
Peroxisomal Alterations in Prostate Cancer: Metabolic Shifts and Clinical Relevance

 , and
, and 
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Data Sources and Search Strategy
2.2. Eligibility Criteria
2.3. Exclusion Criteria
2.4. Study Screening and Selection Process
2.5. Data Extraction and Processing
3. Results
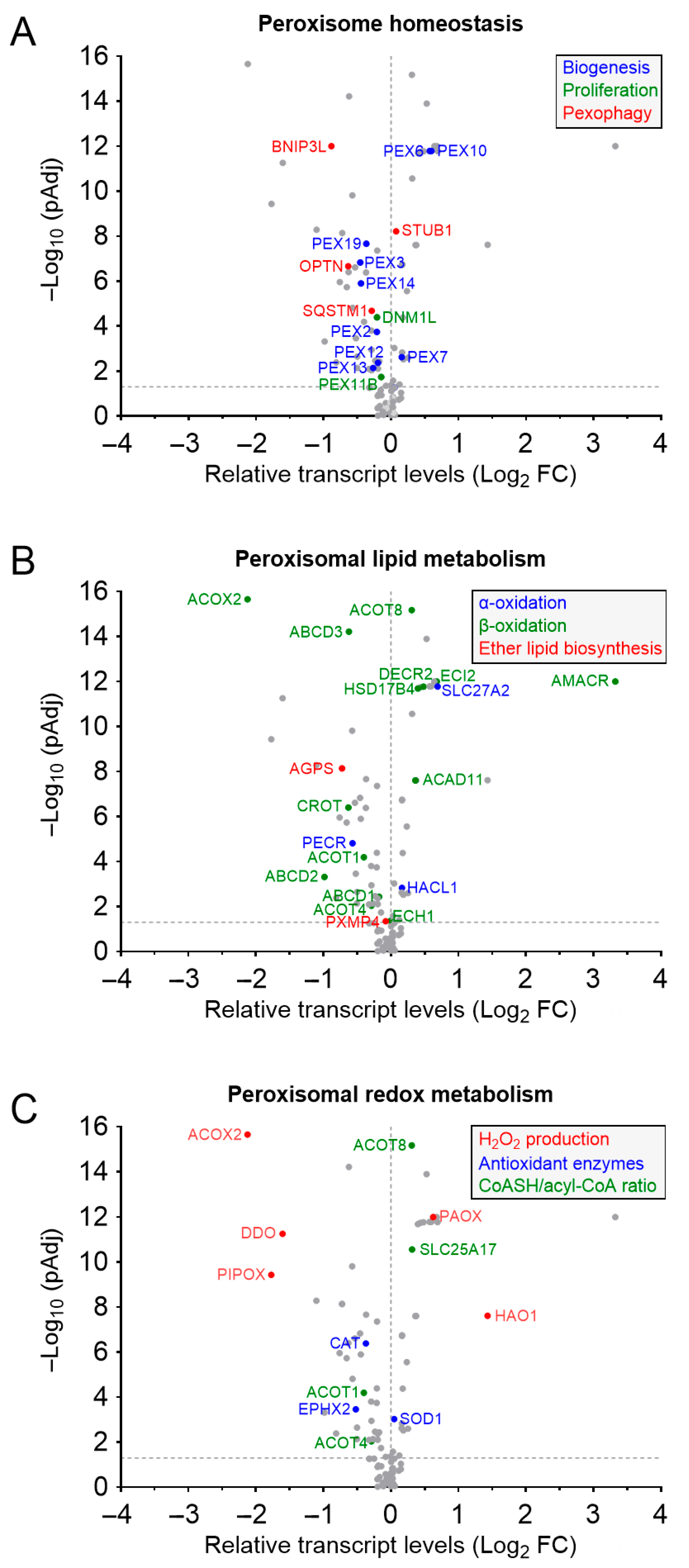
3.1. Peroxisome Abundance and Prostate Cancer
3.2. Peroxisomes, Ether Lipids, and Prostate Cancer
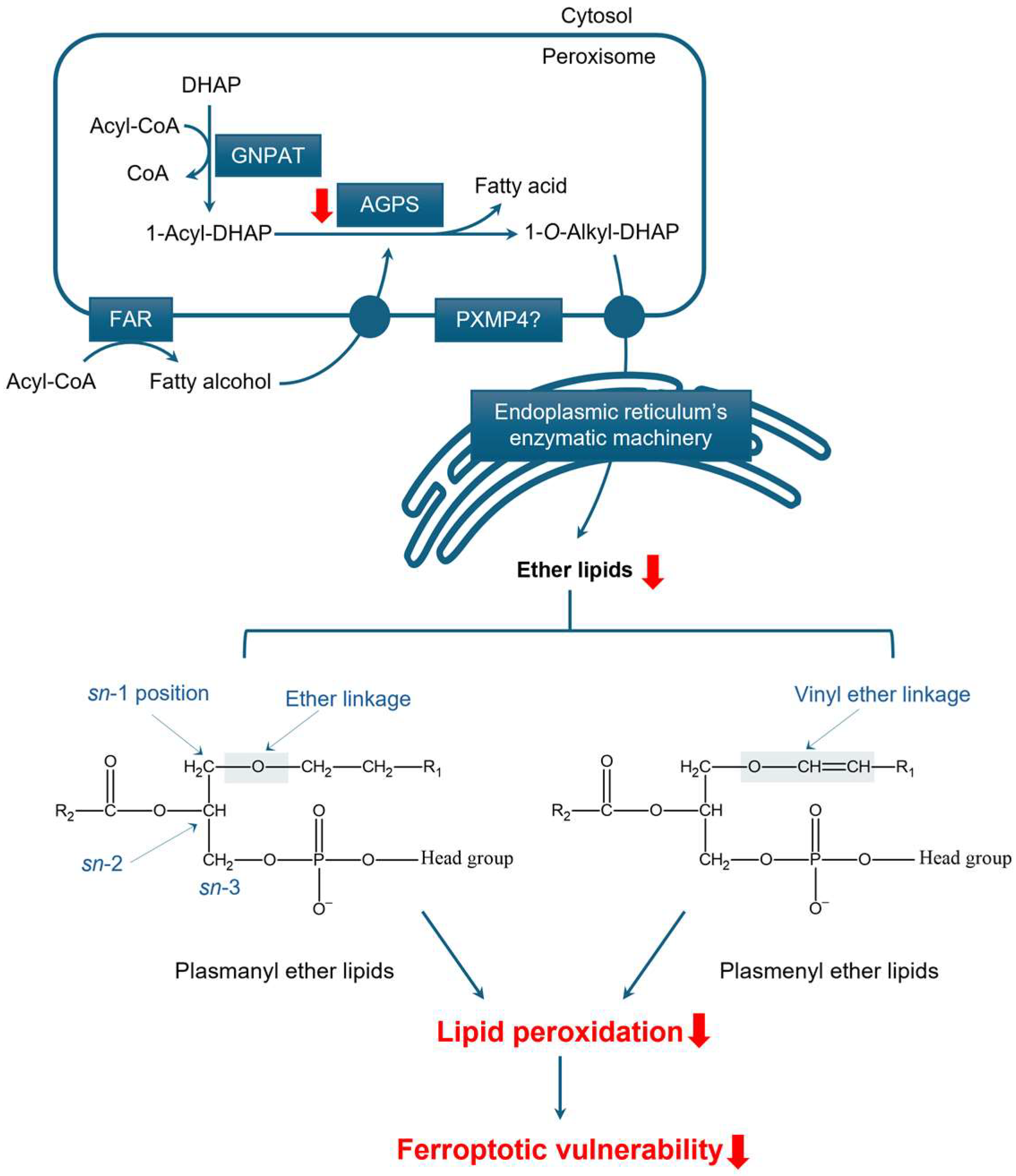
3.2.1. Structure and Function of Ether Lipids
3.2.2. The Role of Peroxisomes in Ether Lipid Synthesis
3.2.3. Peroxisomal Ether Lipid Metabolism in Prostate Cancer
3.3. Peroxisomes, Fatty Acid Oxidation, and Prostate Cancer
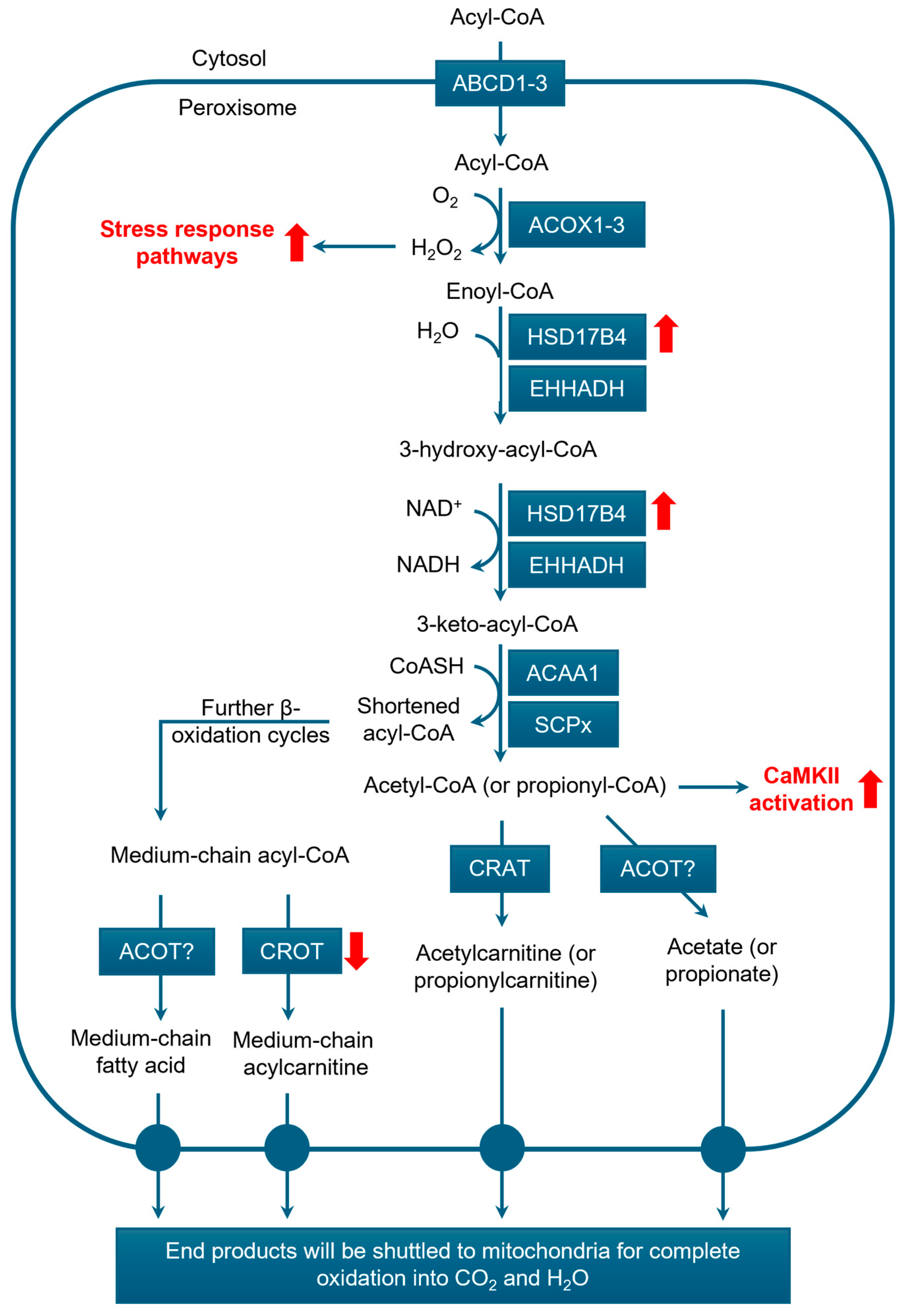
3.3.1. β-Oxidation
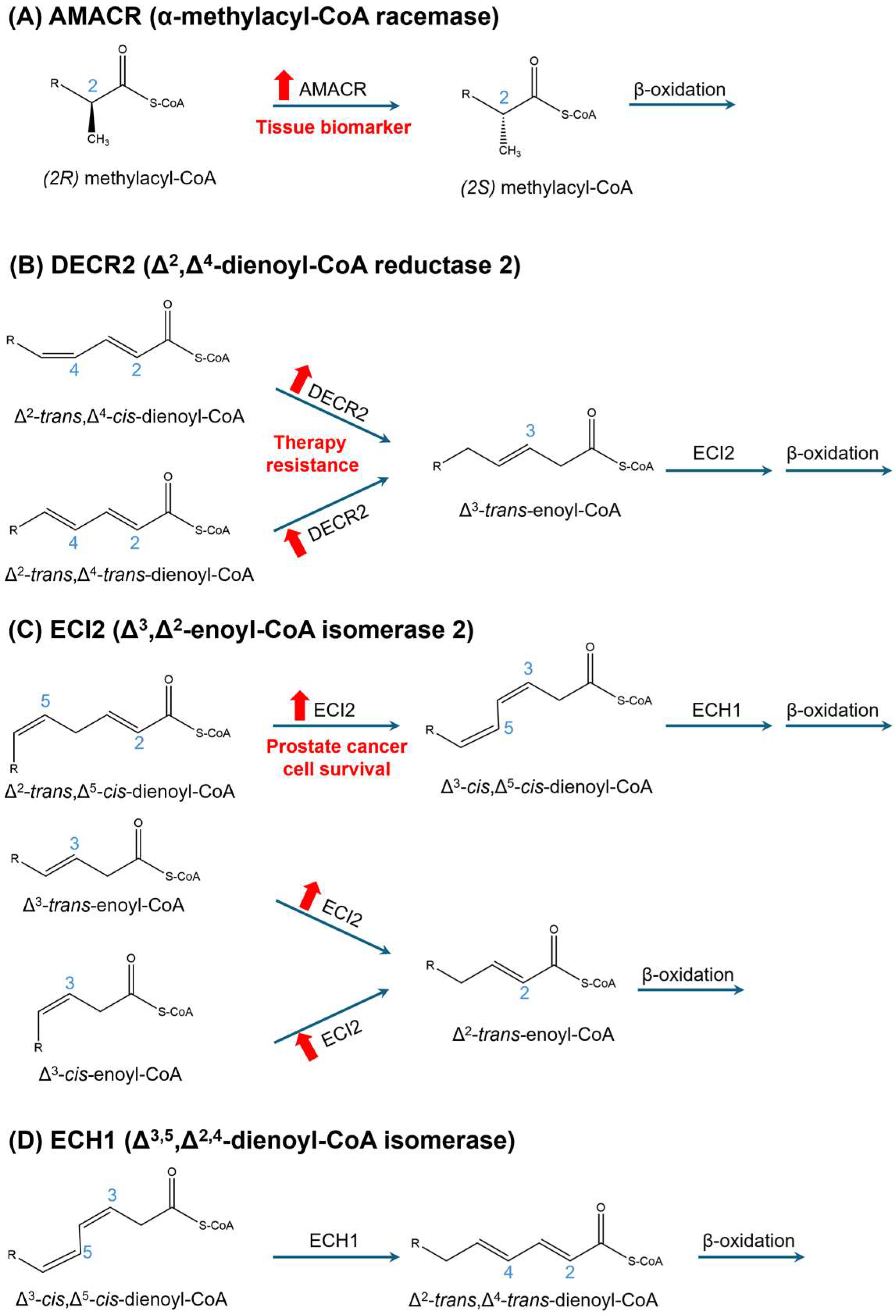
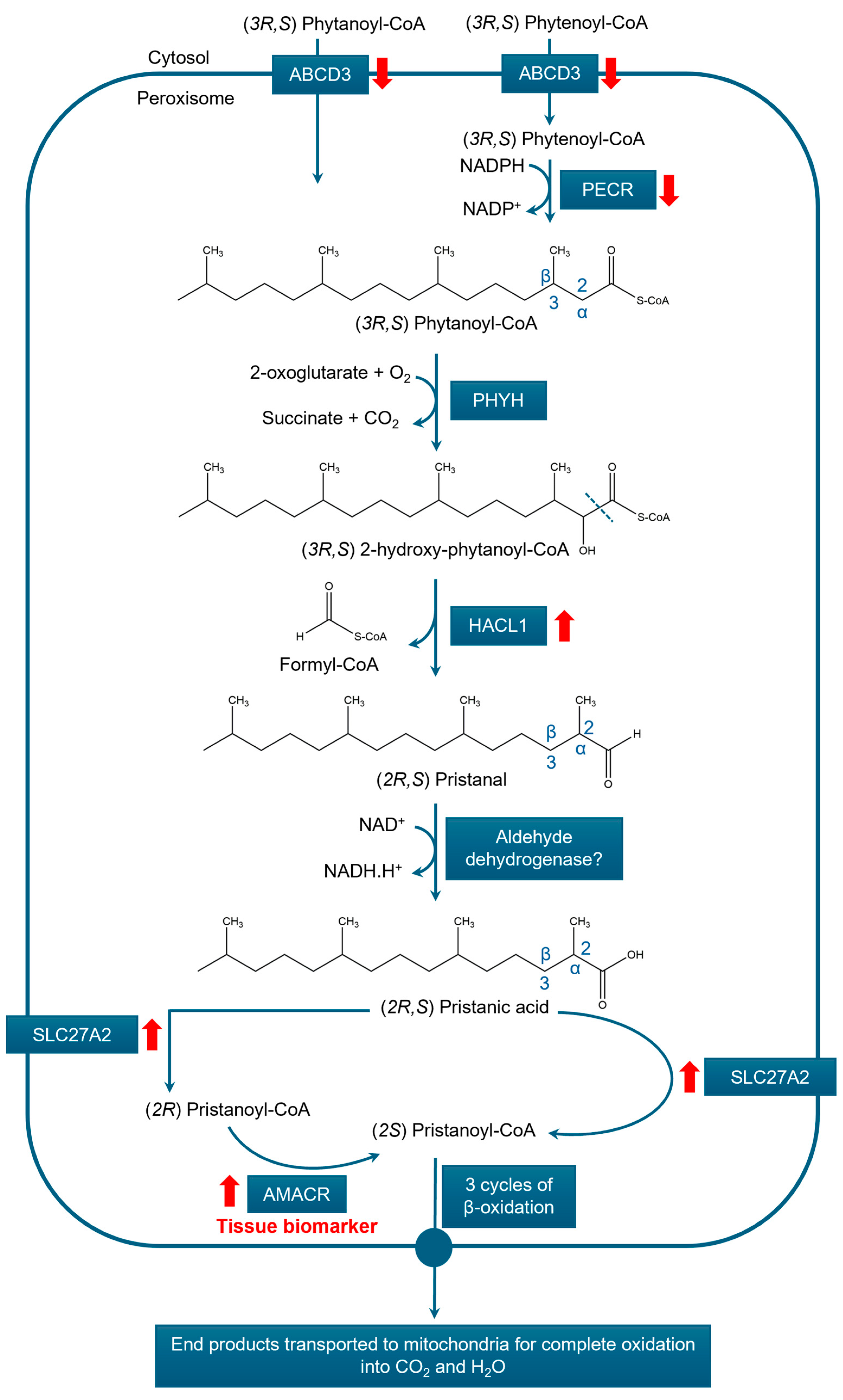
3.3.2. α-Oxidation
3.3.3. Peroxisomal Fatty Acid Oxidation and Prostate Cancer
3.4. Peroxisomes, Redox Homeostasis, and Prostate Cancer
3.4.1. Peroxisomes and Redox Homeostasis
3.4.2. Peroxisomes as Redox Regulators in Prostate Cancer
4. Future Directions
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-acyl-DHAP | 1-acyl-dihydroxyacetone-3-phosphate |
| 3-AT | 3-amino-1,2,4-triazole |
| ABCD | ATP-binding cassette subfamily D |
| ACAA1 | 3-ketoacyl-CoA thiolase 1 |
| ACOT | Acyl-CoA thioesterase |
| AGPS | 1-alkyl-glycerone-3-phosphate synthase |
| AMACR | A-methylacyl coenzyme A racemase |
| AR | Androgen receptor |
| BCFA | Branched-chain fatty acid |
| CAT | Catalase |
| CoASH | Coenzyme A |
| CRAT | Carnitine acetyltransferase |
| CRPC | Castration-resistant prostate cancer |
| DECR2 | 2,4-dienoyl CoA reductase 2 |
| DHA | Docosahexaenoic acid |
| DHAP | Dihydroxyacetone-3-phosphate |
| DHCA | Dihydroxycholestanoic acid |
| ECH1 | Δ3,5,Δ2,4-enoyl-CoA isomerase |
| ECI2 | 2-enoyl-CoA isomerase |
| EPHX2 | Epoxide hydrolase 2 |
| ER | Endoplasmic reticulum |
| FA | Fatty acid |
| FAR | Fatty acyl-CoA reductase |
| FC GNPAT | Fold change Glycerone-3-phosphate O-acyltransferase |
| GSTK1 | Glutathione S-transferase kappa 1 |
| HAO1 | Hydroxyacid oxidase 1 |
| HSD17B4 | Hydroxysteroid 17-beta dehydrogenase 4 |
| MARC2/MTARC2 | Mitochondrial amidoxime-reducing component 2 |
| MCT2 | Monocarboxylate transporter 2 |
| MUFA | Monounsaturated fatty acid |
| PCa | Prostate cancer |
| PEX | Peroxin |
| PIPOX | Pipecolic acid oxidase |
| PMP | Peroxisomal membrane protein |
| PPAR | Peroxisome proliferator-activated receptor |
| PRDX | Peroxiredoxin |
| PSA | Prostate-specific antigen |
| PUFA | Polyunsaturated fatty acid |
| PXMP4 | Peroxisomal membrane protein 4 |
| ROS | Reactive oxygen species |
| SLC25A17 | Solute carrier family 25 member 17 |
| SLC27A2 | Solute carrier family 27 member 2 |
| SOD | Superoxide dismutase |
| TCA | Tricarboxylic acid |
| TCGA | The Cancer Genome Atlas Program |
| THCA | Trihydroxycholestanoic acid |
| TrkA | Tyrosine kinase receptor |
| UALCAN | The University of Alabama at Birmingham cancer data analysis portal |
| VLCFA | Very long-chain fatty acid |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, J.; Lu, W. The significance of mitochondrial dysfunction in cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium homeostasis and cancer: Insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef]
- Bui, S.; Mejia, I.; Díaz, B.; Wang, Y. Adaptation of the Golgi apparatus in cancer cell invasion and metastasis. Front. Cell Dev. Biol. 2021, 9, 806482. [Google Scholar] [CrossRef]
- Jeger, J.L. Endosomes, lysosomes, and the role of endosomal and lysosomal biogenesis in cancer development. Mol. Biol. Rep. 2020, 47, 9801–9810. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; Del Rincón, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Pratama, A.M.; Sharma, M.; Naidu, S.; Bömmel, H.; Prabhuswamimath, S.C.; Madhusudhan, T.; Wihadmadyatami, H.; Bachhuka, A.; Karnati, S. Peroxisomes and PPARs: Emerging role as master regulators of cancer metabolism. Mol. Metab. 2024, 90, 102044. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C. Peroxisomal hydrogen peroxide signaling: A new chapter in intracellular communication research. Curr. Opin. Chem. Biol. 2024, 78, 102426. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Global Cancer Observatory: Cancer Today, version 1.1; International Agency for Research on Cancer: Lyon, France, 2022; Available online: https://gco.iarc.who.int/today (accessed on 7 May 2025).
- Claessens, F.; Helsen, C.; Prekovic, S.; Van den Broeck, T.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Giunchi, F.; Fiorentino, M.; Loda, M. The metabolic landscape of prostate cancer. Eur. Urol. Oncol. 2019, 2, 28–36. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells 1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef]
- Kelly, R.S.; Sinnott, J.A.; Rider, J.R.; Ebot, E.M.; Gerke, T.; Bowden, M.; Pettersson, A.; Loda, M.; Sesso, H.D.; Kantoff, P.W.; et al. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: Results from a nested case-control study. Cancer Metab. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.J.; Zafar, M.B.; Lai, Y.H.; Segall, G.M.; Terris, M.K. Fluorodeoxyglucose positron emission tomography studies in diagnosis and staging of clinically organ-confined prostate cancer. Urology 2001, 57, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Gavane, S.; Tarlinton, L.; Fareedy, S.B.; Doran, M.G.; Zelefsky, M.J.; Osborne, J.R. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate 2014, 74, 1153–1159. [Google Scholar] [CrossRef]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Yifrach, E.; Fischer, S.; Oeljeklaus, S.; Schuldiner, M.; Zalckvar, E.; Warscheid, B. Defining the mammalian peroxisomal proteome. Subcell. Biochem. 2018, 89, 47–66. [Google Scholar] [CrossRef]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef]
