Current Landscape of Preclinical Models for Pediatric Gliomas: Clinical Implications and Future Directions

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Molecular Classification of Malignant Pediatric Brain Tumors
2.1. pHGGs
2.2. DIPG/DMG, H3K27M-Mutant
2.3. Infant-Type Hemispheric Gliomas
2.4. pLGGs
2.5. Oligodendrogliomas
2.6. Ependymomas
2.7. Integration of Molecular and Histological Insights
3. In Vivo Models of pHGG
3.1. Carcinogen-Induced Animal Models
3.2. Oncogenic Virus-Induced Models
3.3. Xenograft Animal Models
4. Immune-Competent Pediatric Brain Tumor Models
4.1. GEMMs
4.2. Viral Delivery Models
4.3. In Utero Electroporation
4.4. Transposon-Mediated Delivery
4.5. Zebrafish Models
4.5.1. Xenograft Models
4.5.2. Syngeneic Models
4.6. Syngeneic Allograft Mouse Models
4.7. Humanized Mouse Models for Pediatric Brain Tumors
5. Large Animal Models for Pediatric Brain Tumor Research
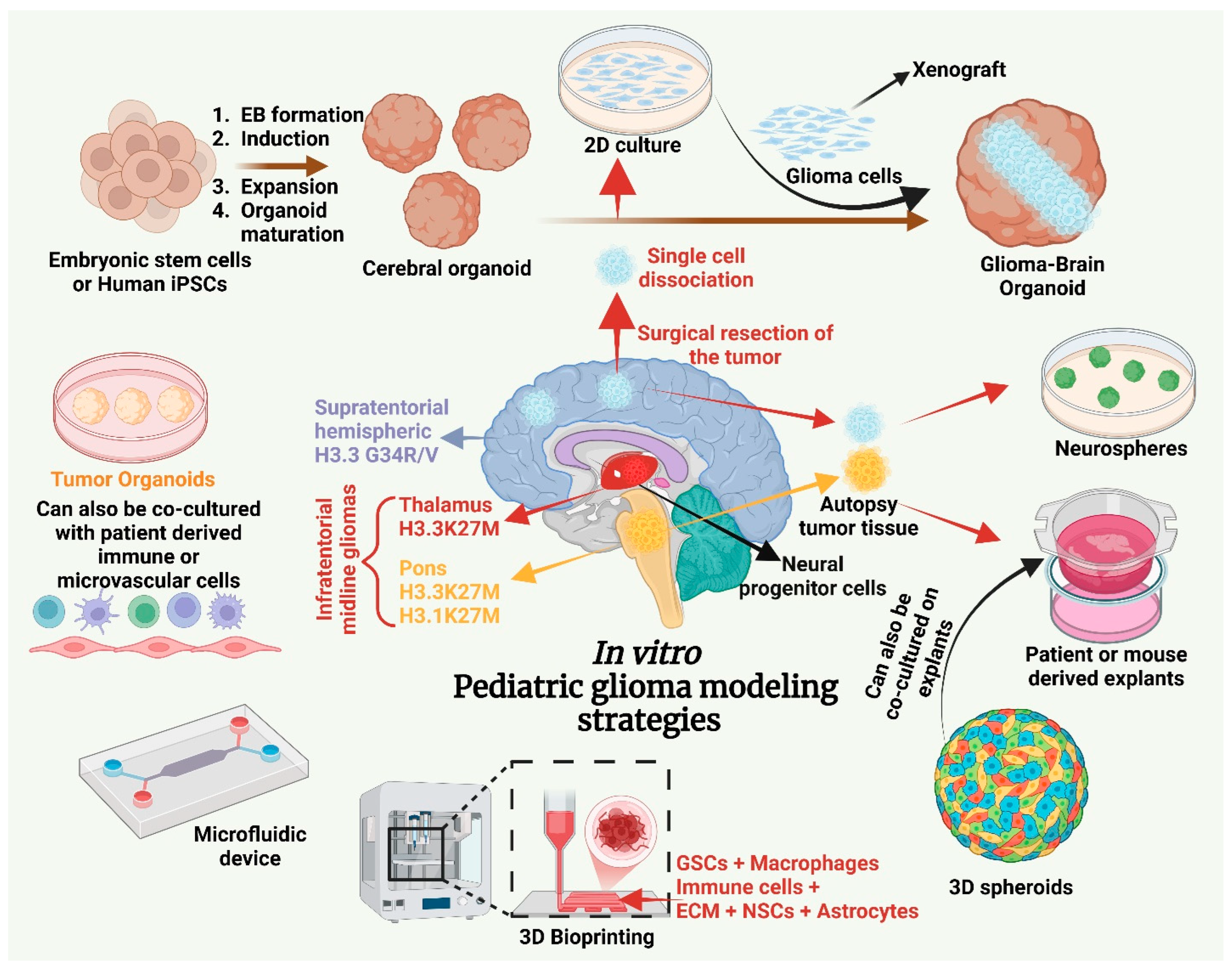
6. In Vitro Models of Pediatric Brain Tumors
6.1. 2D and 3D Cultures
6.2. Brain Organoids
6.3. Types of Brain Organoids
6.3.1. Glioblastoma Organoids (GBOs)
6.3.2. Neoplastic Cerebral Organoids (neoCORs)
6.3.3. Glioblastoma-like Cerebral Organoids (GLICOs)
6.3.4. Tumor-Bearing Organoids (TBOs)
6.3.5. Patient-Derived Organoids (PDOs)
6.3.6. Microglia-Containing Brain Organoids (MiCBOs)
6.3.7. Medulloblastoma Organoids
6.3.8. Expanded Neuroepithelium Organoids (ENOs)
7. Ex Vivo Models
8. Advances in Tumor Detection Methods
9. Future Directions
10. Conclusions and Prospects
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| pHGG | pediatric high-grade glioma |
| DMG | diffuse midline glioma |
| TME | tumor microenvironment |
| BBB | blood–brain barrier |
| NPC | neural precursor cell |
| OPC | oligodendrocyte precursor cell |
| GSC | glioma stem cell |
| BTB | blood–brain tumor barrier |
| PDX | patient-derived xenograft |
| GEMM | genetically engineered mouse model |
| CNS | central nervous system |
| WHO | World Health Organization |
| NF1 | neurofibromatosis type 1 |
| NF2 | neurofibromatosis type 2 |
| DIPG | diffuse intrinsic pontine glioma |
| OS | overall survival |
| pLGG | pediatric low-grade glioma |
| DHG | diffuse hemispheric glioma |
| RTK | receptor tyrosine kinase |
| PA | pilocytic astrocytoma |
| ST-RELA | supratentorial-RELA |
| ST-YAP1 | supratentorial-YAP1 |
| PF-A | posterior fossa-A |
| PF-B | posterior fossa-B |
| SP-EPN | spinal ependymoma |
| MB | medulloblastoma |
| NGS | next-generation sequencing |
| IUE | in utero electroporation |
| SB | Sleeping Beauty |
| ZFN | zinc finger nuclease |
| TALEN | transcription activator-like effector nuclease |
| PXA | pleomorphic xanthoastrocytoma |
| iPSC | induced pluripotent stem cell |
| EB | embryoid body |
| ECM | extracellular matrix |
| NSC | neural stem cell |
| RSV-1 | Rous sarcoma virus-1 |
| Ad12 | human adenovirus 12 |
| CDX | cell line-derived xenograft |
| PDOX | patient-derived orthotopic xenograft |
| GVHD | graft-versus-host disease |
| BTIC | brain tumor-initiating cell |
| PDX-MI | PDX Minimal Information standard |
| PDGF | platelet-derived growth factor |
| TIME | tumor–immune microenvironment |
| PBMC | peripheral blood mononuclear cell |
| HSC | hematopoietic stem cell |
| GBM | glioblastoma multiforme |
| ATRT | atypical teratoid rhabdoid tumor |
| GBO | glioblastoma organoid |
| neoCOR | neoplastic cerebral organoid |
| GLICO | glioblastoma-like cerebral organoid |
| TBO | tumor-bearing organoid |
| PDO | patient-derived organoid |
| MiCBO | microglia-containing brain organoid |
| ETV2 | ETS variant 2 |
| ENO | expanded neuroepithelium organoid |
| MEA | microelectrode array |
| MRI | magnetic resonance imaging |
| DWI | diffusion-weighted imaging |
| ADC | apparent diffusion coefficient |
| MR | magnetic resonance |
| CEST | chemical exchange saturation transfer |
| PET | positron emission tomography |
| CT | computed tomography |
References
- Ostrom, Q.T.; Price, M.; Ryan, K.; Edelson, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Pediatric Brain Tumor Foundation Childhood and Adolescent Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2022, 24, iii1–iii38. [Google Scholar] [CrossRef]
- Subramanian, S.; Ahmad, T. Childhood Brain Tumors. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [PubMed]
- Grigore, F.N.; Yang, S.J.; Chen, C.C.; Koga, T. Pioneering models of pediatric brain tumors. Neoplasia 2023, 36, 100859. [Google Scholar] [CrossRef] [PubMed]
- AlRayahi, J.; Alwalid, O.; Mubarak, W.; Maaz, A.U.R.; Mifsud, W. Pediatric Brain Tumors in the Molecular Era: Updates for the Radiologist. Semin. Roentgenol. 2023, 58, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Popovski, D.; Morris, S.M.; Bondoc, A.; Senthil Kumar, S.; Girard, E.J.; Rutka, J.; Fouladi, M.; Huang, A.; Olson, J.M.; et al. Preclinical pediatric brain tumor models for immunotherapy: Hurdles and a way forward. Neuro Oncol. 2024, 26, 226–235. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- De Cola, A.; Foss, A.; Gilbertson, R.; Pathania, M. Biological, Diagnostic, and Therapeutic Insights from (Epi)Genomic Profiling of Pediatric Brain Tumors. Annu. Rev. Cancer Biol. 2024, 8, 199–226. [Google Scholar] [CrossRef]
