
Genes Encoding Multiple Modulators of the Immune Response Are Methylated in the Prostate Tumor Microenvironment of African Americans

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Data
2.2. Sample Preparation and DNA Methylation Enrichment
2.3. Pathway and Network Analysis
2.4. Global DNA Methylation Analysis of AA and EA TAS Using Long Interspersed Nuclear Element-1 (LINE-1)
2.5. Carcinoma-Associated Fibroblast (CAF) Tissue Culture
2.6. DNA Methylation Analysis
2.7. Tumor Suppressor Gene Analysis
2.8. Global DNA Methylation Analysis Using LINE-1 Assay
3. Results
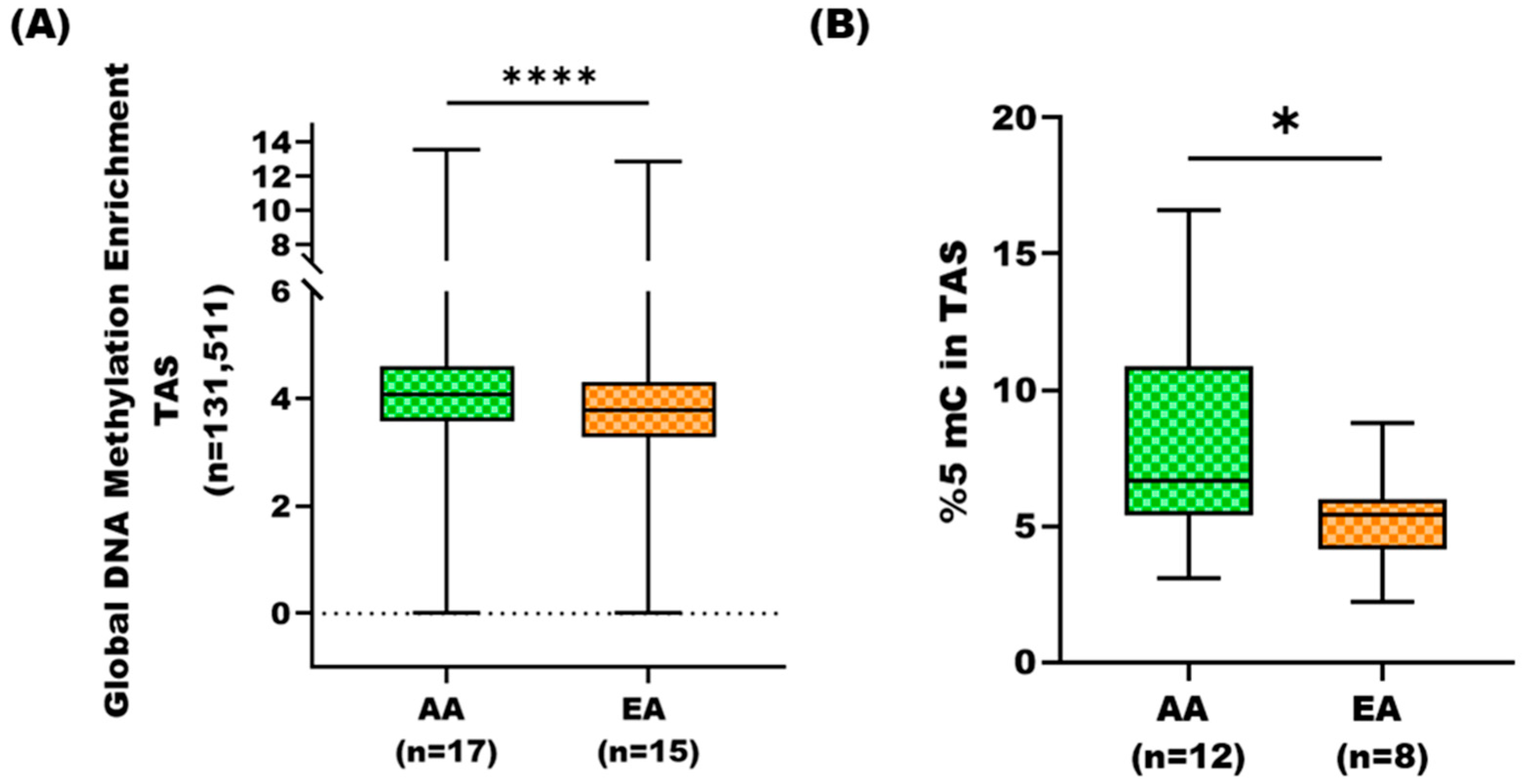
3.1. DNA Methylation Profiles of Tumor-Adjacent Stroma in African American and European American Prostate Cancer Patients
3.2. Global DNA Methylation Analysis of AA TAS and EA TAS Using Long Interspersed Nucleotide Elements Assay
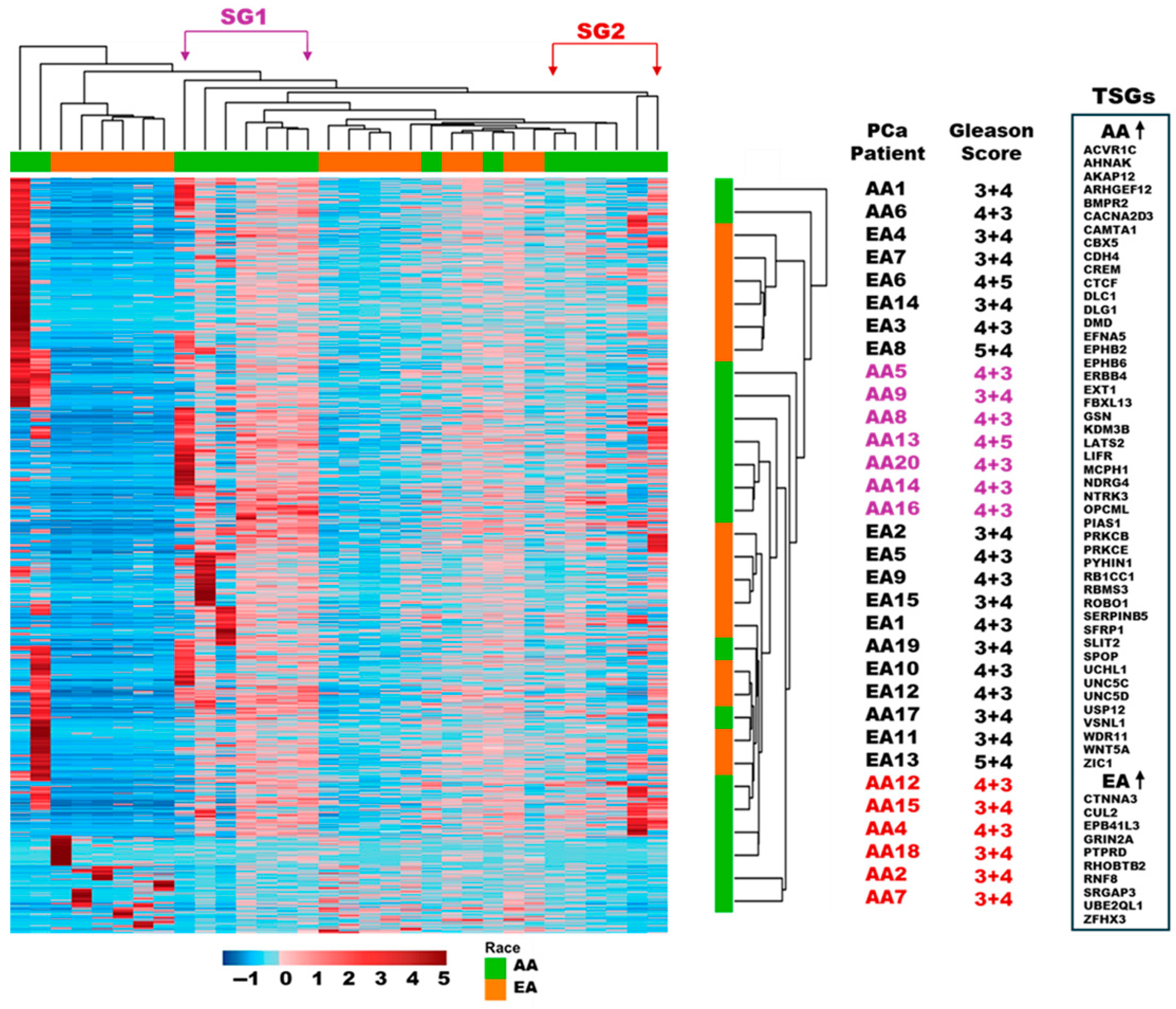
3.3. Differentially Methylated Regions/Genes in the Tumor-Adjacent Stroma of AA vs. EA PCa Patients
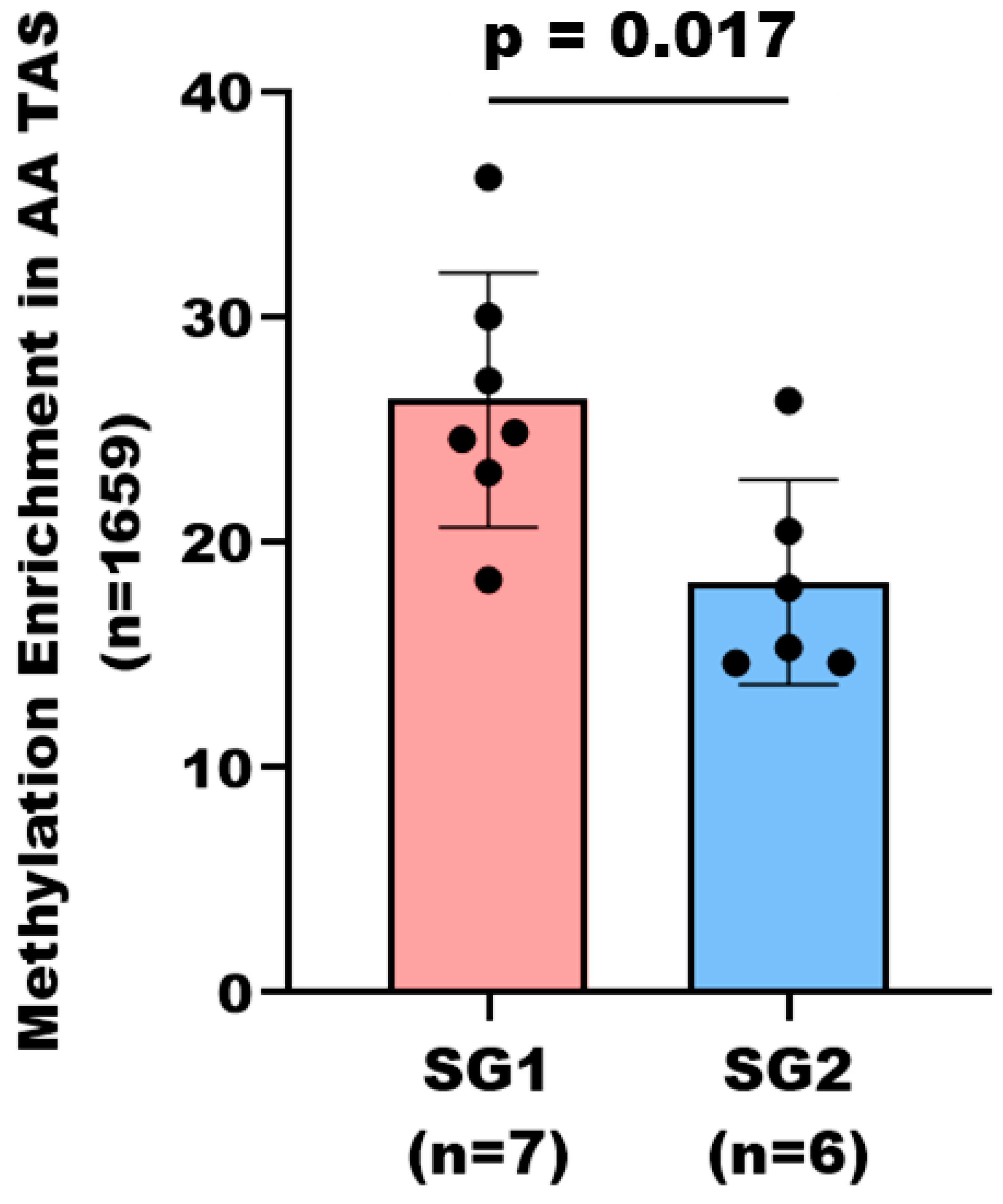
3.4. Subgroup Analysis of DNA Methylation Patterns in AA TAS
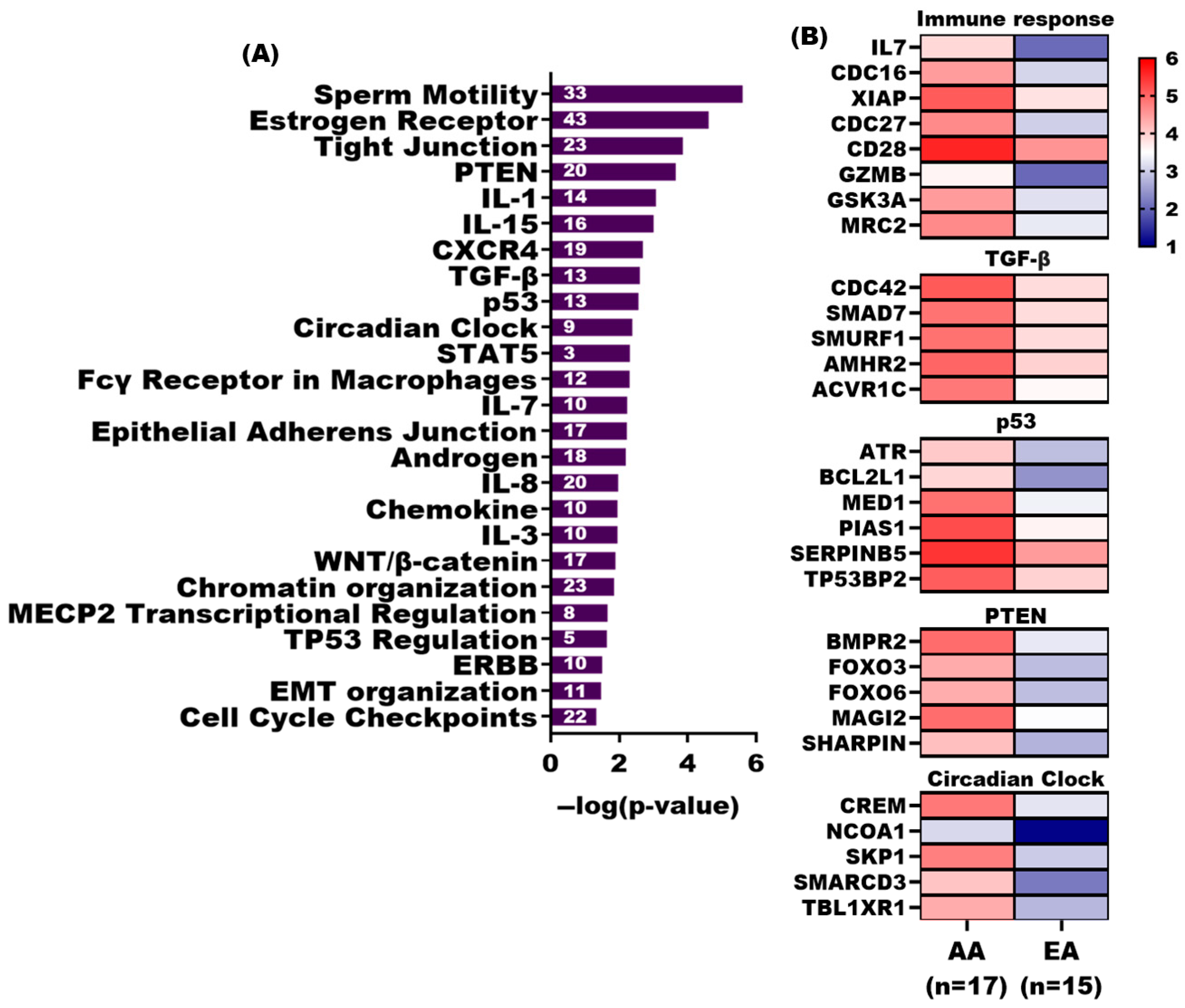
3.5. Pathway Analysis of Differentially Methylated Genes in TAS of AA and EA Patients
3.6. Network Analysis of Differentially Methylated Genes in AA and EA TAS
3.7. Development and Characterization of Carcinoma-Associated Fibroblasts (CAFs) from Primary Prostate Tumors
3.8. Comparative Analysis of DNA Methylation Patterns Between Carcinoma-Associated Fibroblast Derived from Primary Prostate Tumors and Tumor-Adjacent Stroma from PCa Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-AzaC | 5-Azacytidine |
| AA | African American |
| CAFs | carcinoma-associated fibroblasts |
| DMRs | differentially methylated regions |