- Kim, P.K.; Mullen, R.T.; Schumann, U.; Lippincott-Schwartz, J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J. Cell Biol. 2006, 173, 521–532. [Google Scholar] [CrossRef]
- Verhoeven, N.; Oshima, Y.; Cartier, E.; Bippes, C.C.; Neutzner, A.; Boyman, L.; Karbowski, M. Outer mitochondrial membrane E3 Ub ligase MARCH5 controls de novo peroxisome biogenesis. Dev. Cell 2025, 60, 40–50.e5. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, J.; Cao, Z.; Wu, K.; Wang, J.; Guo, Y.; Zhuang, M. Ubiquitin ligase MARCH5 controls the formation of mitochondria-derived pre-peroxisomes. Dev. Cell 2025, 60, 30–39.e3. [Google Scholar] [CrossRef]
- Li, H.; Lismont, C.; Revenco, I.; Hussein, M.A.F.; Costa, C.F.; Fransen, M. The peroxisome-autophagy redox connection: A double-edged sword? Front. Cell Dev. Biol. 2021, 9, 814047. [Google Scholar] [CrossRef] [PubMed]
- Bajdzienko, J.; Bremm, A. Mammalian pexophagy at a glance. J. Cell Sci. 2024, 137, jcs259775. [Google Scholar] [CrossRef]
- Wei, X.; Manandhar, L.; Kim, H.; Chhetri, A.; Hwang, J.; Jang, G.; Park, C.; Park, R. Pexophagy and oxidative stress: Focus on peroxisomal proteins and reactive oxygen species (ROS) signaling pathways. Antioxidants 2025, 14, 126. [Google Scholar] [CrossRef]
- Hussein, M.A.F.; Lismont, C.; Costa, C.F.; Li, H.; Claessens, F.; Fransen, M. Characterization of the peroxisomal proteome and redox balance in human prostate cancer cell lines. Antioxidants 2024, 13, 1340. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, Y.; Li, H.; Wang, T.; Lu, F.; Liu, R.; Xie, G.; Song, L.; Huang, B.; Li, X.; et al. Enzalutamide inhibits PEX10 function and sensitizes prostate cancer cells to ROS activators. Cell Death Dis. 2024, 15, 559. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, B.; Wei, Y.; Gan, H.; Fang, B.; Li, X.; Wu, J.; Bian, X.; Wang, J.; Freedland, S.J.; et al. Neoadjuvant fuzuloparib combined with abiraterone for localized high-risk prostate cancer (FAST-PC): A single-arm phase 2 study. Cell Rep. Med. 2025, 6, 102018. [Google Scholar] [CrossRef]
- Valença, I.; Pértega-Gomes, N.; Vizcaino, J.R.; Henrique, R.M.; Lopes, C.; Baltazar, F.; Ribeiro, D. Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell Mol. Med. 2015, 19, 723–733. [Google Scholar] [CrossRef]
- Valença, I.; Ferreira, A.R.; Correia, M.; Kühl, S.; van Roermund, C.; Waterham, H.R.; Máximo, V.; Islinger, M.; Ribeiro, D. Prostate cancer proliferation is affected by the subcellular localization of MCT2 and accompanied by significant peroxisomal alterations. Cancers 2020, 12, 3152. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaino, J.R.; Felisbino, S.; Warren, A.Y.; Shaw, G.; Kay, J.; Whitaker, H.; Lynch, A.G.; Fryer, L.; Neal, D.E.; et al. Epigenetic and oncogenic regulation of SLC16A7 (MCT2) results in protein over-expression, impacting on signalling and cellular phenotypes in prostate cancer. Oncotarget 2015, 6, 21675–21684. [Google Scholar] [CrossRef]
- Dorninger, F.; Forss-Petter, S.; Wimmer, I.; Berger, J. Plasmalogens, platelet-activating factor and beyond— Ether lipids in signaling and neurodegeneration. Neurobiol. Dis. 2020, 145, 105061. [Google Scholar] [CrossRef]
- Hossain, M.S.; Mawatari, S.; Fujino, T. Biological functions of plasmalogens. Adv. Exp. Med. Biol. 2020, 1299, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Papin, M.; Bouchet, A.M.; Chantôme, A.; Vandier, C. Ether-lipids and cellular signaling: A differential role of alkyl- and alkenyl-ether-lipids? Biochimie 2023, 21, 50–59. [Google Scholar] [CrossRef]
- Jové, M.; Mota-Martorell, N.; Obis, È.; Sol, J.; Martín-Garí, M.; Ferrer, I.; Portero-Otin, M.; Pamplona, R. Ether lipid-mediated antioxidant defense in Alzheimer’s disease. Antioxidants 2023, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Gee, R.; McGroarty, E.; Hsieh, B.; Wied, D.M.; Tolbert, N.E. Glycerol phosphate dehydrogenase in mammalian peroxisomes. Arch. Biochem. Biophys. 1974, 161, 187–193. [Google Scholar] [CrossRef]
- Antonenkov, V.D. Dehydrogenases of the pentose phosphate pathway in rat liver peroxisomes. Eur. J. Biochem. 1989, 183, 75–82. [Google Scholar] [CrossRef]
- Biermann, J.; Just, W.W.; Wanders, R.J.; Van Den Bosch, H. Alkyl-dihydroxyacetone phosphate synthase and dihydroxyacetone phosphate acyltransferase form a protein complex in peroxisomes. Eur. J. Biochem. 1999, 261, 492–499. [Google Scholar] [CrossRef]
- Honsho, M.; Asaoku, S.; Fukumoto, K.; Fujiki, Y. Topogenesis and homeostasis of fatty acyl-CoA reductase 1. J. Biol. Chem. 2013, 288, 34588–34598. [Google Scholar] [CrossRef]
- Honsho, M.; Tanaka, M.; Zoeller, R.A.; Fujiki, Y. Distinct functions of acyl/alkyl dihydroxyacetonephosphate reductase in peroxisomes and endoplasmic reticulum. Front. Cell Dev. Biol. 2020, 8, 855. [Google Scholar] [CrossRef]
- Lodhi, I.J.; Yin, L.; Jensen-Urstad, A.P.L.; Funai, K.; Coleman, T.; Baird, J.H.; Ramahi, M.K.E.; Razani, B.; Song, H.; Fu-Hsu, F.; et al. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARγ activation to decrease diet-induced obesity. Cell Metab. 2012, 16, 189–201. [Google Scholar] [CrossRef]