- Lebrun, L.; Allard-Demoustiez, S.; Gilis, N.; Van Campenhout, C.; Rodesch, M.; Roman, C.; Calo, P.; Lolli, V.; David, P.; Fricx, C.; et al. Clinicopathological and molecular characterization of a case classified by DNA-methylation profiling as “CNS embryonal tumor with BRD4-LEUTX fusion”. Acta Neuropathol. Commun. 2023, 11, 46. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Fangusaro, J.; Bandopadhayay, P. Advances in the classification and treatment of pediatric brain tumors. Curr. Opin. Pediatr. 2021, 33, 26–32. [Google Scholar] [CrossRef]
- Guo, X.; Shi, Y.; Liu, D.; Li, Y.; Chen, W.; Wang, Y.; Wang, Y.; Xing, H.; Xia, Y.; Li, J.; et al. Clinical updates on gliomas and implications of the 5th edition of the WHO classification of central nervous system tumors. Front. Oncol. 2023, 13, 1131642. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Hopman, S.M.; Merks, J.H.; Aalfs, C.M.; Hennekam, R.C. Brain tumors and syndromes in children. Neuropediatrics 2014, 45, 137–161. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Brien, G.L.; Bressan, R.B.; Monger, C.; Gannon, D.; Lagan, E.; Doherty, A.M.; Healy, E.; Neikes, H.; Fitzpatrick, D.J.; Deevy, O.; et al. Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat. Genet. 2021, 53, 1221–1232. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef]
- Hashizume, R.; Smirnov, I.; Liu, S.; Phillips, J.J.; Hyer, J.; McKnight, T.R.; Wendland, M.; Prados, M.; Banerjee, A.; Nicolaides, T.; et al. Characterization of a diffuse intrinsic pontine glioma cell line: Implications for future investigations and treatment. J. Neurooncol. 2012, 110, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Reitman, Z.J.; Lu, S.; Buchan, G.; Gionet, G.; Dubois, F.; Carvalho, D.M.; Shih, J.; Zhang, S.; Greenwald, N.F.; et al. PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat. Commun. 2022, 13, 604. [Google Scholar] [CrossRef]
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.J.; Witt, H.; Korshunov, A.; Read, T.A.; Sun, J.L.; et al. An animal model of MYC-driven medulloblastoma. Cancer Cell 2012, 21, 155–167. [Google Scholar] [CrossRef]
- Filbin, M.; Monje, M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat. Med. 2019, 25, 367–376. [Google Scholar] [CrossRef]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.; Jiang, L.; Samuelsson, E.R.; Marco Salas, S.; Beck, A.; Hack, O.A.; Jeong, D.; Shaw, M.L.; Englinger, B.; LaBelle, J.; et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat. Genet. 2022, 54, 1881–1894. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Damodharan, S.; Shireman, J.M.; Xie, E.; Distler, E.; Kendziorski, C.; Dey, M. Transcriptomic and proteomic spatial profiling of pediatric and adult diffuse midline glioma H3 K27-Altered. Sci. Rep. 2024, 14, 22668. [Google Scholar] [CrossRef]
- Monje, M.; Mahdi, J.; Majzner, R.; Yeom, K.W.; Schultz, L.M.; Richards, R.M.; Barsan, V.; Song, K.W.; Kamens, J.; Baggott, C.; et al. Intravenous and intracranial GD2-CAR T cells for H3K27M(+) diffuse midline gliomas. Nature 2025, 637, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular B7-H3 CAR T Cells for Diffuse Intrinsic Pontine Glioma: Preliminary First-in-Human Bioactivity and Safety. Cancer Discov. 2023, 13, 114–131. [Google Scholar] [CrossRef]
- Baxter, P.A.; Su, J.M.; Onar-Thomas, A.; Billups, C.A.; Li, X.N.; Poussaint, T.Y.; Smith, E.R.; Thompson, P.; Adesina, A.; Ansell, P.; et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: A Pediatric Brain Tumor Consortium study. Neuro Oncol. 2020, 22, 875–885. [Google Scholar] [CrossRef]
- Gallego Perez-Larraya, J.; Garcia-Moure, M.; Labiano, S.; Patino-Garcia, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef]
- Monje, M.; Cooney, T.; Glod, J.; Huang, J.; Peer, C.J.; Faury, D.; Baxter, P.; Kramer, K.; Lenzen, A.; Robison, N.J.; et al. Phase I trial of panobinostat in children with diffuse intrinsic pontine glioma: A report from the Pediatric Brain Tumor Consortium (PBTC-047). Neuro Oncol. 2023, 25, 2262–2272. [Google Scholar] [CrossRef]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.L.; et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 2019, 36, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Caretti, V.; Sewing, A.C.; Lagerweij, T.; Schellen, P.; Bugiani, M.; Jansen, M.H.; van Vuurden, D.G.; Navis, A.C.; Horsman, I.; Vandertop, W.P.; et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol. 2014, 127, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Janowski, M.; Killick-Cole, C.L.; Singleton, W.G.B.; Campbell, E.; Walczak, P.; Khatua, S.; Faltings, L.; Symons, M.; Schneider, J.R.; et al. Childhood Brain Tumors: A Review of Strategies to Translate CNS Drug Delivery to Clinical Trials. Cancers 2023, 15, 857. [Google Scholar] [CrossRef] [PubMed]
- Ausejo-Mauleon, I.; Labiano, S.; de la Nava, D.; Laspidea, V.; Zalacain, M.; Marrodan, L.; Garcia-Moure, M.; Gonzalez-Huarriz, M.; Hervas-Corpion, I.; Dhandapani, L.; et al. TIM-3 blockade in diffuse intrinsic pontine glioma models promotes tumor regression and antitumor immune memory. Cancer Cell 2023, 41, 1911–1926e1918. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Miklja, Z.; Yadav, V.N.; Cartaxo, R.T.; Siada, R.; Thomas, C.C.; Cummings, J.R.; Mullan, B.; Stallard, S.; Paul, A.; Bruzek, A.K.; et al. Everolimus improves the efficacy of dasatinib in PDGFRalpha-driven glioma. J. Clin. Investig. 2020, 130, 5313–5325. [Google Scholar] [CrossRef]
- Hwang, E.I.; Sayour, E.J.; Flores, C.T.; Grant, G.; Wechsler-Reya, R.; Hoang-Minh, L.B.; Kieran, M.W.; Salcido, J.; Prins, R.M.; Figg, J.W.; et al. The current landscape of immunotherapy for pediatric brain tumors. Nat. Cancer 2022, 3, 11–24. [Google Scholar] [CrossRef]
