncol. 2012, 48, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gimenez-Conti, I.B.; Cunningham, J.E.; Collet, A.M.; Luna, M.A.; Lanfranchi, H.E.; Spitz, M.R.; Conti, C.J. Alterations of p53, cyclin D1, Rb, and H-ras in human oral carcinomas related to tobacco use. Cancer 1998, 83, 204–212. [Google Scholar] [CrossRef]
- Milasin, J.; Pujić, N.; Dedović, N.; Nikolić, Z.; Petrović, V.; Dimitrijević, B. High incidence of H-ras oncogene mutations in squamous cell carcinoma of lip vermilion. J. Oral Pathol. Med. 1994, 23, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. MTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. P53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, J.H.; te Riele, H.P. The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl. Cell Differ. 2006, 42, 183–225. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lee, W.H. The retinoblastoma gene: A prototypic and multifunctional tumor suppressor. Exp. Cell Res. 2001, 264, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Corson, T.W.; Gallie, B.L. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosom. Cancer 2007, 46, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Genetic predisposition to cancer. Cancer Detect. Prev. 1984, 7, 1–8. [Google Scholar] [PubMed]
- Perez-Ordoñez, B.; Beauchemin, M.; Jordan, R.C.K. Molecular biology of squamous cell carcinoma of the head and neck. J. Clin. Pathol. 2006, 59, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.-S. Autophagy failure in AD—Locating the primary defect. Neurobiol. Dis. 2011, 41, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Necroptosis: A Specialized Pathway of Programmed Necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L. No PUMA, no death: Implications for p53-dependent apoptosis. Cancer Cell 2003, 4, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.; Littlewood, T. A Matter of Life and Cell Death. Science 1998, 281, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.; Lowe, S.; Cepero, E. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Canto, M.; Kern, S.E. Focus on pancreas cancer. Cancer Cell 2002, 2, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment—TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.-H.; Moustakas, A. Transforming growth factor-β employs HMGA2 to elicit epithelial–mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Asthagiri, A.R.; Parry, D.M.; Butman, J.A.; Kim, H.J.; Tsilou, E.T.; Zhuang, Z.; Lonser, R.R. Neurofibromatosis type 2. Lancet 2009, 373, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. Tumor suppression by LKB1: SIK-ness prevents metastasis. Sci. Signal. 2009, 2, e55. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Bardeesy, N. LKB1; linking cell structure and tumor suppression. Oncogene 2008, 27, 6908–6919. [Google Scholar] [CrossRef] [PubMed]
- Partanen, J.I.; Nieminen, A.I.; Klefstrom, J. 3D view to tumor suppression: Lkb1, polarity and the arrest of oncogenic c-Myc. Cell Cycle 2009, 8, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; DePinho, R.A. Telomeres and telomerase in cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Avilion, A.A.; Lefeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Choudhury, B.; Campbell, P.J.; Feng, B.; Wong, K.K.; Protopopov, A.; O’Neil, J.; Gutierrez, A.; Ivanova, E.; Perna, I.; et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007, 447, 966–971. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, R.C.; Chang, S.; Maser, R.S.; Mohan, R.; Artandi, S.E.; Chin, L.; DePinho, R.A. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2002, 2, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; De Solorzano, C.O.; Knowles, D.; Jones, A.; Chou, W.; Rodriguez, E.G.; Kuo, W.L.; Ljung, B.M.; Chew, K.; Myambol, K.; et al. In situ analyses of genome instability in breast cancer. Nat. Genet. 2004, 36, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, C.M.; Hernandez, J.; Llorca, F.P.; Nuciforo, P.; Mathieu, M.C.; Commo, F.; Delaloge, S.; Sabatier, L.; André, F.; Soria, J.C. DNA damage repair and telomere length in normal breast, preneoplastic lesions, and invasive cancer. Am. J. Clin. Oncol. Cancer Clin. Trials 2010, 33, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M.; Ribatti, D. Angiogenesis in pre-malignant conditions. Eur. J. Cancer 2009, 45, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Coghlin, C. Current and emerging concepts in tumour metastasis. J. Pathol. 2014, 222, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; C Ng, A.H.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Abell, A.N.; Jordan, N.V.; Huang, W.; Prat, A.; Midland, A.A.; Johnson, N.L.; Granger, D.A.; Mieczkowski, P.A.; Perou, C.M.; Gomez, S.M.; et al. MAP3K4/CBP-regulated H2B acetylation controls epithelial-mesenchymal transition in trophoblast stem cells. Cell Stem Cell 2011, 8, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Marin, F.; Cubillo, E.; Stark, H.J.; Fusenig, N.; Nieto, M.A.; Cano, A. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J. Cell Sci. 2004, 117, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, F.; Brabletz, T.; Budczies, J.; Pfeiffer, S.; Jung, A.; Kirchner, T. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. Int. J. Cancer 2007, 121, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two Decades After BRCA: Setting Paradigms in Personalized Cancer Care and Prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro-versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlem; Arnold Constable: London, UK, 1930. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Strati, A.; Adamopoulos, C.; Kotsantis, I.; Psyrri, A.; Lianidou, E.; Papavassiliou, A.G. Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers. Int. J. Mol. Sci. 2025, 26, 1235. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Strauss, D.C.; Thomas, J.M. Transmission of donor melanoma by organ transplantation. Lancet Oncol. 2010, 11, 790–796. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Total Sample | 22 Studies |
|---|---|
| Year of publication | |
| Range min. (first publication) | 2019 |
| Range max. | 2025 |
| Study design | |
| Systematic reviews | 8 (36.36%) |
| Systematic reviews + meta-analysis | 14 (63.63%) |
| Study population | |
| Oral leukoplakia (OL) | 8 (36.36%) |
| OPMD (including OL) | 14 (63.63%) |
| Study | Year | Population | Study Design | Meta-Analysis | Systematic Reviews Guidelines | Study Protocol (Platform: Registration Code) | Risk of Bias Analysis (Tool) | Quality of Evidence Assessment (System) | Funding/ Conflict of Interest (COI) |
|---|---|---|---|---|---|---|---|---|---|
| Cívico-Ortega et al. | 2025 | OPMD (including OL) | SR | YES | MOOSE PRISMA Cochrane PRISMA-P | YES (PROSPERO: CRD42024626482) | Quality in Prognosis Studies (QUIPS) | NO | Funding: no COI: none |
| López-Ansio et al. | 2025 | OPMD (including OL) | SR | YES | MOOSE PRISMA Cochrane PRISMA-P | YES (CRD42024614644) | Quality in Prognosis Studies (QUIPS) | NO | Funding: no COI: none |
| Monteiro et al. | 2024 | OL | SR | YES | PRISMA | YES (PROSPERO: CRD42022329326) | Quality in Prognosis Studies (QUIPS) | NO | Funding: yes COI: none |
| Huang et al. | 2023 | OL | SR | YES | PRISMA | YES (INPLASY: INPLASY202250166) | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: yes COI: none |