- Blankestijn, M.; Bloks, V.W.; Struik, D.; Huijkman, N.; Kloosterhuis, N.; Wolters, J.C.; Wanders, R.J.A.; Vaz, F.M.; Islinger, M.; Kuipers, F.; et al. Mice with a deficiency in peroxisomal membrane protein 4 (PXMP4) display mild changes in hepatic lipid metabolism. Sci. Rep. 2022, 12, 2512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Z.; Li, K.; Xie, G.; Feng, Y.; Wang, Z.; Li, N.; Liu, R.; Ding, Y.; Wang, J.; et al. TrkA promotes MDM2-mediated AGPS ubiquitination and degradation to trigger prostate cancer progression. J. Exp. Clin. Cancer Res. 2024, 43, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, X.; Wan, Z.; Wang, W.; Zhang, J.; Mi, Y.; Li, R.; Xu, Z.; Wang, B.; Li, N.; et al. PXMP4 promotes gastric cancer cell epithelial-mesenchymal transition via the PI3K/AKT signaling pathway. Mol. Biol. Rep. 2024, 51, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, M.; Xiao, H.; Lee, M.; Levin, L.; Leung, Y.; Ho, S. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Prostate 2010, 70, 765–776. [Google Scholar] [CrossRef]
- Wu, M.; Ho, S.M. PMP24, a gene identified by MSRF, undergoes DNA hypermethylation-associated gene silencing during cancer progression in an LNCaP model. Oncogene 2004, 23, 250–259. [Google Scholar] [CrossRef]
- Benjamin, D.I.; Cozzo, A.; Ji, X.; Roberts, L.S.; Louie, S.M.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 14912–14917. [Google Scholar] [CrossRef]
- Phuyal, S.; Skotland, T.; Hessvik, N.P.; Simolin, H.; Øverbye, A.; Brech, A.; Parton, R.G.; Ekroos, K.; Sandvig, K.; Llorente, A. The ether lipid precursor hexadecylglycerol stimulates the release and changes the composition of exosomes derived from PC-3 cells. J. Biol. Chem. 2015, 290, 4225–4237. [Google Scholar] [CrossRef]
- Louie, S.M.; Roberts, L.S.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta 2013, 1831, 1566–1572. [Google Scholar] [CrossRef]
- Stazi, G.; Battistelli, C.; Piano, V.; Mazzone, R.; Marrocco, B.; Marchese, S.; Louie, S.M.; Zwergel, C.; Antonini, L.; Patsilinakos, A.; et al. Development of alkyl glycerone phosphate synthase inhibitors: Structure-activity relationship and effects on ether lipids and epithelial-mesenchymal transition in cancer cells. Eur. J. Med. Chem. 2019, 163, 722–735. [Google Scholar] [CrossRef]
- Brzozowski, J.S.; Jankowski, H.; Bond, D.R.; McCague, S.B.; Munro, B.R.; Predebon, M.J.; Scarlett, C.J.; Skelding, K.A.; Weidenhofer, J. Lipidomic profiling of extracellular vesicles derived from prostate and prostate cancer cell lines. Lipids Health Dis. 2018, 17, 211. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.R.; Carvalho, M.; Aveiro, S.S.; Melo, T.; Domingues, M.R.; Macedo-Silva, C.; Coimbra, N.; Jerónimo, C.; Henrique, R.; De Lourdes Bastos, M.; et al. Comprehensive metabolomics and lipidomics profiling of prostate cancer tissue reveals metabolic dysregulations associated with disease development. J. Proteome. Res. 2022, 21, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, S.; Piao, H.L.; Wang, F.; Yin, P.; Xu, C.; Lu, X.; Ye, G.; Shao, Y.; Yan, M.; et al. Integration of lipidomics and transcriptomics unravels aberrant lipid metabolism and defines cholesteryl oleate as potential biomarker of prostate cancer. Sci. Rep. 2016, 6, 20984. [Google Scholar] [CrossRef]
- Young, R.S.E.; Bowman, A.P.; Tousignant, K.D.; Poad, B.L.J.; Gunter, J.H.; Philp, L.K.; Nelson, C.C.; Ellis, S.R.; Heeren, R.M.A.; Sadowski, M.C.; et al. Isomeric lipid signatures reveal compartmentalized fatty acid metabolism in cancer. J. Lipid Res. 2022, 63, 100223. [Google Scholar] [CrossRef]
- Smith, R.E.; Lespi, P.; Di Luca, M.; Bustos, C.; Marra, F.A.; de Alaniz, M.J.; Marra, C.A. A reliable biomarker derived from plasmalogens to evaluate malignancy and metastatic capacity of human cancers. Lipids 2008, 43, 79–89. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Cui, W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021, 28, 2536–2551. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Vaz, F.M.; Waterham, H.R.; Ferdinandusse, S. Fatty acid oxidation in peroxisomes: Enzymology, metabolic crosstalk with other organelles and peroxisomal disorders. Adv. Exp. Med. Biol. 2020, 1299, 55–70. [Google Scholar] [CrossRef]
- Morito, K.; Ali, H.; Kishino, S.; Tanaka, T. Fatty acid metabolism in peroxisomes and related disorders. Adv. Exp. Med. Biol. 2024, 1470, 31–55. [Google Scholar] [CrossRef]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef]
- Fourcade, S.; Ruiz, M.; Camps, C.; Schlüter, A.; Houten, S.M.; Mooyer, P.A.; Pàmpols, T.; Dacremont, G.; Wanders, R.J.; Giròs, M.; et al. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E211–E221. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Costello, J.L.; Kors, S.; Soupene, E.; Levine, T.P.; Kuypers, F.A.; Schrader, M. The diversity of ACBD proteins—From lipid binding to protein modulators and organelle tethers. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118675. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Kotti, T.J.; Savolainen, K.; Helander, H.M.; Yagi, A.; Novikov, D.K.; Kalkkinen, N.; Conzelmann, E.; Hiltunen, J.K.; Schmitz, W. In mouse alpha-methylacyl-CoA racemase, the same gene product is simultaneously located in mitochondria and peroxisomes. J. Biol. Chem. 2000, 275, 20887–20895. [Google Scholar] [CrossRef]
- Amery, L.; Fransen, M.; De Nys, K.; Mannaerts, G.P.; Van Veldhoven, P.P. Mitochondrial and peroxisomal targeting of 2-methylacyl-CoA racemase in humans. J. Lipid Res. 2000, 41, 1752–1759. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; IJlst, L.; Dacremont, G.; Waterham, H.R.; Wanders, R.J. Subcellular localization and physiological role of alpha-methylacyl-CoA racemase. J. Lipid Res. 2000, 41, 1890–1896. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Van Roermund, C.W.; Wanders, R.J.; Dacremont, G. Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids. J. Lipid Res. 2004, 45, 1104–1111. [Google Scholar] [CrossRef]
- Houten, S.M.; Denis, S.; Argmann, C.A.; Jia, Y.; Ferdinandusse, S.; Reddy, J.K.; Wanders, R.J. Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res. 2012, 53, 1296–1303. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Kostopoulos, P.; Denis, S.; Rusch, H.; Overmars, H.; Dillmann, U.; Reith, W.; Haas, D.; Wanders, R.J.; Duran, M.; et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am. J. Hum. Genet. 2006, 78, 1046–1052. [Google Scholar] [CrossRef]
- Horvath, R.; Lewis-Smith, D.; Douroudis, K.; Duff, J.; Keogh, M.; Pyle, A.; Fletcher, N.; Chinnery, P.F. SCP2 mutations and neurodegeneration with brain iron accumulation. Neurology 2015, 85, 1909–1911. [Google Scholar] [CrossRef]
- Hunt, M.C.; Tillander, V.; Alexson, S.E. Regulation of peroxisomal lipid metabolism: The role of acyl-CoA and coenzyme A metabolizing enzymes. Biochimie 2014, 98, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Westin, M.A.; Hunt, M.C.; Alexson, S.E. Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of beta-oxidation products out of peroxisomes. Cell Mol. Life Sci. 2008, 65, 982–990. [Google Scholar] [CrossRef]
- Fillgrove, K.L.; Anderson, V.E. Orientation of coenzyme A substrates, nicotinamide and active site functional groups in (di)enoyl−coenzyme A reductases. Biochemistry 2000, 39, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef]
- Filppula, S.A.; Yagi, A.I.; Kilpeläinen, S.H.; Novikov, D.; FitzPatrick, D.R.; Vihinen, M.; Valle, D.; Hiltunen, J.K. Delta3,5-delta2,4-dienoyl-CoA isomerase from rat liver. Molecular characterization. J. Biol. Chem. 1998, 273, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Van Veldhoven, P.P.; Subramani, S. Identification of peroxisomal proteins by using M13 phage protein VI phage display: Molecular evidence that mammalian peroxisomes contain a 2,4-dienoyl-CoA reductase. Biochem. J. 1999, 340, 561–568. [Google Scholar] [CrossRef]
- Fan, J.; Li, X.; Issop, L.; Culty, M.; Papadopoulos, V. ACBD2/ECI2-mediated peroxisome-mitochondria interactions in Leydig cell steroid biosynthesis. Mol. Endocrinol. 2016, 30, 763–782. [Google Scholar] [CrossRef]
- Christensen, E.; Hagve, T.A.; Christophersen, B.O. The Zellweger syndrome: Deficient chain-shortening of erucic acid (22:1 (n-9)) and adrenic acid (22:4 (n-6)) in cultured skin fibroblasts. Biochim. Biophys. Acta 1988, 959, 134–142. [Google Scholar] [CrossRef]
- Petroni, A.; Bertagnolio, B.; La Spada, P.; Blasevich, M.; Papini, N.; Govoni, S.; Rimoldi, M.; Galli, C. The beta-oxidation of arachidonic acid and the synthesis of docosahexaenoic acid are selectively and consistently altered in skin fibroblasts from three Zellweger patients versus X-adrenoleukodystrophy, Alzheimer and control subjects. Neurosci. Lett. 1998, 250, 145–148. [Google Scholar] [CrossRef]
- Sprecher, H.; Luthria, D.L.; Mohammed, B.S.; Baykousheva, S.P. Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J. Lipid Res. 1995, 36, 2471–2477. [Google Scholar] [CrossRef]
- Sprecher, H. The roles of anabolic and catabolic reactions in the synthesis and recycling of polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fat. Acids 2002, 67, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Bian, F.; Tomcik, K.; Kelleher, J.K.; Zhang, G.F.; Brunengraber, H. Compartmentation of metabolism of the C12-, C9-, and C5-n-dicarboxylates in rat Liver, investigated by mass isotopomer analysis: Anaplerosis from dodecanedioate. J. Biol. Chem. 2015, 290, 18671–18677. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef]
- Ranea-Robles, P.; Chen, H.; Stauffer, B.; Yu, C.; Bhattacharya, D.; Friedman, S.L.; Puchowicz, M.; Houten, S.M. The peroxisomal transporter ABCD3 plays a major role in hepatic dicarboxylic fatty acid metabolism and lipid homeostasis. J. Inherit. Metab. Dis. 2021, 44, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507. [Google Scholar] [CrossRef]
- Mah, C.Y.; Nguyen, A.D.T.; Niijima, T.; Helm, M.; Dehairs, J.; Ryan, F.J.; Ryan, N.; Quek, L.E.; Hoy, A.J.; Don, A.S.; et al. Peroxisomal β-oxidation enzyme, DECR2, regulates lipid metabolism and promotes treatment resistance in advanced prostate cancer. Br. J. Cancer 2024, 130, 741–754. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Brown, M.; Urbanucci, A.; Tredwell, G.; Lau, C.H.; Barfeld, S.; Hart, C.; Guldvik, I.J.; Takhar, M.; Heemers, H.V.; et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget 2017, 8, 38264–38275. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Poulose, N.; Walker, S.; Mills, I.G. CDK9 inhibition induces a metabolic switch that renders prostate cancer cells dependent on fatty acid oxidation. Neoplasia 2019, 21, 713–720. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Centenera, M.M.; Irani, S.; Sadowski, M.C.; Scott, J.S.; Nguyen, E.V.; Nagarajan, S.R.; Moldovan, M.; Lynn, D.J.; et al. Fatty acid oxidation is an adaptive survival pathway induced in prostate tumors by HSP90 inhibition. Mol. Cancer Res. 2020, 18, 1500–1511. [Google Scholar] [CrossRef]
- Xu, J.; Stolk, J.A.; Zhang, X.; Silva, S.J.; Houghton, R.L.; Matsumura, M.; Vedvick, T.S.; Leslie, K.B.; Badaro, R.; Reed, S.G. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res. 2000, 60, 1677–1682. [Google Scholar]
- Beach, R.; Gown, A.M.; De Peralta-Venturina, M.N.; Folpe, A.L.; Yaziji, H.; Salles, P.G.; Grignon, D.J.; Fanger, G.R.; Amin, M.B. P504S immunohistochemical detection in 405 prostatic specimens including 376 18-gauge needle biopsies. Am. J. Surg. Pathol. 2002, 26, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Wu, C.L.; Woda, B.A.; Dresser, K.; Tretiakova, M.; Fanger, G.R.; Jiang, Z. Expression of alpha-methylacyl-CoA racemase (P504S) in atypical adenomatous hyperplasia of the prostate. Am. J. Surg. Pathol. 2002, 26, 921–925. [Google Scholar] [CrossRef]
- Zha, S.; Ferdinandusse, S.; Denis, S.; Wanders, R.J.; Ewing, C.M.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Alpha-methylacyl-CoA racemase as an androgen-independent growth modifier in prostate cancer. Cancer Res. 2003, 63, 7365–7376. [Google Scholar]
- Kumar-Sinha, C.; Shah, R.B.; Laxman, B.; Tomlins, S.A.; Harwood, J.; Schmitz, W.; Conzelmann, E.; Sanda, M.G.; Wei, J.T.; Rubin, M.A.; et al. Elevated alpha-methylacyl-CoA racemase enzymatic activity in prostate cancer. Am. J. Pathol. 2004, 164, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Zhou, M.; Dhanasekaran, S.M.; Varambally, S.; Barrette, T.R.; Sanda, M.G.; Pienta, K.J.; Ghosh, D.; Chinnaiyan, A.M. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002, 287, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Woda, B.A.; Rock, K.L.; Xu, Y.; Savas, L.; Khan, A.; Pihan, G.; Cai, F.; Babcook, J.S.; Rathanaswami, P.; et al. P504S: A new molecular marker for the detection of prostate carcinoma. Am. J. Surg. Pathol. 2001, 25, 1397–1404. [Google Scholar] [CrossRef]
- Luo, J.; Zha, S.; Gage, W.R.; Dunn, T.A.; Hicks, J.L.; Bennett, C.J.; Ewing, C.M.; Platz, E.A.; Ferdinandusse, S.; Wanders, R.J.; et al. Alpha-methylacyl-CoA racemase: A new molecular marker for prostate cancer. Cancer Res. 2002, 62, 2220–2226. [Google Scholar]
- Lloyd, M.D.; Yevglevskis, M.; Lee, G.L.; Wood, P.J.; Threadgill, M.D.; Woodman, T.J. α-Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog. Lipid Res. 2013, 52, 220–230. [Google Scholar] [CrossRef]
- Carnell, A.J.; Hale, I.; Denis, S.; Wanders, R.J.; Isaacs, W.B.; Wilson, B.A.; Ferdinandusse, S. Design, synthesis, and in vitro testing of alpha-methylacyl-CoA racemase inhibitors. J. Med. Chem. 2007, 50, 2700–2707. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; Mancini, A.; Muzi, P.; Cesare, E.D.; Kirk, R.; Smith, M.; Hughes, S.; Gibson, R.; Lian, L.Y.; et al. Trifluoroibuprofen inhibits α-methylacyl coenzyme A racemase (AMACR/P504S), reduces cancer cell proliferation and inhibits in vivo tumor growth in aggressive prostate cancer models. Anticancer Agents Med. Chem. 2014, 14, 1031–1041. [Google Scholar] [CrossRef]
- Xie, H.; Nie, L.; Zhang, M.; Su, Z.; Chen, X.; Xu, M.; Gong, J.; Chen, N.; Zhou, Q. Suppression of α-methylacyl-coenzyme A racemase by miR200c inhibits prostate adenocarcinoma cell proliferation and migration. Exp. Ther. Med. 2020, 19, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Bu, C.; Cui, B.; Li, N.; Wu, J. Screening of differentially expressed genes and identification of AMACR as a prognostic marker in prostate cancer. Andrologia 2021, 53, e14067. [Google Scholar] [CrossRef]
- Jiang, N.; Zhu, S.; Chen, J.; Niu, Y.; Zhou, L. A-methylacyl-CoA racemase (AMACR) and prostate-cancer risk: A meta-analysis of 4,385 participants. PLoS ONE 2013, 8, e74386. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Kim, K.I. Emerging roles of orphan nuclear receptors in cancer. Annu. Rev. Physiol. 2014, 76, 177–195. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- True, L.; Coleman, I.; Hawley, S.; Huang, C.Y.; Gifford, D.; Coleman, R.; Beer, T.M.; Gelmann, E.; Datta, M.; Mostaghel, E.; et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc. Natl. Acad. Sci. USA 2006, 103, 10991–10996. [Google Scholar] [CrossRef]
- Rasiah, K.K.; Gardiner-Garden, M.; Padilla, E.J.; Möller, G.; Kench, J.G.; Alles, M.C.; Eggleton, S.A.; Stricker, P.D.; Adamski, J.; Sutherland, R.L.; et al. HSD17B4 overexpression, an independent biomarker of poor patient outcome in prostate cancer. Mol. Cell Endocrinol. 2009, 301, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Roberto, D.; Selvarajah, S.; Park, P.C.; Berman, D.; Venkateswaran, V. Functional validation of metabolic genes that distinguish Gleason 3 from Gleason 4 prostate cancer foci. Prostate 2019, 79, 1777–1788. [Google Scholar] [CrossRef]
- Kwon, E.M.; Holt, S.K.; Fu, R.; Kolb, S.; Williams, G.; Stanford, J.L.; Ostrander, E.A. Androgen metabolism and JAK/STAT pathway genes and prostate cancer risk. Cancer Epidemiol. 2012, 36, 347–353. [Google Scholar] [CrossRef]
- Huang, H.; Liu, R.; Huang, Y.; Feng, Y.; Fu, Y.; Chen, L.; Chen, Z.; Cai, Y.; Zhang, Y.; Chen, Y. Acetylation-mediated degradation of HSD17B4 regulates the progression of prostate cancer. Aging 2020, 12, 14699–14717. [Google Scholar] [CrossRef]
- Ko, H.K.; Berk, M.; Chung, Y.M.; Willard, B.; Bareja, R.; Rubin, M.; Sboner, A.; Sharifi, N. Loss of an androgen-inactivating and isoform-specific HSD17B4 splice form enables emergence of castration-resistant prostate cancer. Cell Rep. 2018, 22, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Fulford, M.; Li, C.F.; Boonyaratanakornkit, J.; Sayyah, S. Arachidonic acid activates phosphatidylinositol 3-kinase signaling and induces gene expression in prostate cancer. Cancer Res. 2006, 66, 1427–1433. [Google Scholar] [CrossRef]
- Patel, M.I.; Kurek, C.; Dong, Q. The arachidonic acid pathway and its role in prostate cancer development and progression. J. Urol. 2008, 179, 1668–1675. [Google Scholar] [CrossRef]
- Yang, P.; Cartwright, C.A.; Li, J.; Wen, S.; Prokhorova, I.N.; Shureiqi, I.; Troncoso, P.; Navone, N.M.; Newman, R.A.; Kim, J. Arachidonic acid metabolism in human prostate cancer. Int. J. Oncol. 2012, 41, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qi, H.; Zhang, R.; Zhang, K.; Shi, Z.; Chang, Y.; Chen, L.; Esmaeili, M.; Baniahmad, A.; Hong, W. Docosahexaenoic acid inhibits the growth of hormone-dependent prostate cancer cells by promoting the degradation of the androgen receptor. Mol. Med. Rep. 2015, 12, 3769–3774. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, C.Y.; Kao, C.L.; Jiang, Y.; Liu, C.M. Docosahexaenoic acid inhibits lipopolysaccharide-induced metastatic activities by decreasing inflammation on prostate cancer cell. Pharmazie 2019, 74, 675–679. [Google Scholar] [CrossRef]
- Kudo, Y.; Nakamura, K.; Tsuzuki, H.; Hirota, K.; Kawai, M.; Takaya, D.; Fukuzawa, K.; Honma, T.; Yoshino, Y.; Nakamura, M.; et al. Docosahexaenoic acid enhances the treatment efficacy for castration-resistant prostate cancer by inhibiting autophagy through Atg4B inhibition. Arch. Biochem. Biophys. 2024, 760, 110135. [Google Scholar] [CrossRef]
- Lin, S.C.; Tsai, Y.C.; Chen, Y.L.; Lin, H.K.; Huang, Y.C.; Lin, Y.S.; Cheng, Y.S.; Chen, H.Y.; Li, C.J.; Lin, T.Y.; et al. Un-methylation of NUDT21 represses docosahexaenoic acid biosynthesis contributing to enzalutamide resistance in prostate cancer. Drug Resist. Updat. 2024, 77, 101144. [Google Scholar] [CrossRef]
- Tamarindo, G.H.; Ribeiro, C.F.; Silva, A.D.T.; Castro, A.; Caruso, Í.P.; Souza, F.P.; Taboga, S.R.; Loda, M.; Góes, R.M. The polyunsaturated fatty acid docosahexaenoic affects mitochondrial function in prostate cancer cells. Cancer Metab. 2024, 12, 24. [Google Scholar] [CrossRef]
- Yu, G.; Cheng, C.J.; Lin, S.C.; Lee, Y.C.; Frigo, D.E.; Yu-Lee, L.Y.; Gallick, G.E.; Titus, M.A.; Nutt, L.K.; Lin, S.H. Organelle-derived acetyl-CoA promotes prostate cancer cell survival, migration, and metastasis via activation of calmodulin kinase II. Cancer Res. 2018, 78, 2490–2502. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Revenco, I.; Li, H.; Costa, C.F.; Lenaerts, L.; Hussein, M.A.F.; De Bie, J.; Knoops, B.; Van Veldhoven, P.P.; Derua, R.; et al. Peroxisome-derived hydrogen peroxide modulates the sulfenylation profiles of key redox signaling proteins in Flp-In T-REx 293 cells. Front. Cell Dev. Biol. 2022, 10, 888873. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lismont, C.; Costa, C.F.; Hussein, M.A.F.; Baes, M.; Fransen, M. Enhanced levels of peroxisome-derived H2O2 do not induce pexophagy but impair autophagic flux in HEK-293 and HeLa cells. Antioxidants 2023, 12, 613. [Google Scholar] [CrossRef]
- Karpenko, I.L.; Valuev-Elliston, V.T.; Ivanova, O.N.; Smirnova, O.A.; Ivanov, A.V. Peroxiredoxins—The underrated actors during virus-induced oxidative stress. Antioxidants 2021, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef]
- Costa, C.F.; Lismont, C.; Chornyi, S.; Li, H.; Hussein, M.A.F.; Waterham, H.R.; Fransen, M. Functional analysis of GSTK1 in peroxisomal redox homeostasis in HEK-293 cells. Antioxidants 2023, 12, 1236. [Google Scholar] [CrossRef]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef]
- Rixen, S.; Havemeyer, A.; Tyl-Bielicka, A.; Pysniak, K.; Gajewska, M.; Kulecka, M.; Ostrowski, J.; Mikula, M.; Clement, B. Mitochondrial amidoxime-reducing component 2 (MARC2) has a significant role in N-reductive activity and energy metabolism. J. Biol. Chem. 2019, 294, 17593–17602. [Google Scholar] [CrossRef]
- Rixen, S.; Indorf, P.M.; Kubitza, C.; Struwe, M.A.; Klopp, C.; Scheidig, A.J.; Kunze, T.; Clement, B. Reduction of hydrogen peroxide by human mitochondrial amidoxime reducing component enzymes. Molecules 2023, 28, 6384. [Google Scholar] [CrossRef]
- Islinger, M.; Li, K.W.; Seitz, J.; Völkl, A.; Lüers, G.H. Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes—Evidence for a natural piggyback import mechanism in mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Ruan, Y.; Che, X.; Feng, W. Dual role of PRDX1 in redox-regulation and tumorigenesis: Past and future. Free Radic. Biol. Med. 2024, 210, 120–129. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Gronemeyer, T.; Ofman, R.; Kunze, M.; Grou, C.P.; Almeida, J.A.; Eisenacher, M.; Stephan, C.; Hayen, H.; Schollenberger, L.; et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell Proteom. 2007, 6, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Lüers, G.H.; Li, K.W.; Loos, M.; Völkl, A. Rat liver peroxisomes after fibrate treatment. A survey using quantitative mass spectrometry. J. Biol. Chem. 2007, 282, 23055–23069. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Gavriliuk, L.A.; Pokrovsky, V.S. Oxidative stress and redox-dependent signaling in prostate cancer. Biochemistry 2022, 87, 413–424. [Google Scholar] [CrossRef]
- Baker, A.M.; Oberley, L.W.; Cohen, M.B. Expression of antioxidant enzymes in human prostatic adenocarcinoma. Prostate 1997, 32, 229–233. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Alexander, E.E.; Singh, R.; Shan, A.; Qian, J.; Santella, R.M.; Oberley, L.W.; Yan, T.; Zhong, W.; Jiang, X.; et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer 2000, 89, 123–134. [Google Scholar] [CrossRef]
- Oberley, T.D.; Zhong, W.; Szweda, L.I.; Oberley, L.W. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate 2000, 44, 144–155. [Google Scholar] [CrossRef]
- Miar, A.; Hevia, D.; Muñoz-Cimadevilla, H.; Astudillo, A.; Velasco, J.; Sainz, R.M.; Mayo, J.C. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic. Biol. Med. 2015, 85, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Nowell, S.; McCann, S.E.; Yu, J.; Carter, L.; Lang, N.P.; Kadlubar, F.F.; Ratnasinghe, L.D.; Ambrosone, C.B. Associations between catalase phenotype and genotype: Modification by epidemiologic factors. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, D.; Tian, P.; Shen, K.; Zhu, J.; Feng, M.; Wan, C.; Yang, T.; Chen, L.; Wen, F. The catalase C-262T gene polymorphism and cancer risk: A systematic review and meta-analysis. Medicine 2015, 94, e679. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.D.; Sun, Y.; Chen, N.; Huang, L.; Huang, J.W.; Zhu, M.; Wang, T.; Ji, Y.L. The role of catalase C262T gene polymorphism in the susceptibility and survival of cancers. Sci. Rep. 2016, 6, 26973. [Google Scholar] [CrossRef]
- Tao, Y.; Liu, S.; Lu, J.; Fu, S.; Li, L.; Zhang, J.; Wang, Z.; Hong, M. FOXO3a-ROS pathway is involved in androgen-induced proliferation of prostate cancer cell. BMC Urol. 2022, 22, 70. [Google Scholar] [CrossRef]
- Giginis, F.; Wang, J.; Chavez, A.; Martins-Green, M. Catalase as a novel drug target for metastatic castration-resistant prostate cancer. Am. J. Cancer Res. 2023, 13, 2644–2656. [Google Scholar]
- Sen, S.; Kawahara, B.; Chaudhuri, G. Maintenance of higher H₂O₂ levels, and its mechanism of action to induce growth in breast cancer cells: Important roles of bioactive catalase and PP2A. Free Radic. Biol. Med. 2012, 53, 1541–1551. [Google Scholar] [CrossRef]
- Onumah, O.E.; Jules, G.E.; Zhao, Y.; Zhou, L.; Yang, H.; Guo, Z. Overexpression of catalase delays G0/G1- to S-phase transition during cell cycle progression in mouse aortic endothelial cells. Free Radic. Biol. Med. 2009, 46, 1658–1667. [Google Scholar] [CrossRef]
- Brown, M.R.; Miller, F.J., Jr.; Li, W.G.; Ellingson, A.N.; Mozena, J.D.; Chatterjee, P.; Engelhardt, J.F.; Zwacka, R.M.; Oberley, L.W.; Fang, X.; et al. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ. Res. 1999, 85, 524–533. [Google Scholar] [CrossRef]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Liang, H.Q.; Liu, M.T.; Huang, W.; Yi, P.F.; Yuan, S.Q.; Mei, L.C.; Hu, Y.H.; Cao, Y.Y.; Hao, G.F.; et al. Catalase inhibitors with dual pro-oxidant effect as new therapeutic agents in castration-resistant prostate cancer. Adv. Therap. 2021, 4, 2000164. [Google Scholar] [CrossRef]
- Lin, J.F.; Tsai, T.F.; Yang, S.C.; Lin, Y.C.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Benzyl isothiocyanate induces reactive oxygen species-initiated autophagy and apoptosis in human prostate cancer cells. Oncotarget. 2017, 8, 20220–20234. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Y.; Chen, Y.Y.; Wang, M.S.; Tong, J.J.; Xu, M.; Zhao, C.; Lin, H.Y.; Mei, L.C.; Dong, J.; Zhang, W.L.; et al. A catalase inhibitor: Targeting the NADPH-binding site for castration-resistant prostate cancer therapy. Redox Biol. 2023, 63, 102751. [Google Scholar] [CrossRef]
- Riddell, J.R.; Bshara, W.; Moser, M.T.; Spernyak, J.A.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res. 2011, 71, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Riddell, J.R.; Maier, P.; Sass, S.N.; Moser, M.T.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 stimulates endothelial cell expression of VEGF via TLR4 dependent activation of HIF-1α. PLoS ONE 2012, 7, e50394. [Google Scholar] [CrossRef]
- Park, S.Y.; Yu, X.; Ip, C.; Mohler, J.L.; Bogner, P.N.; Park, Y.M. Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res. 2007, 67, 9294–9303. [Google Scholar] [CrossRef]
- Azam, H.; Veale, C.; Zitzmann, K.; Marcone, S.; Gallagher, W.M.; Prencipe, M. Identification of druggable targets from the interactome of the androgen receptor and serum response factor pathways in prostate cancer. PLoS ONE 2024, 19, e0309491. [Google Scholar] [CrossRef]
- Chhipa, R.R.; Lee, K.S.; Onate, S.; Wu, Y.; Ip, C. Prx1 enhances androgen receptor function in prostate cancer cells by increasing receptor affinity to dihydrotestosterone. Mol. Cancer Res. 2009, 7, 1543–1552. [Google Scholar] [CrossRef]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef]
- Wu, Y.; Chhipa, R.R.; Zhang, H.; Ip, C. The antiandrogenic effect of finasteride against a mutant androgen receptor. Cancer Biol. Ther. 2011, 11, 902–909. [Google Scholar] [CrossRef]
- Dasari, C.; Reddy, K.R.K.; Natani, S.; Murthy, T.R.L.; Bhukya, S.; Ummanni, R. Tumor protein D52 (isoform 3) interacts with and promotes peroxidase activity of Peroxiredoxin 1 in prostate cancer cells implicated in cell growth and migration. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1298–1309. [Google Scholar] [CrossRef]
- Lai, W.; Zhu, W.; Wu, J.; Huang, J.; Li, X.; Luo, Y.; Wang, Y.; Zeng, H.; Li, M.; Qiu, X.; et al. HJURP inhibits sensitivity to ferroptosis inducers in prostate cancer cells by enhancing the peroxidase activity of PRDX1. Redox Biol. 2024, 77, 103392. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Mi, Y.; Ni, J.; Wang, Y.; Ding, L.; Ran, X.; Sun, Q.; Tan, S.Y.; Koeffler, H.P.; Feng, N.; et al. Identification of PRDX5 as A Target for The Treatment of Castration-Resistant Prostate Cancer. Adv. Sci. 2024, 11, e2304939. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Purcell, C.; Kuang, Y.Q.; Charette, N.; Dupré, D.J. Superoxide dismutase 1 regulation of CXCR4-mediated signaling in prostate cancer cells is dependent on cellular oxidative state. Cell Physiol. Biochem. 2015, 37, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.S.; Zhao, H.; Xu, C.X.; Xie, P.B.; Wang, W.; Yang, Y.Y.; Lee, W.H.; Jin, Y.; Zhou, H.Q. Clinical significance of EPHX2 deregulation in prostate cancer. Asian J. Androl. 2021, 23, 109–115. [Google Scholar] [CrossRef]
- Vainio, P.; Gupta, S.; Ketola, K.; Mirtti, T.; Mpindi, J.P.; Kohonen, P.; Fey, V.; Perälä, M.; Smit, F.; Verhaegh, G.; et al. Arachidonic acid pathway members PLA2G7, HPGD, EPHX2, and CYP4F8 identified as putative novel therapeutic targets in prostate cancer. Am. J. Pathol. 2011, 178, 525–536. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Liang, F.; Zhang, L.; Liang, X. Identification of metabolism-associated prostate cancer subtypes and construction of a prognostic risk model. Front. Oncol. 2020, 10, 598801. [Google Scholar] [CrossRef]
- Van Broekhoven, A.; Peeters, M.C.; Debeer, L.J.; Mannaerts, G.P. Subcellular distribution of coenzyme A: Evidence for a separate coenzyme A pool in peroxisomes. Biochem. Biophys. Res. Commun. 1981, 100, 305–312. [Google Scholar] [CrossRef]
- Horie, S.; Isobe, M.; Suga, T. Changes in CoA pools in hepatic peroxisomes of the rat under various conditions. J. Biochem. 1986, 99, 1345–1352. [Google Scholar] [CrossRef]
- Agrimi, G.; Russo, A.; Scarcia, P.; Palmieri, F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem. J. 2012, 443, 241–247. [Google Scholar] [CrossRef]
- Tan, Z.; Deng, Y.; Cai, Z.; He, H.; Tang, Z.; Feng, Y.; Ye, J.; Liu, R.; Cai, S.; Huang, H.; et al. ACOX2 serves as a favorable indicator related to lipid metabolism and oxidative stress for biochemical recurrence in prostate cancer. J. Cancer 2024, 15, 3010–3023. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.P.; Rajendiran, T.M.; Bushra, A.; Asangani, I.A.; Athanikar, J.N.; Yocum, A.K.; Mehra, R.; Siddiqui, J.; Palapattu, G.; Wei, J.T.; et al. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 2013, 15, 491–501. [Google Scholar] [CrossRef]
- Zhao, S.; Li, P.; Wu, W.; Wang, Q.; Qian, B.; Li, X.; Shen, M. Roles of ferroptosis in urologic malignancies. Cancer Cell Int. 2021, 21, 676. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.P.; Verma, S.S.; Shankar, E.; Lin, S.; Gupta, S. Role of solute carrier transporters SLC25A17 and SLC27A6 in acquired resistance to enzalutamide in castration-resistant prostate cancer. Mol. Carcinog. 2022, 61, 397–407. [Google Scholar] [CrossRef]
- Sedelaar, J.P.; Isaacs, J.T. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate 2009, 69, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Li, Y.; Jian, L.; Yang, Y.; Zhao, L.; Wei, M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: Mechanisms and clinical treatment strategies. Mol. Cancer 2022, 21, 177. [Google Scholar] [CrossRef]
- Yu, X.; Liu, R.; Gao, W.; Wang, X.; Zhang, Y. Single-cell omics traces the heterogeneity of prostate cancer cells and the tumor microenvironment. Cell Mol. Biol. Lett. 2023, 28, 38. [Google Scholar] [CrossRef]
- Di Cara, F.; Savary, S.; Kovacs, W.J.; Kim, P.; Rachubinski, R.A. The peroxisome: An up-and-coming organelle in immunometabolism. Trends Cell Biol. 2023, 33, 70–86. [Google Scholar] [CrossRef]
- Yan, Y.; Mao, X.; Zhang, Q.; Ye, Y.; Dai, Y.; Bao, M.; Zeng, Y.; Huang, R.; Mo, Z. Molecular mechanisms, immune cell infiltration, and potential drugs for prostate cancer. Cancer Biomark. 2021, 31, 87–96. [Google Scholar] [CrossRef]
- Cai, Y.; Lin, J.; Wang, Z.; Ma, Y.; Pan, J.; Liu, Y.; Zhao, Z. Identification and validation of a lipid metabolism gene signature for predicting biochemical recurrence of prostate cancer after radical prostatectomy. Front. Oncol. 2022, 12, 1009921. [Google Scholar] [CrossRef]
- Xu, F.; Wang, X.; Huang, Y.; Zhang, X.; Sun, W.; Du, Y.; Xu, Z.; Kou, H.; Zhu, S.; Liu, C.; et al. Prostate cancer cell-derived exosomal IL-8 fosters immune evasion by disturbing glucolipid metabolism of CD8+ T cell. Cell Rep. 2023, 42, 113424. [Google Scholar] [CrossRef] [PubMed]
- Casteels, M.; Croes, K.; Van Veldhoven, P.P.; Mannaerts, G.P. Aminotriazole is a potent inhibitor of alpha-oxidation of 3-methyl-substituted fatty acids in rat liver. Biochem. Pharmacol. 1994, 48, 1973–1975. [Google Scholar] [CrossRef] [PubMed]
- Tephly, T.R.; Hasegawa, E.; Baron, J. Effect of drugs on heme synthesis in the liver. Metabolism 1971, 20, 200–214. [Google Scholar] [CrossRef]
- Koop, D.R. Inhibition of ethanol-inducible cytochrome P450IIE1 by 3-amino-1,2,4-triazole. Chem. Res. Toxicol. 1990, 3, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Fernandes, E.; Lima, J.L. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef]
- Smolyarova, D.D.; Podgorny, O.V.; Bilan, D.S.; Belousov, V.V. A guide to genetically encoded tools for the study of H2O2. FEBS J. 2022, 289, 5382–5395. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussein, M.A.F.; Lismont, C.; Li, H.; Chai, R.; Claessens, F.; Fransen, M. Peroxisomal Alterations in Prostate Cancer: Metabolic Shifts and Clinical Relevance. Cancers 2025, 17, 2243. https://doi.org/10.3390/cancers17132243
Hussein MAF, Lismont C, Li H, Chai R, Claessens F, Fransen M. Peroxisomal Alterations in Prostate Cancer: Metabolic Shifts and Clinical Relevance. Cancers. 2025; 17(13):2243. https://doi.org/10.3390/cancers17132243
Chicago/Turabian StyleHussein, Mohamed A. F., Celien Lismont, Hongli Li, Ruizhi Chai, Frank Claessens, and Marc Fransen. 2025. "Peroxisomal Alterations in Prostate Cancer: Metabolic Shifts and Clinical Relevance" Cancers 17, no. 13: 2243. https://doi.org/10.3390/cancers17132243
APA StyleHussein, M. A. F., Lismont, C., Li, H., Chai, R., Claessens, F., & Fransen, M. (2025). Peroxisomal Alterations in Prostate Cancer: Metabolic Shifts and Clinical Relevance. Cancers, 17(13), 2243. https://doi.org/10.3390/cancers17132243