- Shalita, C.; Hanzlik, E.; Kaplan, S.; Thompson, E.M. Immunotherapy for the treatment of pediatric brain tumors: A narrative review. Transl. Pediatr. 2022, 11, 2040–2056. [Google Scholar] [CrossRef]
- Lin, G.L.; Wilson, K.M.; Ceribelli, M.; Stanton, B.Z.; Woo, P.J.; Kreimer, S.; Qin, E.Y.; Zhang, X.; Lennon, J.; Nagaraja, S.; et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019, 11, eaaw0064. [Google Scholar] [CrossRef]
- Manoharan, N.; Liu, K.X.; Mueller, S.; Haas-Kogan, D.A.; Bandopadhayay, P. Pediatric low-grade glioma: Targeted therapeutics and clinical trials in the molecular era. Neoplasia 2023, 36, 100857. [Google Scholar] [CrossRef]
- Kogiso, M.; Qi, L.; Lindsay, H.; Huang, Y.; Zhao, X.; Liu, Z.; Braun, F.K.; Du, Y.; Zhang, H.; Bae, G.; et al. Xenotransplantation of pediatric low grade gliomas confirms the enrichment of BRAF V600E mutation and preservation of CDKN2A deletion in a novel orthotopic xenograft mouse model of progressive pleomorphic xanthoastrocytoma. Oncotarget 2017, 8, 87455–87471. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Walker, T.R.; Helmy, K.; Nazarian, J.; Albrecht, S.; Hiner, R.L.; Gall, S.; Huse, J.T.; Jabado, N.; et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010, 70, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 117. [Google Scholar] [CrossRef]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef]
- Patel, S.K.; Hartley, R.M.; Wei, X.; Furnish, R.; Escobar-Riquelme, F.; Bear, H.; Choi, K.; Fuller, C.; Phoenix, T.N. Generation of diffuse intrinsic pontine glioma mouse models by brainstem-targeted in utero electroporation. Neuro Oncol. 2020, 22, 381–392. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700e689. [Google Scholar] [CrossRef]
- He, C.; Xu, K.; Zhu, X.; Dunphy, P.S.; Gudenas, B.; Lin, W.; Twarog, N.; Hover, L.D.; Kwon, C.H.; Kasper, L.H.; et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat. Commun. 2021, 12, 4089. [Google Scholar] [CrossRef]
- Li, Z.; Langhans, S.A. In Vivo and Ex Vivo Pediatric Brain Tumor Models: An Overview. Front. Oncol. 2021, 11, 620831. [Google Scholar] [CrossRef]
- Raju, R.R.; AlSawaftah, N.M.; Husseini, G.A. Modeling of brain tumors using in vitro, in vivo, and microfluidic models: A review of the current developments. Heliyon 2024, 10, e31402. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Godbole, S.; Albert, T.K.; Dottermusch, M.; Walter, C.; Ballast, A.; Qin, N.; Olivera, M.B.; Gobel, C.; Neyazi, S.; et al. Mouse models of pediatric high-grade gliomas with MYCN amplification reveal intratumoral heterogeneity and lineage signatures. Nat. Commun. 2023, 14, 7717. [Google Scholar] [CrossRef]
- Simeonova, I.; Huillard, E. In vivo models of brain tumors: Roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell. Mol. Life Sci. 2014, 71, 4007–4026. [Google Scholar] [CrossRef]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Lago, C.; Federico, A.; Leva, G.; Mack, N.L.; Schwalm, B.; Ballabio, C.; Gianesello, M.; Abballe, L.; Giovannoni, I.; Reddel, S.; et al. Patient- and xenograft-derived organoids recapitulate pediatric brain tumor features and patient treatments. EMBO Mol. Med. 2023, 15, e18199. [Google Scholar] [CrossRef]
- Lampis, S.; Galardi, A.; Di Paolo, V.; Di Giannatale, A. Organoids as a new approach for improving pediatric cancer research. Front. Oncol. 2024, 14, 1414311. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Riedel, N.C.; de Faria, F.W.; Alfert, A.; Bruder, J.M.; Kerl, K. Three-Dimensional Cell Culture Systems in Pediatric and Adult Brain Tumor Precision Medicine. Cancers 2022, 14, 5972. [Google Scholar] [CrossRef]
- Cadena, M.; Ning, L.; King, A.; Hwang, B.; Jin, L.; Serpooshan, V.; Sloan, S.A. 3D Bioprinting of Neural Tissues. Adv. Healthc. Mater. 2021, 10, e2001600. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.; Neyazi, S.; Schwark, K.; Trissal, M.; Beck, A.; Labelle, J.; Eder, S.K.; Weiler-Wichtl, L.; Marques, J.G.; de Biagi-Junior, C.A.O.; et al. Effective targeting of PDGFRA-altered high-grade glioma with avapritinib. Cancer Cell 2025, 43, 740–756e748. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Ronsley, R.; Choe, M.; Seidel, K.; Huang, W.; Rawlings-Rhea, S.D.; Beam, M.; Steinmetzer, L.; Wilson, A.L.; Brown, C.; et al. Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: A phase 1 trial. Nat. Med. 2025, 31, 861–868. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Zanzonico, P.B.; Grkovski, M.; Donzelli, M.; Vietri, S.M.; Horan, C.; Serencsits, B.; Prasad, K.; Lyashchenko, S.; Kramer, K.; et al. Theranostic Intratumoral Convection-Enhanced Delivery of (124)I-Omburtamab in Patients with Diffuse Intrinsic Pontine Glioma: Pharmacokinetics and Lesion Dosimetry. J. Nucl. Med. 2024, 65, 1364–1370. [Google Scholar] [CrossRef]
- Ehteda, A.; Khan, A.; Rajakumar, G.; Vanniasinghe, A.S.; Gopalakrishnan, A.; Liu, J.; Tsoli, M.; Ziegler, D.S. Microtubule-Targeting Combined with HDAC Inhibition Is a Novel Therapeutic Strategy for Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2023, 22, 1413–1421. [Google Scholar] [CrossRef]
- Meco, D.; Attina, G.; Mastrangelo, S.; Navarra, P.; Ruggiero, A. Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. Int. J. Mol. Sci. 2023, 24, 1334. [Google Scholar] [CrossRef]
- Rana, J.N.; Mumtaz, S. Prunin: An Emerging Anticancer Flavonoid. Int. J. Mol. Sci. 2025, 26, 2678. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef]
