
Disrupting Cell Cycle Machinery: CREPT Is an Emerging Target in Cancer Therapy


Simple Summary
Abstract

1. Introduction
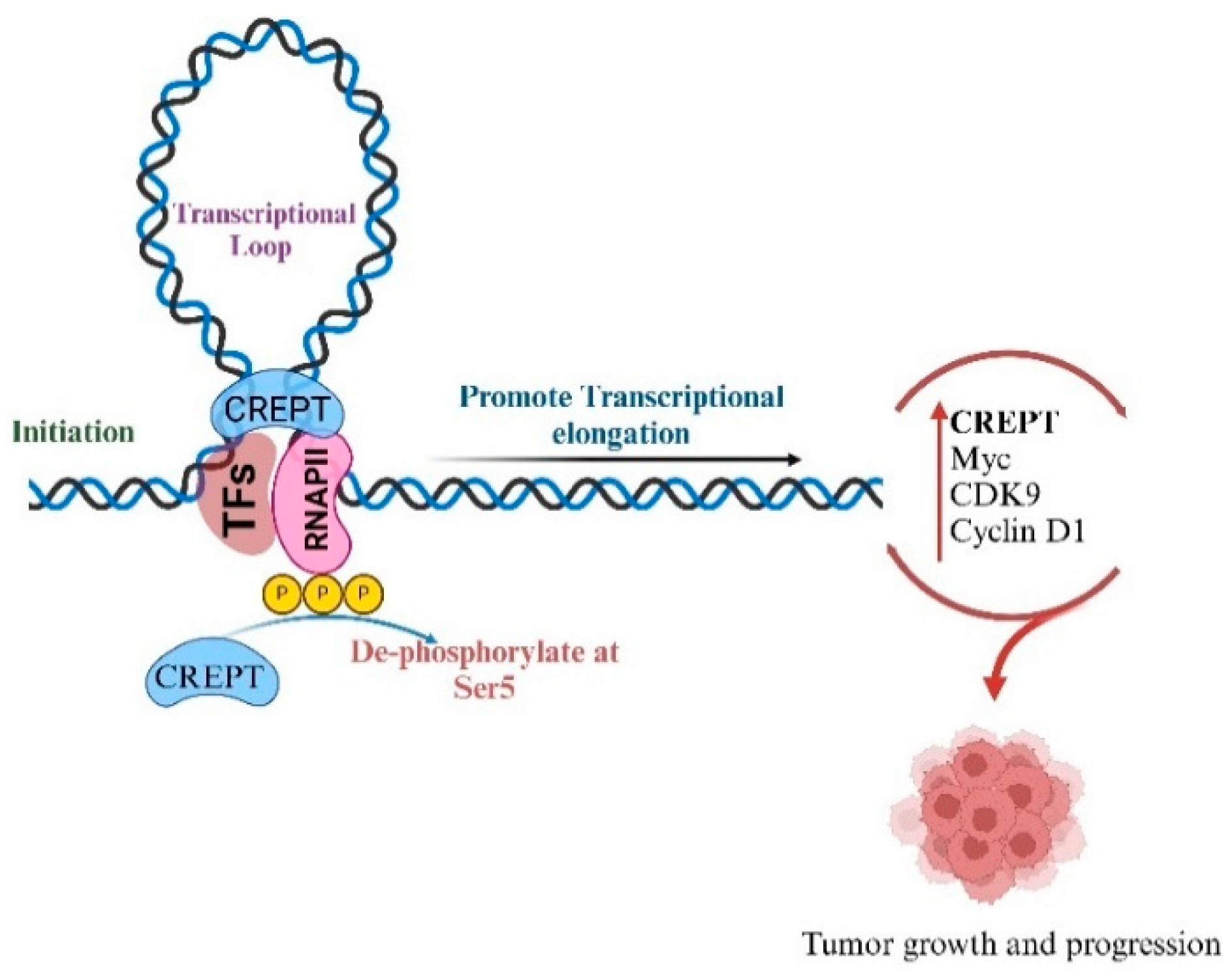
2. Interaction of CREPT with RNA Polymerase II
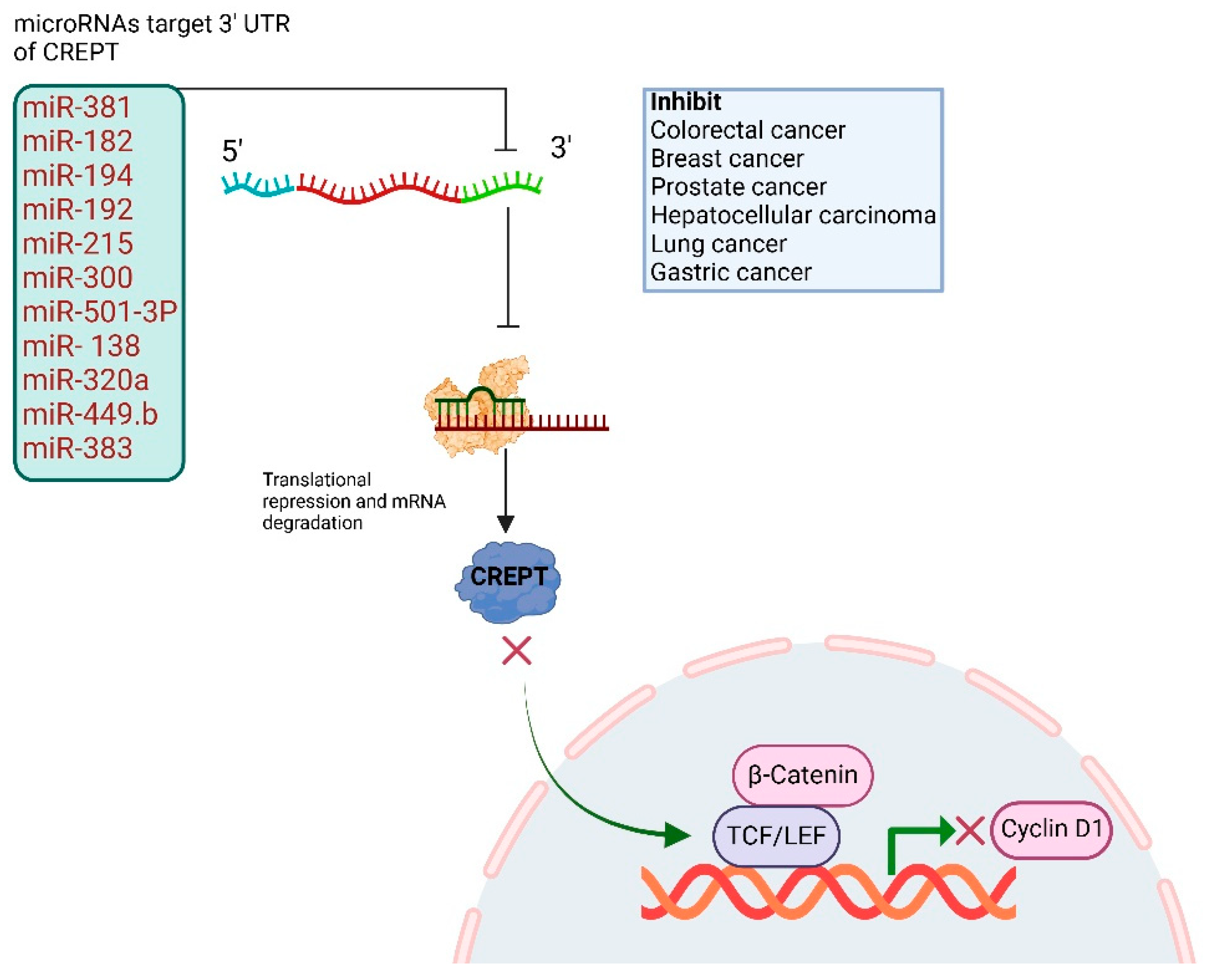
3. CREPT Untranslated Regions and Regulation Through MicroRNAs
4. CREPT’s Role in Cell Cycle: G1/S and G2/M Transition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Study Type | Molecular Pathway | Primary Findings | Ref. |
|---|---|---|---|---|
| Breast cancer | In vitro and in vivo | catenin/TCF4/cyclin D1, miR-138, miR-449b-5p, Wnt/β-catenin | CREPT promotes proliferation/invasion, regulated by miR-138, miR-449b-5p suppresses CREPT, inhibits Wnt signaling, growth/invasion | [11,17] |
| Tumorigenesis | In vitro and in vivo | STAT3, p300, histone acetylation | CREPT enhances STAT3 transcriptional activity via p-STAT3/p300 complex, promoting tumorigenesis | [58] |
| Gastric cancer | In vitro and in vivo | ROS/p53, cell cycle, apoptosis, Aurora B, Cyclin B1, G2/M transition | CREPT knockdown induces cell cycle arrest, apoptosis via ROS/p53, inhibits migration, interacts with Aurora B, regulates Cyclin B1, accelerates G2/M transition | [22,31] |
| Tumorigenesis of multiple cancers | In vitro and in vivo | Wnt/catenin/TCF4, Cyclin D1, RNAPII, chromatin looping, HDAC1, TCF4, Wnt | CREPT acts as co-activator for catenin-TCF4, enhances Wnt signaling, enhances cyclin D1 transcription, competes with HDAC1 for oncogene promoters, promotes oncogene expression | [6,20,59] |
| Colorectal cancer | In vitro and in vivo | Wnt/β-catenin, p300, histone acetylation, Cyclin D3, CDK4/6, miR-383, Wnt/β-catenin, CCND1 | CREPT amplifies Wnt signaling via p300, promotes proliferation/metastasis and cell cycle | [27,30,45] |
| Glioma | In vitro | miR-596, Wnt/beta-catenin | CREPT promotes proliferation/invasion, regulated by miR-596 | [16] |
| Renal cell carcinoma | In vitro, clinical | Cyclin D1, c-myc, RNAPII, Wnt | CREPT overexpression linked to poor prognosis promotes proliferation/cell cycle | [29] |
| Hepatocellular carcinoma | In vitro and in vivo | miR-300 suppresses CREPT/Wnt/β-catenin signaling | CREPT expression promotes cell proliferation and can be targeted by miR-300 in HCC | [7] |
| Diffuse large B-cell lymphoma | In vitro and in vivo and bioinformatics | NF-kB signaling | CREPT promotes proliferation, inhibits apoptosis via NF-kB | [60] |
| Non-small cell lung cancer | In vitro and in vivo | Cyclin D1, RNAPII | CREPT overexpression promotes proliferation, migration, poor prognosis | [10,28] |
| Pancreatic cancer | In vitro, in vivo, and PROTAC | RNAPII, cyclin D1, Wnt, PROTAC | CREPT degradation inhibits proliferation, validates as therapeutic target | [61] |
5. CREPT’s Role in Cell Renewal and Tissue Repair
6. CREPT in Apoptosis Regulation
7. CREPT’s Role in Signaling Pathway
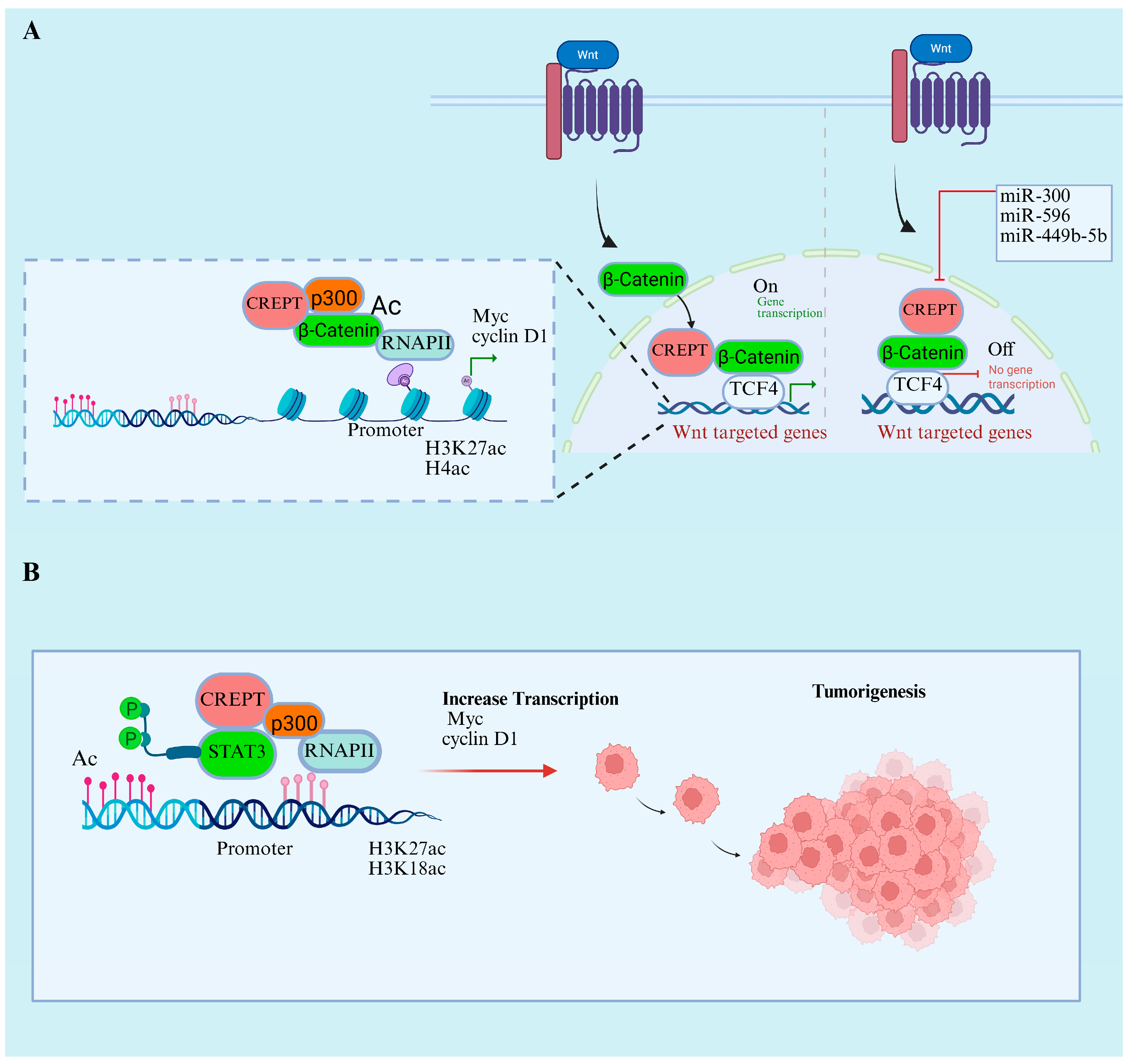
7.1. CREPT’s Role in Wnt/β-Catenin and STAT3 Signaling Pathways
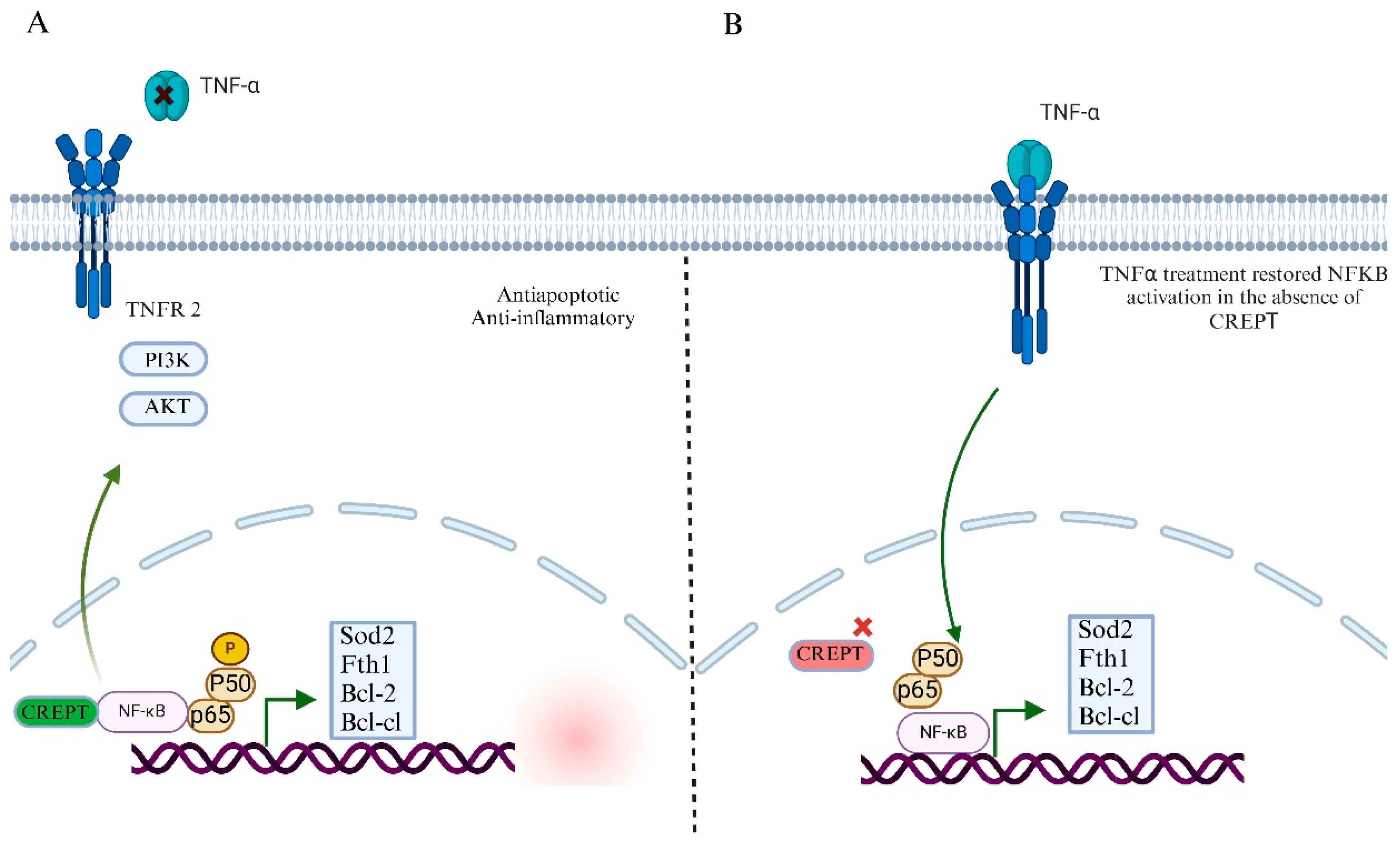
7.2. CREPT’s Role in NF-kB Signaling Pathway and Tumor Microenvironment
8. CREPT’s Role in Metabolic Regulation
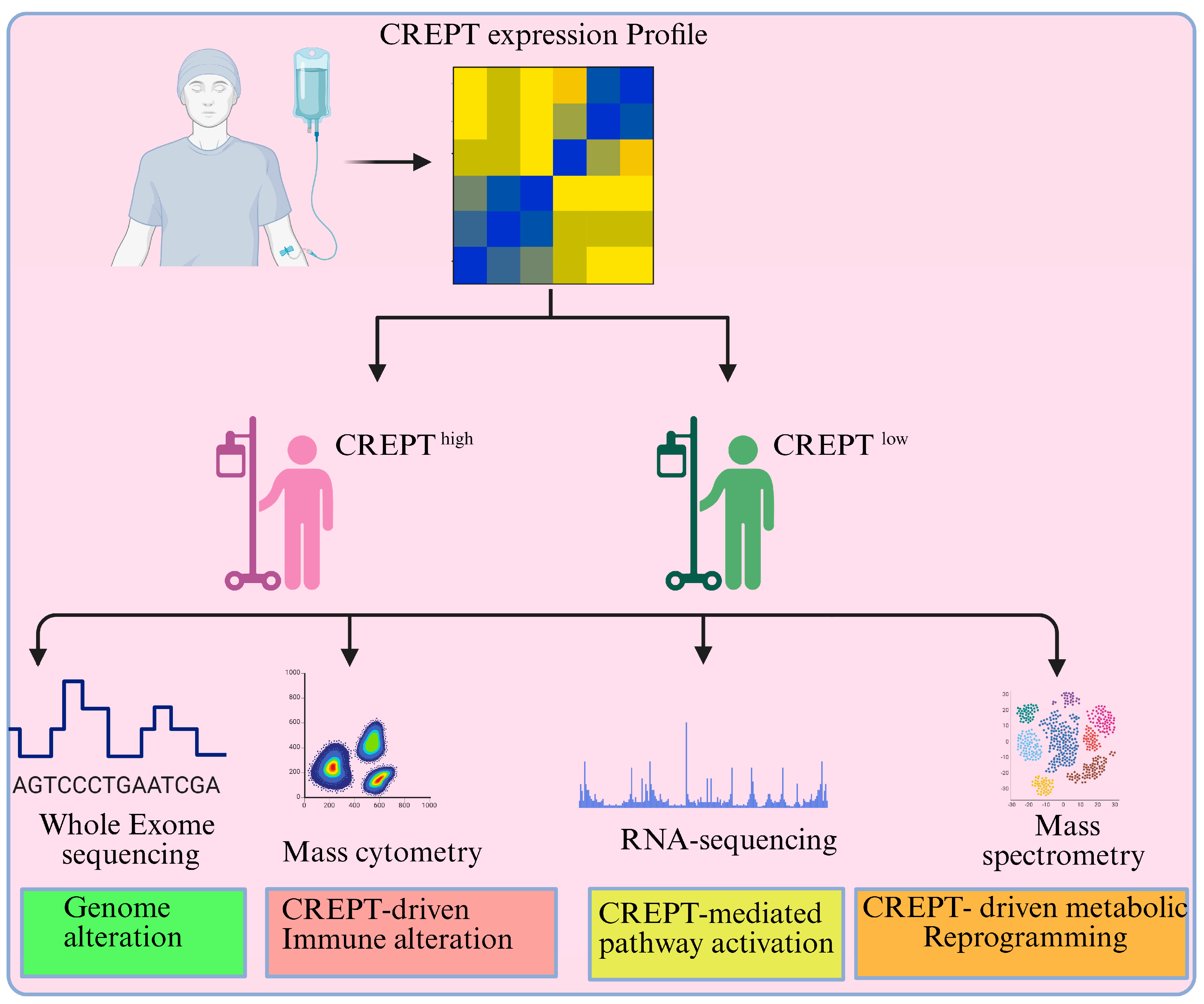
9. Targeting CREPT in Cancer Treatment
10. Future Direction and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choksi, S.P.; Byrnes, L.E.; Konjikusic, M.J.; Tsai, B.W.H.; Deleon, R.; Lu, Q.; Westlake, C.J.; Reiter, J.F. An alternative cell cycle coordinates multiciliated cell differentiation. Nature 2024, 630, 214–221. [Google Scholar] [CrossRef]
- Kiri, S.; Ryba, T. Cancer, metastasis, and the epigenome. Mol. Cancer 2024, 23, 154. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Velez, A.A.; Howard, M. Tumor-suppressor genes, cell cycle regulatory checkpoints, and the skin. N. Am. J. Med. Sci. 2015, 7, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef]
- Lu, D.; Wu, Y.; Wang, Y.; Ren, F.; Wang, D.; Su, F.; Zhang, Y.; Yang, X.; Jin, G.; Hao, X.; et al. CREPT Accelerates Tumorigenesis by Regulating the Transcription of Cell-Cycle-Related Genes. Cancer Cell 2012, 21, 92–104. [Google Scholar] [CrossRef]
- Bai, J.; Gao, Y.; Du, Y.; Yang, X.; Zhang, X. MicroRNA-300 inhibits the growth of hepatocellular carcinoma cells by downregulating CREPT/Wnt/β-catenin signaling. Oncol. Lett. 2019, 18, 3743–3753. [Google Scholar] [CrossRef]
- Kuang, Y.-S.; Wang, Y.; Ding, L.-D.; Yang, L.; Wang, Y.; Liu, S.-H.; Zhu, B.-T.; Wang, X.-N.; Liu, H.-Y.; Li, J.; et al. Overexpression of CREPT confers colorectal cancer sensitivity to fluorouracil. World J. Gastroenterol. 2018, 24, 475–483. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; He, C.; Jiang, G.; Ma, D.; Guan, H.; Lin, Y.; Jia, J.; Duan, X.; Wang, Y.; et al. An oncoprotein CREPT functions as a co-factor in MYC-driven transformation and tumor growth. J. Biol. Chem. 2024, 301, 108030. [Google Scholar] [CrossRef]
- Li, W.; Zheng, G.; Xia, J.; Yang, G.; Sun, J.; Wang, X.; Wen, M.; Sun, Y.; Zhang, Z.; Jin, F. Cell cycle-related and expression-elevated protein in tumor overexpression is associated with proliferation behaviors and poor prognosis in non-small-cell lung cancer. Cancer Sci. 2018, 109, 1012–1023. [Google Scholar] [CrossRef]
- Liang, Z.; Feng, Q.; Xu, L.; Li, S.; Zhou, L. CREPT regulated by miR-138 promotes breast cancer progression. Biochem. Biophys. Res. Commun. 2017, 493, 263–269. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, H.; Li, J.; Ren, F.; Wang, Y.; Qiu, Y.; Li, J.; Li, M.; Wang, Y.; Yang, L.; et al. Microenvironment-induced CREPT expression by cancer-derived small extracellular vesicles primes field cancerization. Theranostics 2024, 14, 662–680. [Google Scholar] [CrossRef]
- Liu, H.; Seynhaeve, A.L.B.; Brouwer, R.W.W.; van Ijcken, W.F.J.; Yang, L.; Wang, Y.; Chang, Z.; Hagen, T.L.M.T. CREPT Promotes Melanoma Progression Through Accelerated Proliferation and Enhanced Migration by RhoA-Mediated Actin Filaments and Focal Adhesion Formation. Cancers 2019, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ma, D.; Chang, Z. Current understanding of CREPT and p15RS, carboxy-terminal domain (CTD)-interacting proteins, in human cancers. Oncogene 2020, 40, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.O.; Bhat, A.Q.; Akhter, Z.; Badsera, N.; Hossain, M.; Showket, F.; Parveen, S.; Dar, M.S.; Tiwari, H.; Kumari, N.; et al. Identification of a novel GSK3β inhibitor involved in abrogating KRas dependent pancreatic tumors in Wnt/beta-catenin and NF-kB dependent manner. Life Sci. 2024, 351, 122840. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Cao, Y.; Jia, D.; Zhao, H.; Zhang, L. CREPT promotes glioma cell proliferation and invasion by activating Wnt/β-catenin pathway and is a novel target of microRNA-596. Biochimie 2019, 162, 116–124. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, X.; He, X.; Ma, W.; Wang, J.; Zhou, Q.; Li, M.; Yu, S. MicroRNA-449b-5p suppresses the growth and invasion of breast cancer cells via inhibiting CREPT-mediated Wnt/β-catenin signaling. Chem. Interact. 2019, 302, 74–82. [Google Scholar] [CrossRef]
- Li, J.; Xu, L.; Wang, J.; Wang, X.; Lin, Y.; Zou, A.Y.; Ren, F.; Wang, Y.; Li, J.; Chang, Z. CREPT is required for the metastasis of triple-negative breast cancer through a co-operational-chromatin loop-based gene regulation. Mol. Cancer 2025, 24, 170. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Li, J.; Wang, Y.; Ren, F.; Zhou, Y.; Wu, Y.; Feng, Y.; Zhou, Y.; Su, F.; et al. p15RS/RPRD1A (p15INK4b-related Sequence/Regulation of Nuclear Pre-mRNA Domain-containing Protein 1A) Interacts with HDAC2 in Inhibition of the Wnt/β-Catenin Signaling Pathway. J. Biol. Chem. 2015, 290, 9701–9713. [Google Scholar] [CrossRef]
- Cao, Y.; Ning, B.; Tian, Y.; Lan, T.; Chu, Y.; Ren, F.; Wang, Y.; Meng, Q.; Li, J.; Jia, B.; et al. CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis. Cancers 2022, 14, 4797. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, H.; Hu, W.; Li, S.; Yu, J. RPRD1B promotes tumor growth by accelerating the cell cycle in endometrial cancer. Oncol. Rep. 2014, 31, 1389–1395. [Google Scholar] [CrossRef]
- Ding, L.; Yang, L.; He, Y.; Zhu, B.; Ren, F.; Fan, X.; Wang, Y.; Li, M.; Li, J.; Kuang, Y.; et al. CREPT/RPRD1B associates with Aurora B to regulate Cyclin B1 expression for accelerating the G2/M transition in gastric cancer. Cell Death Dis. 2018, 9, 1172. [Google Scholar] [CrossRef]
- Wang, M.; Wu, B.; Tang, K.; Wang, X.; Liu, X.; Duan, Y.; Wang, J.; Wang, X.; Wang, Y.; Li, J.; et al. Cell-Cycle-Related and Expression Elevated Protein in Tumor Upregulates the Antioxidant Genes via Activation of NF-κB/Nrf2 in Acute Liver Injury. Toxics 2024, 12, 893. [Google Scholar] [CrossRef]
- Yang, L.; Yang, H.; Chu, Y.; Song, Y.; Ding, L.; Zhu, B.; Zhai, W.; Wang, X.; Kuang, Y.; Ren, F.; et al. CREPT is required for murine stem cell maintenance during intestinal regeneration. Nat. Commun. 2021, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yan, Q.; Zheng, Y.; Li, L.; Zhang, B.; Chang, Z.; Wang, Z.; Tang, H.; Qin, Y.; Guan, X.-Y. Long non-coding RNA NEAT1 mediated RPRD1B stability facilitates fatty acid metabolism and lymph node metastasis via c-Jun/c-Fos/SREBP1 axis in gastric cancer. J. Exp. Clin. Cancer Res. 2022, 41, 287. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Huang, H.; Wang, S.; Xu, H.; Xue, Y.; Huang, Y.; He, J.; Xu, X.; Wu, Z.; Wu, J.; et al. CREPT is a novel predictor of the response to adjuvant therapy or concurrent chemoradiotherapy in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 3301–3310. [Google Scholar] [PubMed]
- Zhang, Y.; Wang, S.; Kang, W.; Liu, C.; Dong, Y.; Ren, F.; Wang, Y.; Zhang, J.; Wang, G.; To, K.F.; et al. CREPT facilitates colorectal cancer growth through inducing Wnt/β-catenin pathway by enhancing p300-mediated β-catenin acetylation. Oncogene 2018, 37, 3485–3500. [Google Scholar] [CrossRef]
- Liu, T.; Li, W.-M.; Wang, W.-P.; Sun, Y.; Ni, Y.-F.; Xing, H.; Xia, J.-H.; Wang, X.-J.; Zhang, Z.-P.; Li, X.-F. Inhibiting CREPT reduces the proliferation and migration of non-small cell lung cancer cells by down-regulating cell cycle related protein. Am. J. Transl. Res. 2016, 8, 2097–2113. [Google Scholar]
- Yin, H.; Cao, Q.; Zhao, H.; Wang, S.; Chen, W.; Zhang, X.; Chang, Z.; Xu, T.; Ye, X. Expression of CREPT is associated with poor prognosis of patients with renal cell carcinoma. Oncol. Lett. 2019, 18, 4789–4797. [Google Scholar] [CrossRef]
- Zheng, G.; Li, W.; Zuo, B.; Guo, Z.; Xi, W.; Wei, M.; Chen, P.; Wen, W.; Yang, A.-G. High expression of CREPT promotes tumor growth and is correlated with poor prognosis in colorectal cancer. Biochem. Biophys. Res. Commun. 2016, 480, 436–442. [Google Scholar] [CrossRef]
- Sun, M.; Si, G.; Sun, H.-S.; Si, F.-C. Inhibition of CREPT restrains gastric cancer growth by regulation of cycle arrest, migration and apoptosis via ROS-regulated p53 pathway. Biochem. Biophys. Res. Commun. 2018, 496, 1183–1190. [Google Scholar] [CrossRef]
- She, Y.; Liang, J.; Chen, L.; Qiu, Y.; Liu, N.; Zhao, X.; Huang, X.; Wang, Y.; Ren, F.; Chang, Z.; et al. CREPT expression correlates with poor prognosis in patients with retroperitoneal leiomyosarcoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6596–6605. [Google Scholar] [PubMed]
- Ni, Z.; Olsen, J.B.; Guo, X.; Zhong, G.; Ruan, E.D.; Marcon, E.; Young, P.; Guo, H.; Li, J.; Moffat, J.; et al. Control of the RNA polymerase II phosphorylation state in promoter regions by CTD interaction domain-containing proteins RPRD1A and RPRD1B. Transcription 2011, 2, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Drobnjak, M.; Osman, I.; I Scher, H.; Fazzari, M.; Cordon-Cardo, C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin. Cancer Res. 2000, 6, 1891–1895. [Google Scholar] [PubMed]
- Cai, Z.; Wang, J.; Li, Y.; Shi, Q.; Jin, L.; Li, S.; Zhu, M.; Wang, Q.; Wong, L.L.; Yang, W.; et al. Overexpressed Cyclin D1 and CDK4 proteins are responsible for the resistance to CDK4/6 inhibitor in breast cancer that can be reversed by PI3K/mTOR inhibitors. Sci. China Life Sci. 2022, 66, 94–109. [Google Scholar] [CrossRef]
- Fang, M.; Wu, H.-K.; Pei, Y.; Zhang, Y.; Gao, X.; He, Y.; Chen, G.; Lv, F.; Jiang, P.; Li, Y.; et al. E3 ligase MG53 suppresses tumor growth by degrading cyclin D1. Signal Transduct. Target. Ther. 2023, 8, 263. [Google Scholar] [CrossRef]
- Tchakarska, G.; Sola, B. The double dealing of cyclin D1. Cell Cycle 2020, 19, 163–178. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Chen, S.; Li, L. Degradation strategy of cyclin D1 in cancer cells and the potential clinical application. Front. Oncol. 2022, 12, 949688. [Google Scholar] [CrossRef]
- Ali, I.; Ruiz, D.G.; Ni, Z.; Johnson, J.R.; Zhang, H.; Li, P.-C.; Khalid, M.M.; Conrad, R.J.; Guo, X.; Min, J.; et al. Crosstalk between RNA Pol II C-Terminal Domain Acetylation and Phosphorylation via RPRD Proteins. Mol. Cell 2019, 74, 1164–1174.e4. [Google Scholar] [CrossRef]
- Siniscalco, D.; Galderisi, U.; Peluso, G.; Finicelli, M. Circulating microRNAs in Cancer: A 5-Year Update with a Focus on Breast and Lung Cancers. Int. J. Mol. Sci. 2024, 25, 3140. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Chaubey, D.; Joshi, D.C.; Ranmale, S.; Pillai, B. Noncoding RNAs: Emerging regulators of behavioral complexity. Wiley Interdiscip. Rev. RNA 2024, 15, e1847. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.L.; Hsieh, A.C. The Untranslated Regions of mRNAs in Cancer. Trends Cancer 2019, 5, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yang, Y.; Saito, M.; Park, C.Y.; Funato, K.; Tabar, V.; Darnell, R.B. NOVA1 acts as an oncogenic RNA-binding protein to regulate cholesterol homeostasis in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 2024, 121, e2314695121. [Google Scholar] [CrossRef]
- Li, J.; Smith, A.R.; Marquez, R.T.; Li, J.; Li, K.; Lan, L.; Wu, X.; Zhao, L.; Ren, F.; Wang, Y.; et al. MicroRNA-383 acts as a tumor suppressor in colorectal cancer by modulating CREPT/RPRD1B expression. Mol. Carcinog. 2018, 57, 1408–1420. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, L.; Wang, Y.; Luo, X. MicroRNA-501–3p restricts prostate cancer growth through regulating cell cycle-related and expression-elevated protein in tumor/cyclin D1 signaling. Biochem. Biophys. Res. Commun. 2019, 509, 746–752. [Google Scholar] [CrossRef]
- Wang, Z. Cell Cycle Progression and Synchronization: An Overview. Methods Mol Biol 2022, 2579, 3–23. [Google Scholar]
- Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2021, 23, 74–88. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Poratti, M.; Marzaro, G. Corrigendum to: Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib [Eur. J. Med. Chem. 172 (2019) 143–153]. Eur. J. Med. Chem. 2020, 195, 112272. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B.; et al. INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov. 2021, 12, 356–371. [Google Scholar] [CrossRef]
- Hao, Y.-H.; Lafita-Navarro, M.C.; Zacharias, L.; Borenstein-Auerbach, N.; Kim, M.; Barnes, S.; Kim, J.; Shay, J.; DeBerardinis, R.J.; Conacci-Sorrell, M. Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun. Signal. 2019, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Wen, N.; Bian, L.; Gong, J.; Meng, Y. Overexpression of cell-cycle related and expression-elevated protein in tumor (CREPT) in malignant cervical cancer. J. Int. Med. Res. 2020, 48, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Shi, Y.; Wang, T.; Kong, X.; Bu, R.; Ren, Y. Elevated CREPT Expression Enhances the Progression of Salivary Gland Adenoid Cystic Carcinoma. J. Hard Tissue Biol. 2021, 30, 273–282. [Google Scholar] [CrossRef]
- Ma, J.; Ren, Y.; Zhang, L.; Kong, X.; Wang, T.; Shi, Y.; Bu, R.; Ahmad, A. Knocking-down of CREPT prohibits the progression of oral squamous cell carcinoma and suppresses cyclin D1 and c-Myc expression. PLoS ONE 2017, 12, e0174309. [Google Scholar] [CrossRef]
- Lu, W.-J.; Peng, W.; Sun, Q.-Q.; Li, Y.-H.; Chen, B.; Yu, L.-T.; Xu, Y.-Z.; Wang, S.-Y.; Zhao, Y.-L. #2714, a novel active inhibitor with potent G2/M phase arrest and antitumor efficacy in preclinical models. Cell Death Discov. 2018, 4, 24. [Google Scholar] [CrossRef]
- Zhai, W.; Ye, X.; Wang, Y.; Feng, Y.; Wang, Y.; Lin, Y.; Ding, L.; Yang, L.; Wang, X.; Kuang, Y.; et al. CREPT/RPRD1B promotes tumorigenesis through STAT3-driven gene transcription in a p300-dependent manner. Br. J. Cancer 2021, 124, 1437–1448. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Duan, X.; Ren, F.; Li, S.; Jin, Z.; Wang, Y.; Feng, Y.; Liu, Z.; Chang, Z. CREPT/RPRD1B, a Recently Identified Novel Protein Highly Expressed in Tumors, Enhances the β-Catenin·TCF4 Transcriptional Activity in Response to Wnt Signaling. J. Biol. Chem. 2014, 289, 22589–22599. [Google Scholar] [CrossRef]
- Xu, L.; Xie, Z.-H.; Li, J.; Tao, S.; Ren, F.-L.; Wang, Y.-Y.; Chang, Z.-J.; Hao, X.-B. RPRD1B/CREPT facilitates the progression of diffuse large B-cell lymphoma by inhibiting apoptosis through the NF-κB signaling pathway. Asian Pac. J. Trop. Biomed. 2024, 14, 307–317. [Google Scholar] [CrossRef]
- Ma, D.; Zou, Y.; Chu, Y.; Liu, Z.; Liu, G.; Chu, J.; Li, M.; Wang, J.; Sun, S.-Y.; Chang, Z. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics 2020, 10, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Li, M.; Duan, X.; Jia, H.; Cao, Y.; Wang, Y.; Ren, F.; Sheng, J.; Xu, J.; Chang, Z. Regulation of revival stem cell differentiation by CREPT/RPRD1B during intestinal regeneration. Cell Biosci. 2025, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E. Apoptosis (programmed cell death) and its signals—A review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’ORazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Travaglino, A.; Russo, D.; Varricchio, S.; Picardi, M.; Mascolo, M. Prognostic value of Bcl2 and p53 in Hodgkin lymphoma: A systematic review and meta-analysis. Pathol. Res. Pract. 2021, 219, 153370. [Google Scholar] [CrossRef]
- Thomas, S.; A Quinn, B.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.-Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2012, 17, 61–75. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2013, 15, 46–61. [Google Scholar] [CrossRef]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-κB, IκB, and IKK: Integral Components of Immune System Signaling. Adv. Exp. Med. Biol. 2019, 1172, 207–226. [Google Scholar] [CrossRef]
- Guler, A.; Hamurcu, Z.; Ulutabanca, H.; Cınar, V.; Nurdinov, N.; Erdem, S.; Ozpolat, B. Flavopiridol Suppresses Cell Proliferation and Migration and Induces Apoptotic Cell Death by Inhibiting Oncogenic FOXM1 Signaling in IDH Wild-Type and IDH-Mutant GBM Cells. Mol. Neurobiol. 2023, 61, 1061–1079. [Google Scholar] [CrossRef]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, C.; Lv, M.; Yu, J.; Cheng, S.; Cui, X.; Wan, X.; Ahmad, M.; Xu, B.; Qin, J.; et al. Trifluoromethyl quinoline derivative targets inhibiting HDAC1 for promoting the acetylation of histone in cervical cancer cells. Eur. J. Pharm. Sci. 2024, 194, 106706. [Google Scholar] [CrossRef] [PubMed]
- Eblen, S.T. Chapter Four—Extracellular-Regulated Kinases: Signaling from Ras to ERK Substrates to Control Biological Outcomes. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: New York, NY, USA, 2018; Volume 138, pp. 99–142. [Google Scholar]
- Diehl, F.F.; Sapp, K.M.; Heiden, M.G.V. The bidirectional relationship between metabolism and cell cycle control. Trends Cell Biol. 2023, 34, 136–149. [Google Scholar] [CrossRef]
- Patidar, P.L.; Motea, E.A.; Fattah, F.J.; Zhou, Y.; Morales, J.C.; Xie, Y.; Garner, H.R.; Boothman, D.A. The Kub5-Hera/RPRD1B interactome: A novel role in preserving genetic stability by regulating DNA mismatch repair. Nucleic Acids Res. 2016, 44, 1718–1731. [Google Scholar] [CrossRef]
- Schnall-Levin, M.; Rissland, O.S.; Johnston, W.K.; Perrimon, N.; Bartel, D.P.; Berger, B. Unusually effective microRNA targeting within repeat-rich coding regions of mammalian mRNAs. Genome Res. 2011, 21, 1395–1403. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, U.; Li, J.; Chang, Z. Disrupting Cell Cycle Machinery: CREPT Is an Emerging Target in Cancer Therapy. Cancers 2025, 17, 2401. https://doi.org/10.3390/cancers17142401
Farooq U, Li J, Chang Z. Disrupting Cell Cycle Machinery: CREPT Is an Emerging Target in Cancer Therapy. Cancers. 2025; 17(14):2401. https://doi.org/10.3390/cancers17142401
Chicago/Turabian StyleFarooq, Umar, Jun Li, and Zhijie Chang. 2025. "Disrupting Cell Cycle Machinery: CREPT Is an Emerging Target in Cancer Therapy" Cancers 17, no. 14: 2401. https://doi.org/10.3390/cancers17142401
APA StyleFarooq, U., Li, J., & Chang, Z. (2025). Disrupting Cell Cycle Machinery: CREPT Is an Emerging Target in Cancer Therapy. Cancers, 17(14), 2401. https://doi.org/10.3390/cancers17142401