| Normando et al. | 2023 | OL | SR | YES | PRISMA PRISMA-P | YES (CRD42020157561) | Joanna Briggs Institute (JBI) tools for Cohort and for Cross-sectional studies | GRADE | Funding: yes COI: none |
| Benito-Ramal et al. | 2023 | OPMD (including OL) | SR | YES | PRISMA | NO | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: none COI: none |
| Kumar et al. | 2023 | OPMD (including OL) | SR | YES | PRISMA | YES (PROSPERO: CRD42020198298) | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: none COI: none |
| Lorenzo-Pouso et al. | 2023 | OPMD (including OL) | SR | YES | PRISMA-P | YES (PROSPERO: CRD42022355931). | Quality in Prognosis Studies (QUIPS) | NO | Funding: yes COI: none |
| Ramos-García et al. | 2022 | OPMD (including OL) | SR | YES | MOOSE PRISMA Cochrane PRISMA-P | YES (CRD42021279108) | Quality in Prognosis Studies (QUIPS) | NO | Funding: none COI: none |
| Iglesias-Velásquez et al. | 2022 | OPMD (including OL) | SR | YES | PRISMA | NO | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: none COI: none |
| Monteiro et al. | 2021 | OL | SR | NO | PRISMA PRISMA-P | YES (CRD42020163464) | Quality in Prognosis Studies (QUIPS) | NO | Funding: yes COI: none |
| Celentano et al. | 2021 | OL | SR | NO | PRISMA | NO | Quality in Prognosis Studies (QUIPS) | NO | Funding: yes COI: none |
| Arroyo et al. | 2021 | OPDM (including OL) | SR | YES | PRISMA | NO | Quality Assessment of Diagnostic Studies-2 (QUADAS-2) | NO | Funding: none COI: none |
| Piyarathne et al. | 2021 | OPDM (including OL) | SR | NO | NO | NO | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: none COI: none |
| Kasradzee et al. | 2020 | OL | SR | NO | PRISMA | YES (PROSPERO: CRD42015026821) | Cochrane Risk of Bias | NO | Funding: none COI: none |
| Morais et al. | 2020 | OPMD (including OL) | SR | NO | PRISMA | NO | REporting recommendations for tumour MARKer prognostic studies (REMARK) statement | NO | Funding: none COI: none |
| Rivera et al. | 2020 | OL | SR | NO | NO | YES (PROSPERO: CRD42018086476) | REporting recommendations for tumour MARKer prognostic studies (REMARK) statement | NO | Funding: none COI: none |
| Ramos García et al. | 2019 | OPMD (including OL) | SR | YES | MOOSE PRISMA Cochrane PRISMA-P | YES (PROSPERO: CRD42019123753) | Quality in Prognosis Studies (QUIPS) | NO | Funding: none COI: none |
| Villa et al. | 2019 | OL | SR | NO | PRISMA | NO | Quality in Prognosis Studies (QUIPS) | NO | Funding: yes COI: none |
| Saluja et al. | 2019 | OPMD (including OL) | SR | YES | PRISMA | NO | REporting recommendations for tumour MARKer prognostic studies (REMARK) statement | NO | Funding: none COI: none |
| Venugopal et al. | 2016 | OPMD (including OL) | SR | NO | NO | NO | Quality Assessment of Diagnostic Studies | NO | Funding: none COI: none |
| Smith et al. | 2009 | OPMD (including OL) | SR | YES | NO | NO | Newcastle–Ottawa Quality Assessment Scale (NOS) | NO | Funding: none COI: none |
| Hallmark of cancer: Sustaining proliferative signaling | |
| Cyclin D1 | 1 study |
| EGFR | 1 study |
| Ki-67 | 1 study |
| Hallmark of cancer: Evading growth suppressors | |
| p53 | 5 studies |
| pRb | 1 study |
| p27 | 1 study |
| p16 | 1 study |
| Hallmark of cancer: Resisting cell death | |
| CYFRA21 | 1 study |
| Hallmark of cancer: Enabling replicative inmortality | |
| BMI1 | 1 study |
| Hallmark of cancer: Angiogenesis | |
| No evidences | 0 studies |
| Hallmark of cancer: Activating invasion and metastasis | |
| Podoplanin | 5 studies |
| ALDH1 | 2 studies |
| CEA | 1 study |
| B-Catenin | 1 study |
| E-Cadherin | 1 study |
| Twist | 1 study |
| MMP9 | 1 study |
| Hallmark of cancer: Deregulating cellular energetics | |
| LDH | 2 studies |
| Hallmark of cancer: Avoiding immune destruction | |
| No evidences | 0 studies |
| Hallmark of cancer: Genome instability and mutation | |
| No evidences | 0 studies |
| Hallmark of cancer: Tumor-promoting inflammation and tumor microenvironment | |
| Interleukin 6 | 3 studies |
| TNF-α | 3 studies |
| Interleukin 1β | 2 studies |
| CD133 | 2 studies |
| Biomarker | Study | Year | Population | Design | Key Results |
|---|---|---|---|---|---|
| Hallmark: Sustaining proliferative signaling | |||||
| EGFR | Cívico-Ortega et al. | 2025 | OPMD (including OL) | SR + MTA | EGFR upregulation was found to be significantly associated with an elevated malignant transformation risk of OPMD (RR = 2.17, 95% CI = 1.73–2.73, p < 0.001). Subgroup analyses demonstrated that OLs also preserved significant results (RR = 1.85, 95% CI = 1.31–2.59; p < 0.001). |
| Ki-67 | Normando et al. | 2023 | OL | SR + MTA | The expression of Ki-67 progressively increased from normal mucosa to OL and oral cancer (p < 0.001). Authors suggested that OL patients overexpressing Ki-67 may have a higher risk of developing OSCC. Furthermore, the expression of Ki-67 also increased from hyperplasia to dysplasia in OLs. Ki-67 is the biomarker for which there is the greatest scientific evidence in the malignant transformation of OLs. |
| Cyclin D1 | Ramos-García et al. | 2019 | OPMD (including OL) | SR + MTA | CCND1/cyclin D1 upregulation was significantly associated with higher OPMD malignant transformation risk (RR = 2.31, 95% CI = 1.46 to 3.64, p < 0.001). Furthermore, the subgroup mta specifically confirmed significant results for OL ((RR = 1.86, 95% CI = 1.13 to 3.06; p = 0.01). |
| Hallmark: Evading growth suppressors | |||||
| pRb | Lopez-Ansio et al. | 2025 | OPMD (including OL) | SR + MTA | The loss of pRb expression was significantly associated with a higher malignant transformation risk of OPMDs (RR = 1.92, 95% CI = 1.25 to 2.94, p = 0.003). The leukoplakia subgroup retained this significant association (p = 0.006), being the OPMD where the loss of pRb expression showed the best predictive value for oral cancer development (RR = 2.00, 95% CI = 1.22 to 3.29). |
| p53 | Normando et al. | 2023 | OL | SR + MTA | The expression of p53 progressively increased from normal mucosa to OL and oral cancer (p < 0.005). Authors suggested that OL patients overexpressing p53 may have a higher risk of developing oral cancer. Furthermore, the expression of p53 also increased from hyperplasia to dysplasia in OLs. p53 is the biomarker for which there is the greatest scientific evidence in the malignant transformation of OLs. |
| Ramos García et al. | 2022 | OPMD (including OL) | SR + MTA | p53 overexpression was significantly associated with a higher risk of malignant transformation in patients with OL (RR = 2.22, 95% CI = 1.35–3.64, p = 0.002). | |
| Monteiro et al. | 2021 | OL | SR | p53 was the most frequently reported protein with significant results in multivariable analyses (p < 0.005). However, no stratified statistical data for OL were presented. | |
| Celentano et al. | 2021 | OL | SR | The authors identified that the loss of p53 expression is the most promising biomarker for predicting malignant transformation of OLs, acting as an independent predictive factor for progression to oral cancer in the primary-level studies systematically reviewed. | |
| Smith et al. | 2009 | OPMD (including OL) | SR + MTA | The risk for cancer progression in p53 positive cases was not significant (RR = 0.96, 95% CI = 0.65 to 1.42; p = 0.27). Nevertheless, it should be noted that the results of the present meta-analysis do not derive from a large sample size (n = 6, primary level studies). | |
| p16 | Lorenzo-Pouso et al. | 2023 | OPMD (including OL) | SR + MTA | CDKN2A/p16IN expression was significantly associated with malignant development (RR = 2.01, 95% CI = 1.36 to 2.96; p < 0.001), including OL. |
| p27 | Villa et al. | 2019 | OL | SR | It was identified that the overexpression of p27 is the most promising biomarker for predicting the malignant transformation of OLs. p27 acted as an independent predictive factor for progression to oral cancer in the systematically reviewed primary-level studies. |
| Hallmark: Resisting cell death | |||||
| CYFRA21 | Arroyo et al. | 2021 | OPMD (including OL) | SR + MTA | The salivary expression of CYFRA21 presented significant differences between oral cancer and OPMD, which included OL (MD = 9.31, 95% CI = 9.014 to 9.619; p < 0.001). |
| Hallmark: Enabling replicative immortality | |||||
| BMI1 | Saluja et al. | 2019 | OPMD (including OL) | SR + MTA | Bmi1 was considered alongside ALDH1 and CD133. The subgroup meta-analysis for OL showed that this combination of biomarkers was significantly associated with higher risk of malignant transformation (RR = 3.19, 95% CI = 2.55 to 3.98). |
| Hallmark: Angiogenesis | |||||
| No evidences | - | - | - | - | - |
| Hallmark: Activating invasion and metastasis | |||||
| Podoplanin | Monteiro et al. | 2024 | OPMD (including OL) | SR + MTA | A high expression of podoplanin is significantly associated with an increased risk of oral cancer development in patients with OL (HR = 3.72, 95% CI = 2.40 to 5.76; p < 0.001) and could serve as a biomarker for oral malignancy of this OPMD. |
| Monteiro et al. | 2021 | OL | SR | Podoplanin and p53 were the most frequently reported proteins with significant results in multivariable analyses (p < 0.005). However, no stratified statistical data for OL were presented. | |
| Celentano et al. | 2021 | OL | SR | The authors identified that the overexpression of podoplanin is the most promising biomarker for predicting the malignant transformation of OL. Podoplanin acted as an independent predictive factor for progression to oral cancer in the systematically reviewed primary-level studies. | |
| Rivera et al. | 2020 | OL | SR | The overexpression of podoplanin (HR = 8.7, 95% CI = 1.8 to 41.6; p = 0.007) in OL was associated with a higher risk of malignant transformation. | |
| Villa et al. | 2019 | OL | SR | It was identified that the overexpression of podoplanin is the most promising biomarker for predicting the malignant transformation of OLs. Podoplanin acted as an independent predictive factor for progression to oral cancer in the systematically reviewed primary-level studies. | |
| CEA | Arroyo et al. | 2021 | OPMD (including OL) | SR + MTA | The salivary expression of CEA presented significant differences between oral cancer and OPMD, which included OL (MD = 25.85, 95% CI = 13.215 to 38.492; p < 0.001). Based on these results it was concluded that CEA harbored diagnostic value when differentiating oral cancer from OPMD. |
| β-catenin | Morais et al. | 2020 | OPMD (including OL) | SR | The results showed a possible value of β-catenin expression between OL with and without dysplasia (p < 0.001). |
| E-cadherin | Morais et al. | 2020 | OPMD (including OL) | SR | Significant differences in E-cadherin immuno-expression were observed between normal epithelium and epithelial dysplasia (p < 0.001). The results showed a possible value of E-cadherin in the prediction of risk of malignant transformation of oral epithelium. |
| Twist | Morais et al. | 2020 | OPMD (including OL) | SR | Significant differences in Twist immuno-expression were observed between normal epithelium and epithelial dysplasia (p < 0.001). The results showed a possible value of Twist in the prediction of risk of malignant transformation of oral epithelium. |
| ALDH1 | Rivera et al. | 2020 | OPMD (including OL) | SR | The overexpression of aldehyde dehydrogenase 1 (ALDH1A1) (HR = 4.2, 95% CI = 2.0 to 8.9; p < 0.001) in OL was associated with a higher risk of malignant transformation. |
| Saluja et al. | 2019 | OPMD (including OL) | SR + MTA | ALDH1 was considered alongside Bmi1 and CD133. The subgroup meta-analysis for OL showed that this combination of biomarkers was significantly associated with higher risk of malignant transformation (RR = 3.19, 95% CI = 2.55 to 3.98). | |
| MMP-9 | Venugopal et al. | 2016 | OPMD (including OL) | SR | The biomarker expression in serum was statistically significant between the OL and healthy control group (p < 0.001) and showed an increase in the progression from OL to oral cancer (p < 0.01). |
| Hallmark: Deregulating cellular energetics | |||||
| LDH | Kumar et al. | 2023 | OPMD (including OL) | SR + MTA | The salivary lactate dehydrogenase (LDH) levels were higher in patients with OL than in healthy control (p < 0.001). This expression biomarker was also higher in patients with head and neck cancer than in patients with OL (p < 0.001), thus indicating that LDH could be useful in early detection of oral cancer. |
| Iglesias-Velásquez et al. | 2022 | OPMD (including OL) | SR + MTA | Salivary lactate dehydrogenase (LDH) was significantly higher in OL patients than in healthy control (SMD = 11.67, 95% CI = 1.01 to 22.33; p = 0.03), though lower than in oral cancer patients (SMD = 5.62, 95% CI = 2.14 to 9.11; p = 0.002). | |
| Hallmark: Avoiding immune destruction | |||||
| No evidences | - | - | - | - | - |
| Hallmark: Genome instability and mutation | |||||
| No evidences | - | - | - | - | - |
| Hallmark: Tumor-promoting inflammation and tumor microenvironment | |||||
| IL-6 | Huang et al. | 2023 | OL | SR + MTA | Salivary interleukin 6 (IL-6) levels were higher in OL than in healthy controls (SMD = −1.07, 95% CI = −1.86 to −0.28) and in oral cancer compared to OL (SMD = −1.01, 95% CI = −1.80 to −0.22). These findings suggest a rising trend in IL-6 levels in OL patients. |
| Piyarathne et al. | 2021 | OPMD (including OL) | SR | Salivary interleukin 6 (IL-6) levels were significantly elevated in patients with OL (p < 0.05), suggesting an altered immune response. Compared to OL patients and healthy controls, individuals with oral cancer showed markedly higher concentrations of IL-6 (p = 0.012). IL-6 expression also increased in parallel with the severity of epithelial dysplasia. | |
| Kasradzee et al. | 2020 | OL | SR | Salivary interleukin 6 (IL-6) levels in saliva showed an increase in oral cancer cases in comparison to healthy controls, patients with OL, smokers, and alcohol consumers (p ≤ 0.05). | |
| IL-1β | Piyarathne et al. | 2021 | OPMD (including OL) | SR | Compared to OL patients and healthy controls, individuals with oral cancer showed markedly higher concentrations of IL-1β (p < 0.001). |
| Kasradzee et al. | 2020 | OL | SR | Salivary concentrations of salivary Interleukin 1-beta (IL-1β) were significantly elevated in patients with oral cancer compared to those with OL and healthy controls (p < 0.005). | |
| TNF-α | Huang et al. | 2023 | OL | SR + MTA | Tumor necrosis factor alpha (TNF-α) levels had a higher expression in OL than in healthy controls (SMD = −0.83, 95% CI = −1.61 to −0.05) and in oral cancer compared to OL (SMD = −0.86, 95% CI = −1.58 to −0.13). These findings suggest a rising trend in TNF-α levels in OL patients. |
| Benito-Ramal et al. | 2023 | OPMD (including OL) | SR + MTA | The difference in tumor necrosis factor alpha (TNF-α) salivary concentration is statistically significant between the OPMD and healthy control (MD = 21.58, 95% CI = 12.72 to 30.45; p < 0.001). However, no stratified results specifically for OL were provided. | |
| Kasradzee et al. | 2020 | OL | SR | Salivary tumor necrosis factor alpha (TNF-α) levels were significantly higher in patients with OL compared to healthy control (p < 0.005). | |
| CD133 | Rivera et al. | 2020 | OL | SR | The overexpression CD133(HR = 2.9, 95% CI = 1.5 to 5.6; p = 0.002) in OL was associated with a higher risk of malignant transformation. |
| Saluja et al. | 2019 | OPMD (including OL) | SR + MTA | CD133 was considered alongside Bmi1 and ALDH1. The subgroup meta-analysis for OL showed that this combination of biomarkers was significantly associated with higher risk of malignant transformation (RR = 3.19, 95% CI = 2.55 to 3.98). | |
| Biomarker | Main Oncogenic Mechanisms Involved in OL Malignant Transformation |
|---|---|
| EGFR | Activation of important molecular signalling pathways (such as MAPK or PI3K/Akt/mTOR). |
| Cyclin D1 | Regulator of the G1/S transition of the cell cycle and uncontrolled proliferation gain. It can be activated by the above molecular signalling pathways or by CCND1 gene amplification. |
| p53 | TP53, known as the guardian of the genome, is mutated early, contributing to a decrease in DNA damage repair and resistance to apoptosis promotion. |
| pRb | pRb is a tumor suppressor protein that inhibits cell proliferation by controlling the cell cycle transition from G1 to S, inducing G1 arrest through the sequestration of E2F transcription factors. This prevents the activation of target genes involved in proliferation. |
| P16 | It is another supressor protein, which exerts a negative regulation of cell proliferation by inhibiting progression through the cell cycle by binding to cyclin-dependent kinases (CDK) 4 or 6 and blocking the action of cyclin D1. |
| Podoplanin | A transmembrane protein involved in the reorganisation of the actin cytoskeleton and the acquisition of a migratory phenotype through the epithelial–mesenchymal transition phenomenon. |
| Cytokines | Cytokines and proinflamatory factors (i.e., IL-6, IL-1β, and TNF-α) generate a pro-oncogenic microenvironment that activates oncogenic pathways such as NF-κB, favoring uncontrolled proliferation and the aquisition of other relevants hallmarks of cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Ruiz, I.; Samayoa-Descamps, V.; Guagua-Cortez, K.A.; González-Moles, M.Á.; Ramos-García, P. Hallmarks of Cancer Expression in Oral Leukoplakia: A Scoping Review of Systematic Reviews and Meta-Analyses. Cancers 2025, 17, 2427. https://doi.org/10.3390/cancers17152427
González-Ruiz I, Samayoa-Descamps V, Guagua-Cortez KA, González-Moles MÁ, Ramos-García P. Hallmarks of Cancer Expression in Oral Leukoplakia: A Scoping Review of Systematic Reviews and Meta-Analyses. Cancers. 2025; 17(15):2427. https://doi.org/10.3390/cancers17152427
Chicago/Turabian StyleGonzález-Ruiz, Isabel, Valerie Samayoa-Descamps, Karen Andrea Guagua-Cortez, Miguel Ángel González-Moles, and Pablo Ramos-García. 2025. "Hallmarks of Cancer Expression in Oral Leukoplakia: A Scoping Review of Systematic Reviews and Meta-Analyses" Cancers 17, no. 15: 2427. https://doi.org/10.3390/cancers17152427
APA StyleGonzález-Ruiz, I., Samayoa-Descamps, V., Guagua-Cortez, K. A., González-Moles, M. Á., & Ramos-García, P. (2025). Hallmarks of Cancer Expression in Oral Leukoplakia: A Scoping Review of Systematic Reviews and Meta-Analyses. Cancers, 17(15), 2427. https://doi.org/10.3390/cancers17152427