- Furst, L.M.; Roussel, E.M.; Leung, R.F.; George, A.M.; Best, S.A.; Whittle, J.R.; Firestein, R.; Faux, M.C.; Eisenstat, D.D. The Landscape of Pediatric High-Grade Gliomas: The Virtues and Pitfalls of Pre-Clinical Models. Biology 2024, 13, 424. [Google Scholar] [CrossRef]
- Foss, A.; Pathania, M. Pediatric Glioma Models Provide Insights into Tumor Development and Future Therapeutic Strategies. Dev. Neurosci. 2024, 46, 22–43. [Google Scholar] [CrossRef]
- McNicholas, M.; De Cola, A.; Bashardanesh, Z.; Foss, A.; Lloyd, C.B.; Hebert, S.; Faury, D.; Andrade, A.F.; Jabado, N.; Kleinman, C.L.; et al. A Compendium of Syngeneic, Transplantable Pediatric High-Grade Glioma Models Reveals Subtype-Specific Therapeutic Vulnerabilities. Cancer Discov. 2023, 13, 1592–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Peng, P.; Zhang, X.; Mania-Farnell, B.; Xi, G.; Wan, F. Advanced Pediatric Diffuse Pontine Glioma Murine Models Pave the Way towards Precision Medicine. Cancers 2021, 13, 1114. [Google Scholar] [CrossRef]
- Chen, F.; Becker, A.J.; LoTurco, J.J. Contribution of tumor heterogeneity in a new animal model of CNS tumors. Mol. Cancer Res. 2014, 12, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Bandopadhayay, P.; Jabado, N. The Power of Human Cancer Genetics as Revealed by Low-Grade Gliomas. Annu. Rev. Genet. 2019, 53, 483–503. [Google Scholar] [CrossRef]
- Thomas, D.L. 2021 updates to the World Health Organization classification of adult-type and pediatric-type diffuse gliomas: A clinical practice review. Chin. Clin. Oncol. 2023, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Ocasio, J.K.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019, 10, 5829. [Google Scholar] [CrossRef]
- Jones, C.; Perryman, L.; Hargrave, D. Paediatric and adult malignant glioma: Close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012, 9, 400–413. [Google Scholar] [CrossRef]
- Scherer, H.J. Structural Development in Gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Cohen, K.J.; Pollack, I.F.; Zhou, T.; Buxton, A.; Holmes, E.J.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Hamilton, R.L.; Lavey, R.S.; et al. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro Oncol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Gojo, J.; Englinger, B.; Jiang, L.; Hubner, J.M.; Shaw, M.L.; Hack, O.A.; Madlener, S.; Kirchhofer, D.; Liu, I.; Pyrdol, J.; et al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell 2020, 38, 44–59e49. [Google Scholar] [CrossRef]
- Jessa, S.; Mohammadnia, A.; Harutyunyan, A.S.; Hulswit, M.; Varadharajan, S.; Lakkis, H.; Kabir, N.; Bashardanesh, Z.; Hebert, S.; Faury, D.; et al. K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas. Nat. Genet. 2022, 54, 1865–1880. [Google Scholar] [CrossRef]
- Jones, C.; Baker, S.J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat. Rev. Cancer 2014, 14, 651–661. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Zhang, M.; Piao, Y.; Chen, L.; Liang, H.; Wei, Y.; Hu, Z.; Zhao, L.; Teng, L.; et al. H3 K27M-mutant diffuse midline gliomas in different anatomical locations. Hum. Pathol. 2018, 78, 89–96. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Sweha, S.R.; Chung, C.; Natarajan, S.K.; Panwalkar, P.; Pun, M.; Ghali, A.; Bayliss, J.; Pratt, D.; Shankar, A.; Ravikumar, V.; et al. Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Sci. Transl. Med. 2021, 13, eabf7860. [Google Scholar] [CrossRef]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349e339. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537e525. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Bush, K.; Klein, R.H.; Cervantes, V.; Lewis, N.; Naqvi, A.; Carcaboso, A.M.; Lechpammer, M.; Knoepfler, P.S. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun. Biol. 2020, 3, 363. [Google Scholar] [CrossRef] [PubMed]
- Ishi, Y.; Takamiya, S.; Seki, T.; Yamazaki, K.; Hida, K.; Hatanaka, K.C.; Ishida, Y.; Oda, Y.; Tanaka, S.; Yamaguchi, S. Prognostic role of H3K27M mutation, histone H3K27 methylation status, and EZH2 expression in diffuse spinal cord gliomas. Brain Tumor Pathol. 2020, 37, 81–88. [Google Scholar] [CrossRef]
- Alturkustani, M. Diagnostic Insights into Pediatric Pleomorphic Xanthoastrocytoma through DNA Methylation Class and Pathological Diagnosis Analysis. Diagnostics 2023, 13, 3464. [Google Scholar] [CrossRef]
- Mathkour, M.; Banerjee, S.; Werner, C.; Hanna, J.; Abou-Al-Shaar, H.; Dindial, R.; Scullen, T.; Boehm, L.; Tubbs, R.S.; Ware, M.L. Cerebellar pleomorphic xanthoastrocytoma in the setting of neurofibromatosis type-I: Does it portend a different prognosis? A case report and systematic review. Clin. Neurol. Neurosurg. 2021, 200, 106346. [Google Scholar] [CrossRef]
- Gene-Olaciregui, N.; Perez-Somarriba, M.; Santa-Maria, V.; Cruz, O.; Gomez-Gonzalez, S.; Castaneda, A.; Sunol, M.; Rovira, C.; Muchart, J.; Hinojosa, J.; et al. Clinical and Molecular Evolution of an ALK-Driven Infant-Type Hemispheric Glioma Treated Sequentially With Second- and Third-Generation Anaplastic Lymphoma Kinase Inhibitors. JCO Precis. Oncol. 2023, 7, e2200547. [Google Scholar] [CrossRef]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323e312. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Bandopadhayay, P.; Cooney, T.M. Diffuse intrinsic pontine glioma: Insights into oncogenesis and opportunities for targeted therapy. Pediatr. Hematol. Oncol. J. 2023, 8, 73–79. [Google Scholar] [CrossRef]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Finkelstein, S.D.; Woods, J.; Burnham, J.; Holmes, E.J.; Hamilton, R.L.; Yates, A.J.; Boyett, J.M.; Finlay, J.L.; Sposto, R.; et al. Expression of p53 and prognosis in children with malignant gliomas. N. Engl. J. Med. 2002, 346, 420–427. [Google Scholar] [CrossRef]
- Warren, K.E. Beyond the Blood:Brain Barrier: The Importance of Central Nervous System (CNS) Pharmacokinetics for the Treatment of CNS Tumors, Including Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Barravecchia, I.; Teis, R.; Cruz, J.; Mumby, R.; Ziemke, E.K.; Espinoza, C.E.; Krishnamoorthy, V.; Magnuson, B.; Ljungman, M.; et al. Targeting DNA Repair and Survival Signaling in Diffuse Intrinsic Pontine Gliomas to Prevent Tumor Recurrence. Mol. Cancer Ther. 2024, 23, 24–34. [Google Scholar] [CrossRef]
- Boschert, T.; Kromer, K.; Lerner, T.; Lindner, K.; Haltenhof, G.; Tan, C.L.; Jahne, K.; Poschke, I.; Bunse, L.; Eisele, P.; et al. H3K27M neoepitope vaccination in diffuse midline glioma induces B and T cell responses across diverse HLA loci of a recovered patient. Sci. Adv. 2024, 10, eadi9091. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Deland, L.; Keane, S.; Bontell, T.O.; Fagman, H.; Sjogren, H.; Lind, A.E.; Caren, H.; Tisell, M.; Nilsson, J.A.; Ejeskar, K.; et al. Novel TPR::ROS1 Fusion Gene Activates MAPK, PI3K and JAK/STAT Signaling in an Infant-type Pediatric Glioma. Cancer Genom. Proteom. 2022, 19, 711–726. [Google Scholar] [CrossRef]
- Baker, S.J.; Ellison, D.W.; Gutmann, D.H. Pediatric gliomas as neurodevelopmental disorders. Glia 2016, 64, 879–895. [Google Scholar] [CrossRef]
- Cahill, D.P.; Louis, D.N.; Cairncross, J.G. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol. 2015, 4, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Bruzek, A.K.; Zureick, A.H.; McKeever, P.E.; Garton, H.J.L.; Robertson, P.L.; Mody, R.; Koschmann, C.J. Molecular characterization reveals NF1 deletions and FGFR1-activating mutations in a pediatric spinal oligodendroglioma. Pediatr. Blood Cancer 2017, 64, e26346. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Tihan, T.; Lin, D.; McDonald, W.; Nigro, J.; Feuerstein, B.; Jackson, S.; Cohen, K.; Burger, P.C. Clinicopathologic features of pediatric oligodendrogliomas: A series of 50 patients. Am. J. Surg. Pathol. 2014, 38, 1058–1070. [Google Scholar] [CrossRef]
- Goel, N.J.; Abdullah, K.G.; Lang, S.S. Outcomes and Prognostic Factors in Pediatric Oligodendroglioma: A Population-Based Study. Pediatr. Neurosurg. 2018, 53, 24–35. [Google Scholar] [CrossRef]
- Fukuoka, K.; Kanemura, Y.; Shofuda, T.; Fukushima, S.; Yamashita, S.; Narushima, D.; Kato, M.; Honda-Kitahara, M.; Ichikawa, H.; Kohno, T.; et al. Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol. Commun. 2018, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, A.M.; Calzadilla, A.J.; Grimaldo, E.; Donson, A.M.; Amani, V.; Pierce, A.M.; Steiner, J.; Kargar, S.; Serkova, N.J.; Bertrand, K.C.; et al. Development of Chromosome 1q+ Specific Treatment for Highest Risk Pediatric Posterior Fossa Ependymoma. Clin. Cancer Res. 2024, 30, 1544–1554. [Google Scholar] [CrossRef]
- Bayliss, J.; Mukherjee, P.; Lu, C.; Jain, S.U.; Chung, C.; Martinez, D.; Sabari, B.; Margol, A.S.; Panwalkar, P.; Parolia, A.; et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med. 2016, 8, 366ra161. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Nobre, L.; Zapotocky, M.; Ramaswamy, V.; Ryall, S.; Bennett, J.; Alderete, D.; Balaguer Guill, J.; Baroni, L.; Bartels, U.; Bavle, A.; et al. Outcomes of BRAF V600E Pediatric Gliomas Treated With Targeted BRAF Inhibition. JCO Precis. Oncol. 2020, 4, 561–571. [Google Scholar] [CrossRef]
- Schreck, K.C.; Langat, P.; Bhave, V.M.; Li, T.; Woodward, E.; Pratilas, C.A.; Eberhart, C.G.; Bi, W.L. Integrated molecular and clinical analysis of BRAF-mutant glioma in adults. NPJ Precis. Oncol. 2023, 7, 23. [Google Scholar] [CrossRef]
- Bouffet, E.; Hansford, J.R.; Garre, M.L.; Hara, J.; Plant-Fox, A.; Aerts, I.; Locatelli, F.; van der Lugt, J.; Papusha, L.; Sahm, F.; et al. Dabrafenib plus Trametinib in Pediatric Glioma with BRAF V600 Mutations. N. Engl. J. Med. 2023, 389, 1108–1120. [Google Scholar] [CrossRef]
- Malbari, F. Pediatric Neuro-Oncology. Neurol. Clin. 2021, 39, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Brabetz, S.; Leary, S.E.S.; Grobner, S.N.; Nakamoto, M.W.; Seker-Cin, H.; Girard, E.J.; Cole, B.; Strand, A.D.; Bloom, K.L.; Hovestadt, V.; et al. A biobank of patient-derived pediatric brain tumor models. Nat. Med. 2018, 24, 1752–1761. [Google Scholar] [CrossRef]
- Cancer, M.; Hutter, S.; Holmberg, K.O.; Rosen, G.; Sundstrom, A.; Tailor, J.; Bergstrom, T.; Garancher, A.; Essand, M.; Wechsler-Reya, R.J.; et al. Humanized Stem Cell Models of Pediatric Medulloblastoma Reveal an Oct4/mTOR Axis that Promotes Malignancy. Cell Stem Cell 2019, 25, 855–870e811. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Rosiene, J.; Che, A.; Becker, A.; LoTurco, J. Tracking and transforming neocortical progenitors by CRISPR/Cas9 gene targeting and piggyBac transposase lineage labeling. Development 2015, 142, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Cogels, M.M.; Rouas, R.; Ghanem, G.E.; Martinive, P.; Awada, A.; Van Gestel, D.; Krayem, M. Humanized Mice as a Valuable Pre-Clinical Model for Cancer Immunotherapy Research. Front. Oncol. 2021, 11, 784947. [Google Scholar] [CrossRef]
- Dobson, T.H.W.; Gopalakrishnan, V. Preclinical Models of Pediatric Brain Tumors-Forging Ahead. Bioengineering 2018, 5, 81. [Google Scholar] [CrossRef]
- du Chatinier, A.; Meel, M.H.; Das, A.I.; Metselaar, D.S.; Waranecki, P.; Bugiani, M.; Breur, M.; Simonds, E.F.; Lu, E.D.; Weiss, W.A.; et al. Generation of immunocompetent syngeneic allograft mouse models for pediatric diffuse midline glioma. Neurooncol. Adv. 2022, 4, vdac079. [Google Scholar] [CrossRef]
- Funato, K.; Smith, R.C.; Saito, Y.; Tabar, V. Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell 2021, 28, 894–905e897. [Google Scholar] [CrossRef]
- Graber, P.; Dolman, M.E.M.; Jung, M.; Kavallaris, M. Ex Vivo Modeling of the Tumor Microenvironment to Develop Therapeutic Strategies for Gliomas. Adv. Ther. 2024, 7, 2300442. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Amankulor, N.M.; Helmy, K.Y.; Becher, O.J.; Holland, E.C. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl. Oncol. 2009, 2, 89–95. [Google Scholar] [CrossRef]
- Hermans, E.; Hulleman, E. Patient-Derived Orthotopic Xenograft Models of Pediatric Brain Tumors: In a Mature Phase or Still in Its Infancy? Front. Oncol. 2019, 9, 1418. [Google Scholar] [CrossRef]
- Hicks, W.H.; Bird, C.E.; Pernik, M.N.; Haider, A.S.; Dobariya, A.; Abdullah, K.G.; Aoun, S.G.; Bentley, R.T.; Cohen-Gadol, A.A.; Bachoo, R.M.; et al. Large Animal Models of Glioma: Current Status and Future Prospects. Anticancer Res. 2021, 41, 5343–5353. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.O.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro Oncol. 2012, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Higginbottom, S.L.; Tomaskovic-Crook, E.; Crook, J.M. Considerations for modelling diffuse high-grade gliomas and developing clinically relevant therapies. Cancer Metastasis Rev. 2023, 42, 507–541. [Google Scholar] [CrossRef]
- Basheer, F.; Dhar, P.; Samarasinghe, R.M. Zebrafish Models of Paediatric Brain Tumours. Int. J. Mol. Sci. 2022, 23, 9920. [Google Scholar] [CrossRef]
- Casey, M.J.; Chan, P.P.; Li, Q.; Zu, J.F.; Jette, C.A.; Kohler, M.; Myers, B.R.; Stewart, R.A. A simple and scalable zebrafish model of Sonic hedgehog medulloblastoma. Cell Rep. 2024, 43, 114559. [Google Scholar] [CrossRef]
- Modzelewska, K.; Boer, E.F.; Mosbruger, T.L.; Picard, D.; Anderson, D.; Miles, R.R.; Kroll, M.; Oslund, W.; Pysher, T.J.; Schiffman, J.D.; et al. MEK Inhibitors Reverse Growth of Embryonal Brain Tumors Derived from Oligoneural Precursor Cells. Cell Rep. 2016, 17, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- van Bree, N.; Oppelt, A.S.; Lindstrom, S.; Zhou, L.; Boutin, L.; Coyle, B.; Swartling, F.J.; Johnsen, J.I.; Brautigam, L.; Wilhelm, M. Development of an orthotopic medulloblastoma zebrafish model for rapid drug testing. Neuro Oncol. 2025, 27, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Casey, M.J.; Stewart, R.A. Pediatric Cancer Models in Zebrafish. Trends Cancer 2020, 6, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Swenberg, J.A. Chemical- and virus-induced brain tumors. Natl. Cancer Inst. Monogr. 1977, 46, 3–10. [Google Scholar]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef]
- Adey, W.R.; Byus, C.V.; Cain, C.D.; Higgins, R.J.; Jones, R.A.; Kean, C.J.; Kuster, N.; MacMurray, A.; Stagg, R.B.; Zimmerman, G.; et al. Spontaneous and nitrosourea-induced primary tumors of the central nervous system in Fischer 344 rats chronically exposed to 836 MHz modulated microwaves. Radiat. Res. 1999, 152, 293–302. [Google Scholar] [CrossRef]
- Zook, B.C.; Simmens, S.J. Neurogenic tumors in rats induced by ethylnitrosourea. Exp. Toxicol. Pathol. 2005, 57, 7–14. [Google Scholar] [CrossRef]
- Bulnes, S.; Murueta-Goyena, A.; Lafuente, J.V. Differential exposure to N-ethyl N-nitrosourea during pregnancy is relevant to the induction of glioma and PNSTs in the brain. Neurotoxicol. Teratol. 2021, 86, 106998. [Google Scholar] [CrossRef]
- Barth, R.F.; Kaur, B. Rat brain tumor models in experimental neuro-oncology: The C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol. 2009, 94, 299–312. [Google Scholar] [CrossRef]
- Cuatico, W.; Cho, J.R.; Spiegelman, S. Molecular evidence for a viral etiology of human CNS tumors. Acta Neurochir. 1976, 35, 149–160. [Google Scholar] [CrossRef]
- Rabotti, G.F.; Raine, W.A. Brain Tumours Induced in Hamsters Inoculated Intracerebrally at Birth with Rous Sarcoma Virus. Nature 1964, 204, 898–899. [Google Scholar] [CrossRef]
- Ogawa, K.; Hamaya, K.; Fujii, Y.; Matsuura, K.; Endo, T. Tumor induction by adenovirus type 12 and its target cells in the central nervous system. Gan 1969, 60, 383–392. [Google Scholar] [PubMed]
- Ogawa, K. Embryonal neuroepithelial tumors induced by human adenovirus type 12 in rodents. 2. Tumor induction in the central nervous system. Acta Neuropathol. 1989, 78, 232–244. [Google Scholar] [CrossRef]
- Wippold, F.J., 2nd; Lammle, M.; Anatelli, F.; Lennerz, J.; Perry, A. Neuropathology for the neuroradiologist: Palisades and pseudopalisades. AJNR Am. J. Neuroradiol. 2006, 27, 2037–2041. [Google Scholar] [PubMed]
- Shapiro, J.A.; Gaonkar, K.S.; Spielman, S.J.; Savonen, C.L.; Bethell, C.J.; Jin, R.; Rathi, K.S.; Zhu, Y.; Egolf, L.E.; Farrow, B.K.; et al. OpenPBTA: The Open Pediatric Brain Tumor Atlas. Cell Genom. 2023, 3, 100340. [Google Scholar] [CrossRef]
- Dong, J.; Li, L.; Liang, S.; Zhao, S.; Zhang, B.; Meng, Y.; Zhang, Y.; Li, S. Differentiation Between Ependymoma and Medulloblastoma in Children with Radiomics Approach. Acad. Radiol. 2021, 28, 318–327. [Google Scholar] [CrossRef]
- Doerfler, W. Epigenetic mechanisms in human adenovirus type 12 oncogenesis. Semin. Cancer Biol. 2009, 19, 136–143. [Google Scholar] [CrossRef]
- Shih, R.Y.; Koeller, K.K. Embryonal Tumors of the Central Nervous System: From the Radiologic Pathology Archives. Radiographics 2018, 38, 525–541. [Google Scholar] [CrossRef]
- Roussel, M.F.; Stripay, J.L. Modeling pediatric medulloblastoma. Brain Pathol. 2020, 30, 703–712. [Google Scholar] [CrossRef]
- Smith, K.S.; Xu, K.; Mercer, K.S.; Boop, F.; Klimo, P.; DeCupyere, M.; Grenet, J.; Robinson, S.; Dunphy, P.; Baker, S.J.; et al. Patient-derived orthotopic xenografts of pediatric brain tumors: A St. Jude resource. Acta Neuropathol. 2020, 140, 209–225. [Google Scholar] [CrossRef]
- Yu, L.; Baxter, P.A.; Voicu, H.; Gurusiddappa, S.; Zhao, Y.; Adesina, A.; Man, T.K.; Shu, Q.; Zhang, Y.J.; Zhao, X.M.; et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol. 2010, 12, 580–594. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Golbourn, B.J.; Halbert, M.E.; Halligan, K.; Varadharajan, S.; Krug, B.; Mbah, N.E.; Kabir, N.; Stanton, A.J.; Locke, A.L.; Casillo, S.M.; et al. Loss of MAT2A compromises methionine metabolism and represents a vulnerability in H3K27M mutant glioma by modulating the epigenome. Nat. Cancer 2022, 3, 629–648. [Google Scholar] [CrossRef]
- Rygaard, J.; Povsen, C.O. Heterotransplantation of a human malignant tumour to “nude” mice. 1969. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 604–606, discussion 607–608. [Google Scholar] [CrossRef]
- Xu, J.; Margol, A.; Asgharzadeh, S.; Erdreich-Epstein, A. Pediatric brain tumor cell lines. J. Cell. Biochem. 2015, 116, 218–224. [Google Scholar] [CrossRef]
- Wakefield, L.; Agarwal, S.; Tanner, K. Preclinical models for drug discovery for metastatic disease. Cell 2023, 186, 1792–1813. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, Y.J.; Lin, Q.; Yu, L.; Liu, Z.; Lindsay, H.; Kogiso, M.; Rao, P.; Li, X.N.; Lu, X. Cytogenetic landscape of paired neurospheres and traditional monolayer cultures in pediatric malignant brain tumors. Neuro Oncol. 2015, 17, 965–977. [Google Scholar] [CrossRef]
- Jin, K.T.; Du, W.L.; Lan, H.R.; Liu, Y.Y.; Mao, C.S.; Du, J.L.; Mou, X.Z. Development of humanized mouse with patient-derived xenografts for cancer immunotherapy studies: A comprehensive review. Cancer Sci. 2021, 112, 2592–2606. [Google Scholar] [CrossRef]
- Qi, L.; Baxter, P.; Kogiso, M.; Zhang, H.; Braun, F.K.; Lindsay, H.; Zhao, S.; Xiao, S.; Abdallah, A.S.; Suarez, M.; et al. Direct Implantation of Patient Brain Tumor Cells into Matching Locations in Mouse Brains for Patient-Derived Orthotopic Xenograft Model Development. Cancers 2024, 16, 1716. [Google Scholar] [CrossRef]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef]
- Shu, Q.; Wong, K.K.; Su, J.M.; Adesina, A.M.; Yu, L.T.; Tsang, Y.T.; Antalffy, B.C.; Baxter, P.; Perlaky, L.; Yang, J.; et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 2008, 26, 1414–1424. [Google Scholar] [CrossRef]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef]
- Blanchard, Z.; Brown, E.A.; Ghazaryan, A.; Welm, A.L. PDX models for functional precision oncology and discovery science. Nat. Rev. Cancer 2025, 25, 153–166. [Google Scholar] [CrossRef]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef]
- Pathania, M.; Yan, L.D.; Bordey, A. A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology 2010, 58, 865–876. [Google Scholar] [CrossRef]
- Breunig, J.J.; Levy, R.; Antonuk, C.D.; Molina, J.; Dutra-Clarke, M.; Park, H.; Akhtar, A.A.; Kim, G.B.; Hu, X.; Bannykh, S.I.; et al. Ets Factors Regulate Neural Stem Cell Depletion and Gliogenesis in Ras Pathway Glioma. Cell Rep. 2015, 12, 258–271. [Google Scholar] [CrossRef]
- Zhu, Y.; Guignard, F.; Zhao, D.; Liu, L.; Burns, D.K.; Mason, R.P.; Messing, A.; Parada, L.F. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005, 8, 119–130. [Google Scholar] [CrossRef]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef]
- Kwon, C.H.; Zhao, D.; Chen, J.; Alcantara, S.; Li, Y.; Burns, D.K.; Mason, R.P.; Lee, E.Y.; Wu, H.; Parada, L.F. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008, 68, 3286–3294. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Roncari, L.; Shannon, P.; Wu, X.; Lau, N.; Karaskova, J.; Gutmann, D.H.; Squire, J.A.; Nagy, A.; Guha, A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001, 61, 3826–3836. [Google Scholar] [PubMed]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155e147. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Han, J.; Fang, D.; Gan, H.; Zhang, Z. A lesson learned from the H3.3K27M mutation found in pediatric glioma: A new approach to the study of the function of histone modifications in vivo? Cell Cycle 2013, 12, 2546–2552. [Google Scholar] [CrossRef]
- Lu, V.M.; Alvi, M.A.; McDonald, K.L.; Daniels, D.J. Impact of the H3K27M mutation on survival in pediatric high-grade glioma: A systematic review and meta-analysis. J. Neurosurg. Pediatr. 2019, 23, 308–316. [Google Scholar] [CrossRef]
- Weidenhammer, L.B.; Liu, H.Q.; Luo, L.; Williams, N.T.; Deland, K.; Kirsch, D.G.; Reitman, Z.J. Inducing primary brainstem gliomas in genetically engineered mice using RCAS/TVA retroviruses and Cre/loxP recombination. STAR Protoc. 2023, 4, 102094. [Google Scholar] [CrossRef]
- Misuraca, K.L.; Barton, K.L.; Chung, A.; Diaz, A.K.; Conway, S.J.; Corcoran, D.L.; Baker, S.J.; Becher, O.J. Pax3 expression enhances PDGF-B-induced brainstem gliomagenesis and characterizes a subset of brainstem glioma. Acta Neuropathol. Commun. 2014, 2, 134. [Google Scholar] [CrossRef]
- Saito, T. In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 2006, 1, 1552–1558. [Google Scholar] [CrossRef]
- Messinger, D.; Harris, M.K.; Cummings, J.R.; Thomas, C.; Yang, T.; Sweha, S.R.; Woo, R.; Siddaway, R.; Burkert, M.; Stallard, S.; et al. Therapeutic targeting of prenatal pontine ID1 signaling in diffuse midline glioma. Neuro Oncol. 2023, 25, 54–67. [Google Scholar] [CrossRef]
- Saito, T.; Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001, 240, 237–246. [Google Scholar] [CrossRef]
- Sato, Y.; Kasai, T.; Nakagawa, S.; Tanabe, K.; Watanabe, T.; Kawakami, K.; Takahashi, Y. Stable integration and conditional expression of electroporated transgenes in chicken embryos. Dev. Biol. 2007, 305, 616–624. [Google Scholar] [CrossRef]
- Schwark, K.; Messinger, D.; Cummings, J.R.; Bradin, J.; Kawakibi, A.; Babila, C.M.; Lyons, S.; Ji, S.; Cartaxo, R.T.; Kong, S.; et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Pre-clinical models and precision medicine. Front. Oncol. 2022, 12, 922928. [Google Scholar] [CrossRef] [PubMed]
- Calinescu, A.A.; Nunez, F.J.; Koschmann, C.; Kolb, B.L.; Lowenstein, P.R.; Castro, M.G. Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J. Vis. Exp. 2015, e52443. [Google Scholar] [CrossRef]
- Wiesner, S.M.; Decker, S.A.; Larson, J.D.; Ericson, K.; Forster, C.; Gallardo, J.L.; Long, C.; Demorest, Z.L.; Zamora, E.A.; Low, W.C.; et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009, 69, 431–439. [Google Scholar] [CrossRef]
- Faisal, S.M.; Clewner, J.E.; Stack, B.; Varela, M.L.; Comba, A.; Abbud, G.; Motsch, S.; Castro, M.G.; Lowenstein, P.R. Spatiotemporal Insights into Glioma Oncostream Dynamics: Unraveling Formation, Stability, and Disassembly Pathways. Adv. Sci. 2024, 11, e2309796. [Google Scholar] [CrossRef]
- Koschmann, C.; Calinescu, A.A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Banerjee, K.; Mujeeb, A.A.; Hartlage, C.S.; Nunez, F.M.; Nunez, F.J.; Alghamri, M.S.; Kadiyala, P.; Carney, S.; Barissi, M.N.; et al. H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J. Clin. Investig. 2022, 132, e154229. [Google Scholar] [CrossRef]
- Garcia-Fabiani, M.B.; Kadiyala, P.; Lowenstein, P.R.; Castro, M.G. An Optimized Protocol for In Vivo Analysis of Tumor Cell Division in a Sleeping Beauty-Mediated Mouse Glioma Model. STAR Protoc. 2020, 1, 100044. [Google Scholar] [CrossRef]
- Nunez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef]
- Mendez, F.; Kadiyala, P.; Nunez, F.J.; Carney, S.; Nunez, F.M.; Gauss, J.C.; Ravindran, R.; Pawar, S.; Edwards, M.; Garcia-Fabiani, M.B.; et al. Therapeutic Efficacy of Immune Stimulatory Thymidine Kinase and fms-like Tyrosine Kinase 3 Ligand (TK/Flt3L) Gene Therapy in a Mouse Model of High-Grade Brainstem Glioma. Clin. Cancer Res. 2020, 26, 4080–4092. [Google Scholar] [CrossRef]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Nunez, F.J.; Liu, Y.; Yu, M.; Taher, A.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef] [PubMed]
- Alghamri, M.S.; Banerjee, K.; Mujeeb, A.A.; Mauser, A.; Taher, A.; Thalla, R.; McClellan, B.L.; Varela, M.L.; Stamatovic, S.M.; Martinez-Revollar, G.; et al. Systemic Delivery of an Adjuvant CXCR4-CXCL12 Signaling Inhibitor Encapsulated in Synthetic Protein Nanoparticles for Glioma Immunotherapy. ACS Nano 2022, 16, 8729–8750. [Google Scholar] [CrossRef] [PubMed]
- Comba, A.; Faisal, S.M.; Dunn, P.J.; Argento, A.E.; Hollon, T.C.; Al-Holou, W.N.; Varela, M.L.; Zamler, D.B.; Quass, G.L.; Apostolides, P.F.; et al. Spatiotemporal analysis of glioma heterogeneity reveals COL1A1 as an actionable target to disrupt tumor progression. Nat. Commun. 2022, 13, 3606. [Google Scholar] [CrossRef]
- Andrade, A.F.; Annett, A.; Karimi, E.; Topouza, D.G.; Rezanejad, M.; Liu, Y.; McNicholas, M.; Gonzalez Santiago, E.G.; Llivichuzhca-Loja, D.; Gehlhaar, A.; et al. Immune landscape of oncohistone-mutant gliomas reveals diverse myeloid populations and tumor-promoting function. Nat. Commun. 2024, 15, 7769. [Google Scholar] [CrossRef]
- Ross, J.L.; Velazquez Vega, J.; Plant, A.; MacDonald, T.J.; Becher, O.J.; Hambardzumyan, D. Tumour immune landscape of paediatric high-grade gliomas. Brain 2021, 144, 2594–2609. [Google Scholar] [CrossRef]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652e636. [Google Scholar] [CrossRef]
- Lubanszky, E.; Hawkins, C. Modeling the Landscape of Histone-Mutant Pediatric High-Grade Gliomas: A Study in Partner Alterations. Cancer Discov. 2023, 13, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Ng, P.K.S.; Jeong, K.J.; Shao, S.H.; Ju, Z.; Leonard, P.G.; Hua, X.; Vellano, C.P.; Woessner, R.; Sahni, N.; et al. Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA targeted therapies. Nat. Commun. 2018, 9, 4583. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Pasqualini, C.; Kozaki, T.; Bruschi, M.; Nguyen, T.H.H.; Minard-Colin, V.; Castel, D.; Grill, J.; Ginhoux, F. Modeling the Interaction between the Microenvironment and Tumor Cells in Brain Tumors. Neuron 2020, 108, 1025–1044. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Srivastava, R.; Labani-Motlagh, A.; Chen, A.; Bohorquez, J.A.; Qin, B.; Dodda, M.; