
CD19-ReTARGTPR: A Novel Fusion Protein for Physiological Engagement of Anti-CMV Cytotoxic T Cells Against CD19-Expressing Malignancies

 , ,
, ,  and
and
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Lines and Transfectants
2.3. Primary Patient-Derived B-CLL Cells
2.4. Ex Vivo Expansion of Anti-CMVpp65 CD8pos T Cells
2.5. Construction, Production, and Purification of CD19-ReTARGTPR
2.6. SDS-PAGE Analysis
2.7. CD19 Binding Assays
2.8. In Vitro Cytotoxicity Assays
2.9. Assessment of Activation-Induced Cell Death (AICD)
2.10. Cytokine Secretion Analysis
2.11. Statistical Analysis
3. Results
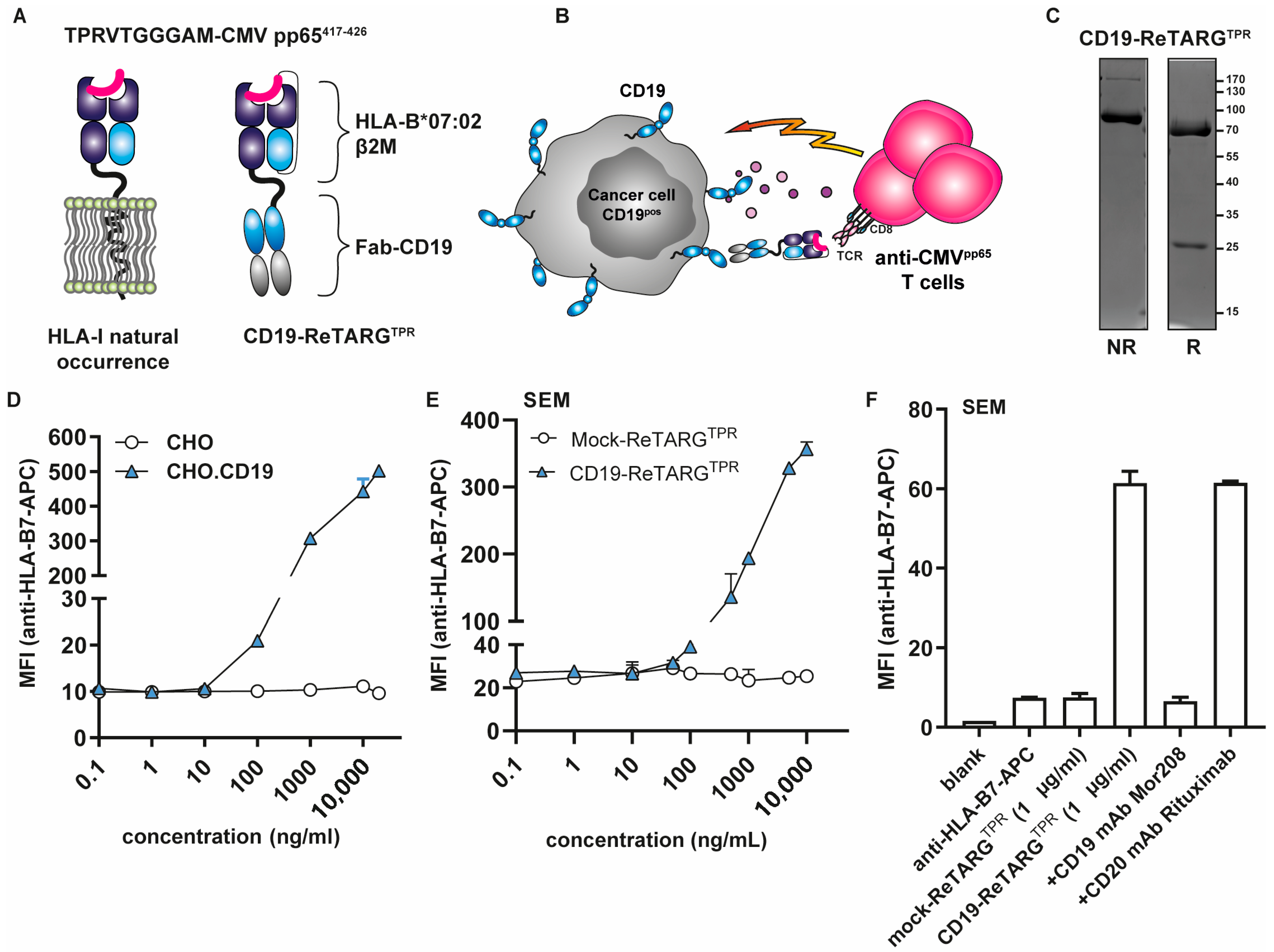
3.1. Construction, Production, and Purification of CD19-ReTARGTPR
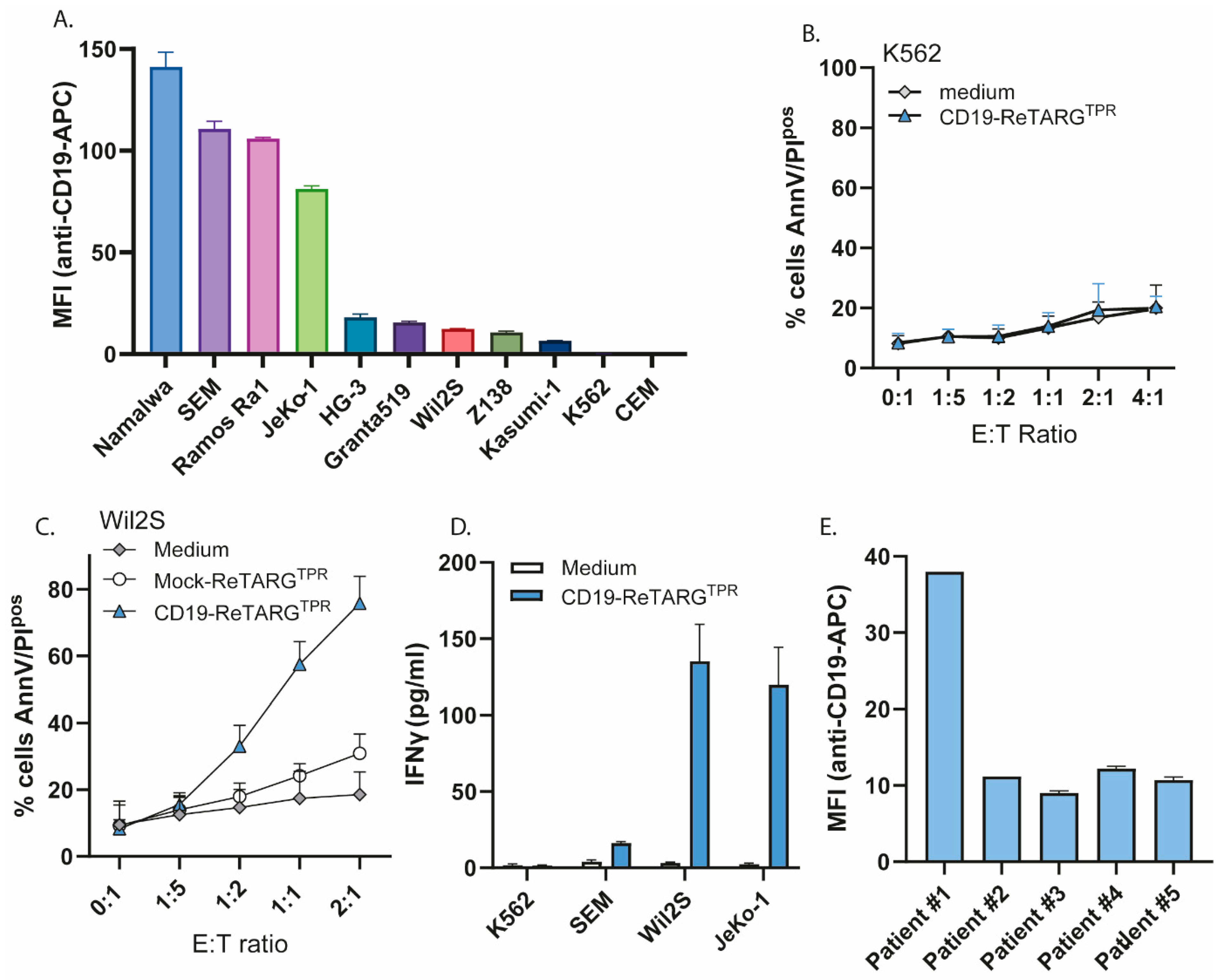
3.2. CD19-ReTARGTPR Selectively Binds to CD19pos Cancer Cells
3.3. CD19-ReTARGTPR Selectively Redirects the Cytotoxic Activity of Anti-CMVpp65 CD8pos T Cells Towards CD19pos Cancer Cell Lines and Patient-Derived CLL Cells
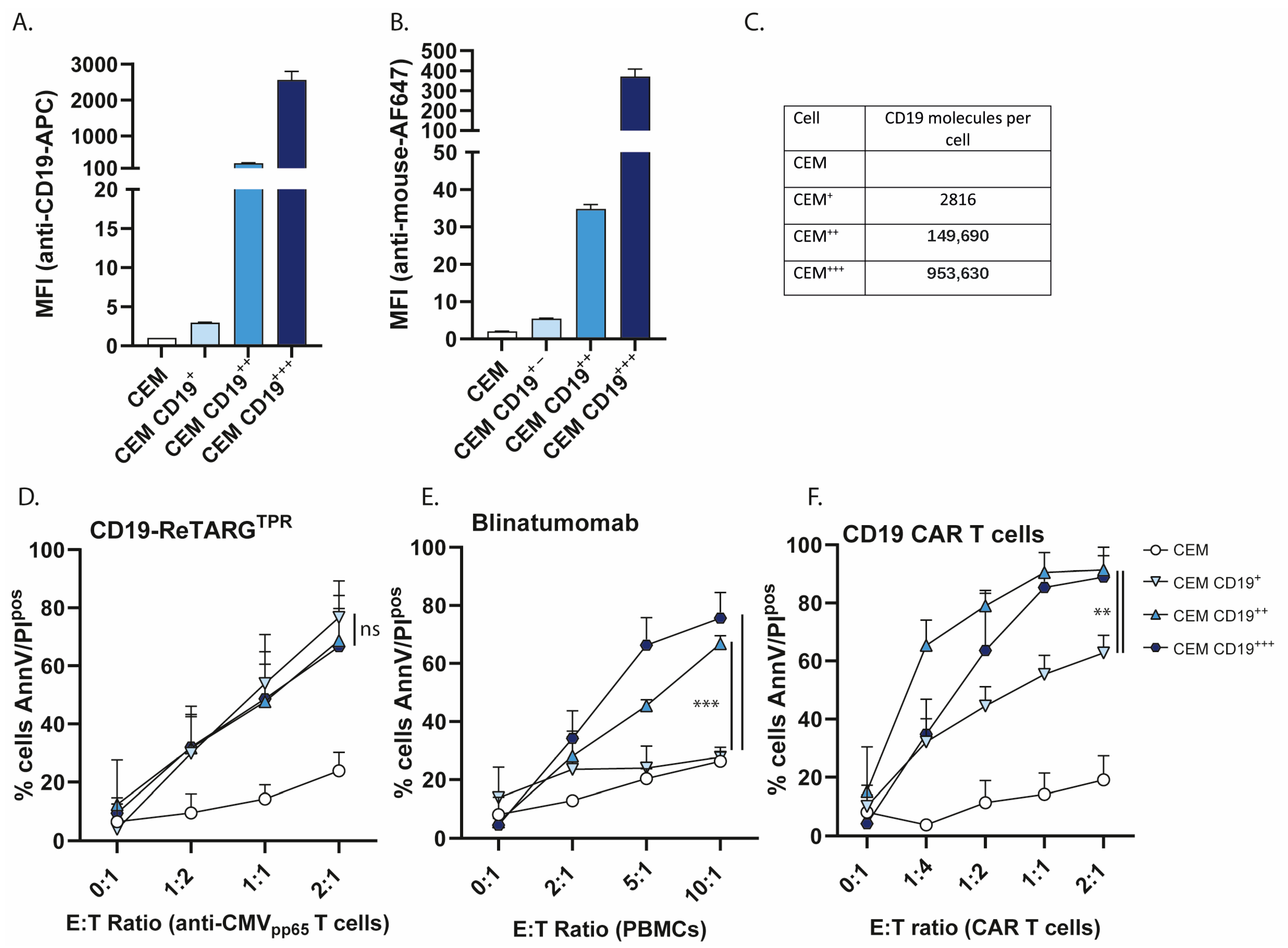
3.4. CD19-ReTARGTPR Retains Efficacy Against Cancer Cells with Low CD19 Expression
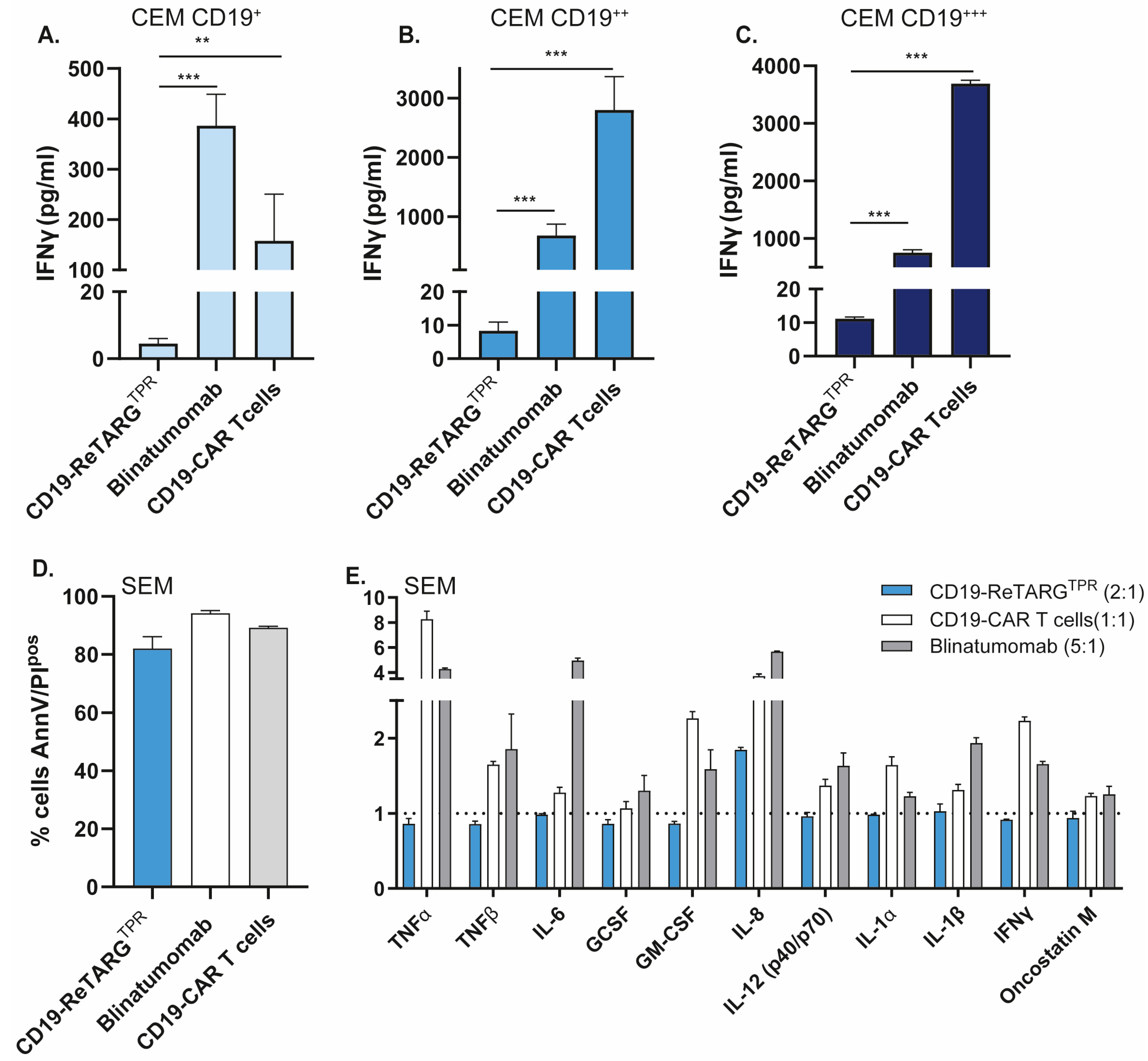
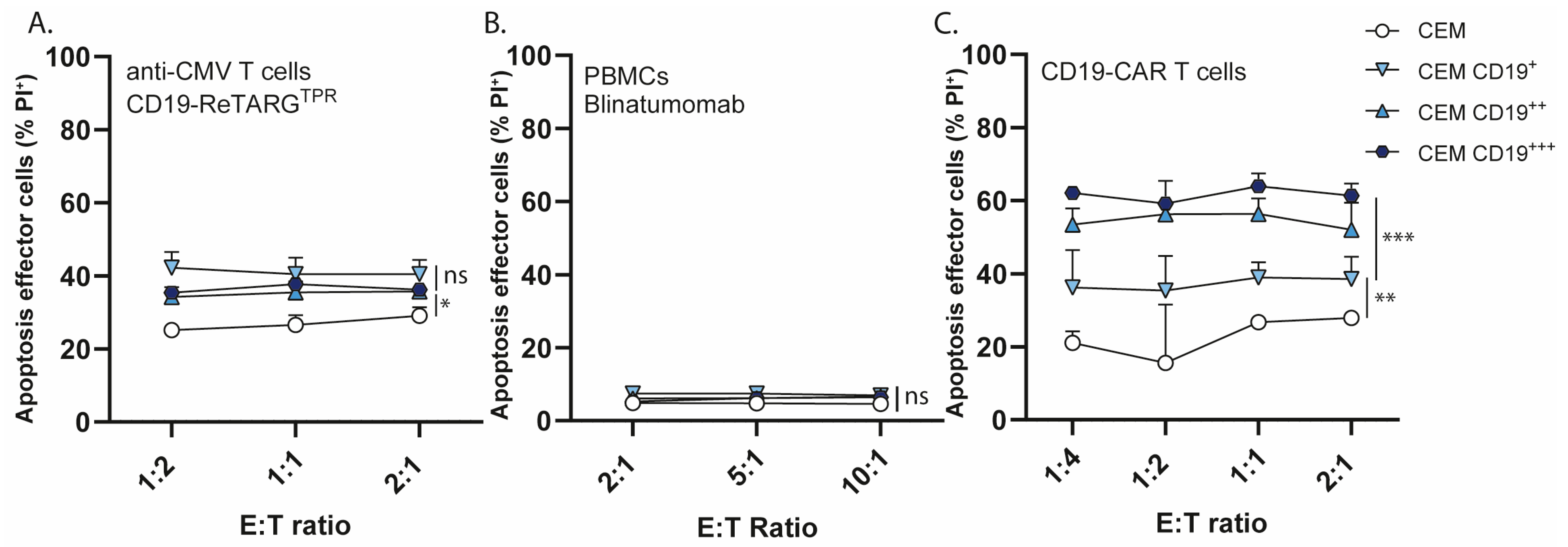
3.5. CD19-ReTARGTPR Induces Effective Lysis of CD19-Expressing Cancer B Cells with Reduced Proinflammatory Cytokine Release Compared to Blinatumomab and CD19 CAR T Cells
3.6. CD19-ReTARGTPR Induces Minimal Activation-Induced Cell Death in Redirected Anti-CMV CD8pos T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICD | activation-induced cell death |
| BiTE | bispecific T cell engager |
| CAR | chimeric antigen receptor |
| CLL | chronic lymphocytic leukemia |
| CMV | cytomegalovirus |
| CRS | cytokine release syndrome |
| CTL | cytotoxic CD8pos T cell |
| E:T | effector to target |
| ICANS | immune effector cell-associated neurotoxicity syndrome |
| IL2 | interleukin 2 |
| irAEs | immune-related adverse events |
| NR | non-reducing |
| PBMCs | peripheral blood mononuclear cells |
| pHLA-I | peptide-human leukocyte antigen class I |
| R | reducing |
| TCR | T cell receptor |
| TME | tumor microenvironment |
| TPR | TPRVTGGAM |
Appendix A
Appendix A.1
Appendix A.2

References
- Mirfakhraie, R.; Dehaghi, B.K.; Ghorbi, M.D.; Ghaffari-Nazari, H.; Mohammadian, M.; Salimi, M.; Ardakani, M.T.; Parkhideh, S. All about blinatumomab: The bispecific T cell engager immunotherapy for B cell acute lymphoblastic leukemia. Hematol. Transfus. Cell Ther. 2024, 46, 192–200. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Lan, H.; Wu, J.; Xiao, Y. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front. Immunol. 2023, 14, 1101495. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L. Moss PAH Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; van Dommelen, S.; Shellam, G.R. Hill AB Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; Beck, R.; de Craen, A.J.M.; Slagboom, E.P.; Westendorp, R.G.J. Pawelec G Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J. Gen. Virol. 2011, 92, 2746–2756. [Google Scholar] [CrossRef]
- Wills, M.R.; Okecha, G.; Weekes, M.P.; Gandhi, M.K.; Sissons, P.J.G.; Carmichael, A.J. Identification of naive or antigen-experienced human CD8(+) T cells by expression of costimulation and chemokine receptors: Analysis of the human cytomegalovirus-specific CD8(+) T cell response. J. Immunol. 2002, 168, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Wijeyesinghe, S.; Stolley, J.M.; Nelson, C.E.; Davis, R.L.; Manlove, L.S.; Pennell, C.A.; Blazar, B.R.; Chen, C.C.; Geller, M.A.; et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 2019, 10, 567. [Google Scholar] [CrossRef]
- Mackus, W.J.M.; Frakking, F.N.J.; Grummels, A.; Gamadia, L.E.; De Bree, G.J.; Hamann, D.; Van Lier, R.A.W.; Van Oers, M.H.J. Expansion of CMV-specific CD8+CD45RA+CD27- T cells in B-cell chronic lymphocytic leukemia. Blood 2003, 102, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Britsch, I.; van Wijngaarden, A.P.; Helfrich, W. Applications of Anti-Cytomegalovirus T Cells for Cancer (Immuno)Therapy. Cancers 2023, 15, 3767. [Google Scholar] [CrossRef]
- Samplonius, D.F.; van Wijngaarden, A.P.; Koll, L.; Ke, X.; Helfrich, W. Enhancing the Anticancer Activity of a Carcinoma-Directed Peptide-HLA-I Fusion Protein by Armoring with Mutein IFNα. Int. J. Mol. Sci. 2025, 26, 3178. [Google Scholar] [CrossRef]
- Britsch, I.; van Wijngaarden, A.P.; Ke, X.; Hendriks, M.A.J.M.; Samplonius, D.F.; Ploeg, E.M. Helfrich W Novel Fab-peptide-HLA-I fusion proteins for redirecting pre-existing anti-CMV T cell immunity to selective eliminate carcinoma cells. Oncoimmunology 2023, 12, 2207868. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Ruella, M.; Maus, M.V. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362. [Google Scholar] [CrossRef]
- von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Chen, D.; Liu, G.; Zhang, H.; Wang, X.; Wu, Z.; Wu, Y.; Xu, Q.; Yu, F. Activation-induced cell death in CAR-T cell therapy. Hum. Cell 2022, 35, 441–447. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Cosenza, M.; Sacchi, S.; Pozzi, S. Cytokine Release Syndrome Associated with T-Cell-Based Therapies for Hematological Malignancies: Pathophysiology, Clinical Presentation, and Treatment. Int. J. Mol. Sci. 2021, 22, 7652. [Google Scholar] [CrossRef]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Xue, L.; Yi, Y.; Xu, Q.; Wang, L.; Yang, X.; Zhang, Y.; Hua, X.; Chai, X.; Yang, J.; Chen, Y.; et al. Chimeric antigen receptor T cells self-neutralizing IL6 storm in patients with hematologic malignancy. Cell Discov. 2021, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- te Raa, G.D.; Pascutti, M.F.; García-Vallejo, J.J.; Reinen, E.; Remmerswaal, E.B.M.; ten Berge, I.J.M.; van Lier, R.A.W.; Eldering, E.; van Oers, M.H.J.; Tonino, S.H.; et al. CMV-specific CD8+ T-cell function is not impaired in chronic lymphocytic leukemia. Blood 2014, 123, 717–724. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Wang, X.; Diamond, D.J.; Forman, S.J.; Nakamura, R. Development of CMV-CD19 bi-specific CAR T cells with post-infusion in vivo boost using an anti-CMV vaccine. Int. J. Hematol. 2021, 114, 544–553. [Google Scholar] [CrossRef]
- Caruana, I.; Weber, G.; Ballard, B.C.; Wood, M.S.; Savoldo, B.; Dotti, G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin. Cancer Res. 2015, 21, 2952–2962. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, M.; Köse, M.C.; Duray, E.; Einsele, H.; Beguin, Y.; Caers, J. Bispecific, T-Cell-Recruiting Antibodies in B-Cell Malignancies. Front. Immunol. 2020, 11, 762. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815. [Google Scholar] [CrossRef]
- Arnold, D.E.; Maude, S.L.; Callahan, C.A.; DiNofia, A.M.; Grupp, S.A. Heimall JR Subcutaneous immunoglobulin replacement following CD19-specific chimeric antigen receptor T-cell therapy for B-cell acute lymphoblastic leukemia in pediatric patients. Pediatr. Blood Cancer 2020, 67, e28092. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Giralt, S.; Torgerson, T.R.; Lazarus, H.M. CAR-T–and a side order of IgG, to go?—Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. 2019, 38, 100596. [Google Scholar] [CrossRef]
- Wat, J.; Barmettler, S. Hypogammaglobulinemia After Chimeric Antigen Receptor (CAR) T-Cell Therapy: Characteristics, Management, and Future Directions. J. Allergy Clin. Immunol. Pract. 2022, 10, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef]
- Watanabe, K.; Terakura, S.; Martens, A.C.; van Meerten, T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Xiong, Y.; Wu, D.; Nölle, V.; Schmitz, S.; Haso, W.; Kaiser, A.; Dropulic, B.; Orentas, R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 2017, 5, 42. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Y.; Song, Z.; Liu, J.; Luo, Z.; Yu, G.; Wang, J. Novel and effective tandem CD38 and CD19 targeting CAR-T cells inhibit hematological tumor immune escape. Cell Immunol. 2025, 411–412, 104950. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Shi, M.; Wang, J.; Huang, H.; Liu, D.; Cheng, H.; Wang, X.; Chen, W.; Yan, Z.; Sang, W.; Qi, K.; et al. Bispecific CAR T cell therapy targeting BCMA and CD19 in relapsed/refractory multiple myeloma: A phase I/II trial. Nat. Commun. 2024, 15, 3371. [Google Scholar] [CrossRef]
- Bachiller, M.; Barceló-Genestar, N.; Rodriguez-Garcia, A.; Alserawan, L.; Dobaño-López, C.; Giménez-Alejandre, M.; Castellsagué, J.; Colell, S.; Otero-Mateo, M.; Antoñana-Vildosola, A.; et al. ARI0003: Co-transduced CD19/BCMA dual-targeting CAR-T cells for the treatment of non-Hodgkin lymphoma. Mol. Ther. 2025, 33, 317–335. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Gedeon, P.C.; Sanchez-Perez, L.; Verla, T.; Alvarez-Breckenridge, C.; Choi, B.D.; Fecci, P.E.; Sampson, J.H. Are BiTEs the “missing link” in cancer therapy? Oncoimmunology 2015, 4, e1008339. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mazas, A.; Nunes, J.M.; PGAE HLA Consortium of the 18th International HLA and Immunogenetics Workshop. The most frequent HLA alleles around the world: A fundamental synopsis. Best. Pract. Res. Clin. Haematol. 2024, 37, 101559. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Ko, D.-H.; Jang, J.-Y.; Kim, Y.R.; Heo, J.Y.; Kim, Y.-S. Harnessing SARS-CoV-2-specific CD8+ T cells to kill target tumor cells for cancer immunotherapy. Cancer Commun. 2024, 44, 173–177. [Google Scholar] [CrossRef]
- Scholten, K.B.J.; Turksma, A.W.; Ruizendaal, J.J.; van den Hende, M.; van der Burg, S.H.; Heemskerk, M.H.M.; Meijer, C.J.L.M.; Hooijberg, E. Generating HPV specific T helper cells for the treatment of HPV induced malignancies using TCR gene transfer. J. Transl. Med. 2011, 9, 147. [Google Scholar] [CrossRef]
- van der Wulp, W.; Remst, D.F.G.; Kester, M.G.D.; Hagedoorn, R.S.; Parren, P.W.H.I.; van Kasteren, S.I.; Schuurman, J.; Hoeben, R.C.; Ressing, M.E.; Bleijlevens, B.; et al. Antibody-mediated delivery of viral epitopes to redirect EBV-specific CD8+ T-cell immunity towards cancer cells. Cancer Gene Ther. 2024, 31, 58–68. [Google Scholar] [CrossRef]
- Mous, R.; Savage, P.; Remmerswaal, E.B.M.; van Lier, R.A.W.; Eldering, E.; van Oers, M.H.J. Redirection of CMV-specific CTL towards B-CLL via CD20-targeted HLA/CMV complexes. Leukemia 2006, 20, 1096–1102. [Google Scholar] [CrossRef]
- Schütz, C.; Varela, J.C.; Perica, K.; Haupt, C.; Oelke, M.; Schneck, J.P. Antigen-specific T cell Redirectors: A nanoparticle based approach for redirecting T cells. Oncotarget 2016, 7, 68503–68512. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Wijngaarden, A.P.; Britsch, I.; Peipp, M.; Samplonius, D.F.; Helfrich, W. CD19-ReTARGTPR: A Novel Fusion Protein for Physiological Engagement of Anti-CMV Cytotoxic T Cells Against CD19-Expressing Malignancies. Cancers 2025, 17, 2300. https://doi.org/10.3390/cancers17142300
van Wijngaarden AP, Britsch I, Peipp M, Samplonius DF, Helfrich W. CD19-ReTARGTPR: A Novel Fusion Protein for Physiological Engagement of Anti-CMV Cytotoxic T Cells Against CD19-Expressing Malignancies. Cancers. 2025; 17(14):2300. https://doi.org/10.3390/cancers17142300
Chicago/Turabian Stylevan Wijngaarden, Anne Paulien, Isabel Britsch, Matthias Peipp, Douwe Freerk Samplonius, and Wijnand Helfrich. 2025. "CD19-ReTARGTPR: A Novel Fusion Protein for Physiological Engagement of Anti-CMV Cytotoxic T Cells Against CD19-Expressing Malignancies" Cancers 17, no. 14: 2300. https://doi.org/10.3390/cancers17142300
APA Stylevan Wijngaarden, A. P., Britsch, I., Peipp, M., Samplonius, D. F., & Helfrich, W. (2025). CD19-ReTARGTPR: A Novel Fusion Protein for Physiological Engagement of Anti-CMV Cytotoxic T Cells Against CD19-Expressing Malignancies. Cancers, 17(14), 2300. https://doi.org/10.3390/cancers17142300