| d0 | before treatment |
| d1 | 1 day after treatment |
| d10 | 10 days post-treatment |
| EA | European American |
| MACS | model-based analysis of ChIP-Seq |
| MBD-seq | methyl-binding domain sequencing |
| PCa | prostate cancer |
| SG1 | subgroup 1 |
| SG2 | subgroup 2 |
| TAS | tumor-adjacent stroma |
| TSGs | tumor suppressor genes |
| UCI | University of California Irvine |
References
- McGinley, K.F.; Tay, K.J.; Moul, J.W. Prostate cancer in men of African origin. Nat. Rev. Urol. 2016, 13, 99–107. [Google Scholar] [CrossRef]
- Thompson, I.; Tangen, C.; Tolcher, A.; Crawford, E.; Eisenberger, M.; Moinpour, C. Association of African-American ethnic background with survival in men with metastatic prostate cancer. J. Natl. Cancer Inst. 2001, 93, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Powell, I.J.; Bollig-Fischer, A. Minireview: The molecular and genomic basis for prostate cancer health disparities. Mol. Endocrinol. 2013, 27, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Hayward, S.W.; Wang, Y.Z. Role of stroma in carcinogenesis of the prostate. Differentiation 2002, 70, 473–485. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef]
- Gonzalez, L.O.; Eiro, N.; Fraile, M.; Beridze, N.; Escaf, A.R.; Escaf, S.; Fernandez-Gomez, J.M.; Vizoso, F.J. Prostate Cancer Tumor Stroma: Responsibility in Tumor Biology, Diagnosis and Treatment. Cancers 2022, 14, 4412. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef]
- Owens, B.M. Inflammation, Innate Immunity, and the Intestinal Stromal Cell Niche: Opportunities and Challenges. Front. Immunol. 2015, 6, 319. [Google Scholar] [CrossRef]
- Hergovits, S.; Mais, C.; Haan, C.; Costa-Pereira, A.P.; Hermanns, H.M. Oncostatin M induces RIG-I and MDA5 expression and enhances the double-stranded RNA response in fibroblasts. J. Cell Mol. Med. 2017, 21, 3087–3099. [Google Scholar] [CrossRef]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef]
- Chao, C.R.; Slezak, J.; Siegmund, K.; Cannavale, K.; Shu, Y.H.; Chien, G.W.; Chen, X.F.; Shi, F.; Song, N.; Van Den Eeden, S.K.; et al. Genome-wide methylation profiling of diagnostic tumor specimens identified DNA methylation markers associated with metastasis among men with untreated localized prostate cancer. Cancer Med. 2023, 12, 18837–18849. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Falasca, M.; Oon, C.E. Cancer-Associated Fibroblasts: Epigenetic Regulation and Therapeutic Intervention in Breast Cancer. Cancers 2020, 12, 2949. [Google Scholar] [CrossRef]
- Zhang, M.W.; Fujiwara, K.; Che, X.; Zheng, S.; Zheng, L. DNA methylation in the tumor microenvironment. J. Zhejiang Univ. Sci. B 2017, 18, 365–372. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Pidsley, R.; Niranjan, B.; Papargiris, M.; Pereira, B.A.; Richards, M.; Teng, L.; Norden, S.; Ryan, A.; Frydenberg, M.; et al. Alterations in the methylome of the stromal tumour microenvironment signal the presence and severity of prostate cancer. Clin. Epigenetics 2020, 12, 48. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zheng, L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Kinseth, M.A.; Jia, Z.; Rahmatpanah, F.; Sawyers, A.; Sutton, M.; Wang-Rodriguez, J.; Mercola, D.; McGuire, K.L. Expression differences between African American and Caucasian prostate cancer tissue reveals that stroma is the site of aggressive changes. Int. J. Cancer 2014, 134, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rahmatpanah, F.; Robles, G.; Lilly, M.; Keane, T.; Kumar, V.; Mercola, D.; Randhawa, P.; McClelland, M. RNA expression differences in prostate tumors and tumor-adjacent stroma between Black and White Americans. Oncotarget 2021, 12, 1457–1469. [Google Scholar] [CrossRef]
- Alldredge, J.; Randall, L.; De Robles, G.; Agrawal, A.; Mercola, D.; Liu, M.; Randhawa, P.; Edwards, R.; McClelland, M.; Rahmatpanah, F. Transcriptome Analysis of Ovarian and Uterine Clear Cell Malignancies. Front. Oncol. 2020, 10, 598579. [Google Scholar] [CrossRef]
- Feng, J.; Liu, T.; Zhang, Y. Using MACS to identify peaks from ChIP-Seq data. Curr. Protoc. Bioinformatics 2011, 34, 2–14. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Tuxhorn, J.A.; Wheeler, T.M.; Frolov, A.; Scardino, P.T.; Ohori, M.; Wheeler, M.; Spitler, J.; Rowley, D.R. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 4792–4801. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Barrett, R.L.; Pure, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, Y.; Sawyers, A.; Yao, H.; Rahmatpanah, F.; Xia, X.Q.; Xu, Q.; Pio, R.; Turan, T.; Koziol, J.A.; et al. Diagnosis of prostate cancer using differentially expressed genes in stroma. Cancer Res. 2011, 71, 2476–2487. [Google Scholar] [CrossRef]
- Kumar, V.; Randhawa, P.; Bilodeau, R.; Mercola, D.; McClelland, M.; Agrawal, A.; Nguyen, J.; Castro, P.; Ittmann, M.M.; Rahmatpanah, F. Spatial Profiling of the Prostate Cancer Tumor Microenvironment Reveals Multiple Differences in Gene Expression and Correlation with Recurrence Risk. Cancers 2022, 14, 4923. [Google Scholar] [CrossRef]
- Yang, N.; Zhang, L.; Kazazian, H.H., Jr. L1 retrotransposon-mediated stable gene silencing. Nucleic Acids Res. 2005, 33, e57. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.E. Epigenetic Modulators and the New Immunotherapies. N. Engl. J. Med. 2016, 374, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Tian, L.; Bian, T.; Zhao, H.; Tao, J.; Feng, L.; Liu, Q.; Hou, H. The role of CD28 in the prognosis of young lung adenocarcinoma patients. BMC Cancer 2020, 20, 910. [Google Scholar] [CrossRef]
- Mareboina, M.; Bakhl, K.; Agioti, S.; Yee, N.S.; Georgakopoulos-Soares, I.; Zaravinos, A. Comprehensive Analysis of Granzymes and Perforin Family Genes in Multiple Cancers. Biomedicines 2025, 13, 408. [Google Scholar] [CrossRef]
- Fujio, K.; Watanabe, M.; Ueki, H.; Li, S.A.; Kinoshita, R.; Ochiai, K.; Futami, J.; Watanabe, T.; Nasu, Y.; Kumon, H. A vaccine strategy with multiple prostatic acid phosphatase-fused cytokines for prostate cancer treatment. Oncol. Rep. 2015, 33, 1585–1592. [Google Scholar] [CrossRef]
- Wilson, C.; Wilson, T.; Johnston, P.G.; Longley, D.B.; Waugh, D.J. Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in prostate cancer cells. Mol. Cancer Ther. 2008, 7, 2649–2661. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: Beta-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43. [Google Scholar] [CrossRef]
- Sartor, O.; Armstrong, A.J.; Ahaghotu, C.; McLeod, D.G.; Cooperberg, M.R.; Penson, D.F.; Kantoff, P.W.; Vogelzang, N.J.; Hussain, A.; Pieczonka, C.M.; et al. Survival of African-American and Caucasian men after sipuleucel-T immunotherapy: Outcomes from the PROCEED registry. Prostate Cancer Prostatic Dis. 2020, 23, 517–526. [Google Scholar] [CrossRef]
- Dasari, S.S.; Archer, M.; Mohamed, N.E.; Tewari, A.K.; Figueiro, M.G.; Kyprianou, N. Circadian Rhythm Disruption as a Contributor to Racial Disparities in Prostate Cancer. Cancers 2022, 14, 5116. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tobilla, P.; Solorzano, S.R.; Salido-Guadarrama, I.; Gonzalez-Covarrubias, V.; Morales-Montor, G.; Diaz-Otanez, C.E.; Rodriguez-Dorantes, M. SFRP1 repression in prostate cancer is triggered by two different epigenetic mechanisms. Gene 2016, 593, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, K.; Xie, H.; Wang, D.; Zhang, P.; Wei, T.; Yan, Y.; Pan, Y.; Ye, W.; Chen, H.; et al. SPOP mutation induces DNA methylation via stabilizing GLP/G9a. Nat. Commun. 2021, 12, 5716. [Google Scholar] [CrossRef] [PubMed]
- Das, P.M.; Thor, A.D.; Edgerton, S.M.; Barry, S.K.; Chen, D.F.; Jones, F.E. Reactivation of epigenetically silenced HER4/ERBB4 results in apoptosis of breast tumor cells. Oncogene 2010, 29, 5214–5219. [Google Scholar] [CrossRef]
- Denu, R.A.; Burkard, M.E. Analysis of the “centrosome-ome” identifies MCPH1 deletion as a cause of centrosome amplification in human cancer. Sci. Rep. 2020, 10, 11921. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, C.; Niu, Y.; Li, C.; Li, X.; Shang, Y.; Zhang, Y.; Zhang, Y.; Zhang, Y.; Zeng, Y. RBM3 suppresses stemness remodeling of prostate cancer in bone microenvironment by modulating N6-methyladenosine on CTNNB1 mRNA. Cell Death Dis. 2023, 14, 91. [Google Scholar] [CrossRef]
- Liu, W.; Gong, J.; Hu, J.; Hu, T.; Sun, Y.; Du, J.; Sun, C.; Guan, M.; Jiang, H.; Lu, Y. Quantitative assessment of AKAP12 promoter methylation in human prostate cancer using methylation-sensitive high-resolution melting: Correlation with Gleason score. Urology 2011, 77, 1006.e1–1006.e7. [Google Scholar] [CrossRef]
- Kagey, J.D.; Kapoor-Vazirani, P.; McCabe, M.T.; Powell, D.R.; Vertino, P.M. Long-term stability of demethylation after transient exposure to 5-aza-2’-deoxycytidine correlates with sustained RNA polymerase II occupancy. Mol. Cancer Res. 2010, 8, 1048–1059. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Shakya, R.; Gonda, T.; Quante, M.; Salas, M.; Kim, S.; Brooks, J.; Hirsch, S.; Davies, J.; Cullo, A.; Olive, K.; et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013, 73, 885–896. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Jennings, T.S.K.; Ueta, L.; Nguyen, J.; Song, L.; McClelland, M.; Chu, W.; Lilly, M.; Ittmann, M.; Castro, P.; et al. Genes Encoding Multiple Modulators of the Immune Response Are Methylated in the Prostate Tumor Microenvironment of African Americans. Cancers 2025, 17, 2399. https://doi.org/10.3390/cancers17142399
Kumar V, Jennings TSK, Ueta L, Nguyen J, Song L, McClelland M, Chu W, Lilly M, Ittmann M, Castro P, et al. Genes Encoding Multiple Modulators of the Immune Response Are Methylated in the Prostate Tumor Microenvironment of African Americans. Cancers. 2025; 17(14):2399. https://doi.org/10.3390/cancers17142399
Chicago/Turabian StyleKumar, Vinay, Tara Sinta Kartika Jennings, Lucas Ueta, James Nguyen, Liankun Song, Michael McClelland, Weiping Chu, Michael Lilly, Michael Ittmann, Patricia Castro, and et al. 2025. "Genes Encoding Multiple Modulators of the Immune Response Are Methylated in the Prostate Tumor Microenvironment of African Americans" Cancers 17, no. 14: 2399. https://doi.org/10.3390/cancers17142399
APA StyleKumar, V., Jennings, T. S. K., Ueta, L., Nguyen, J., Song, L., McClelland, M., Chu, W., Lilly, M., Ittmann, M., Castro, P., Kalebasty, A. R., Mercola, D., Yazdanpanah, O., Zi, X., & Rahmatpanah, F. (2025). Genes Encoding Multiple Modulators of the Immune Response Are Methylated in the Prostate Tumor Microenvironment of African Americans. Cancers, 17(14), 2399. https://doi.org/10.3390/cancers17142399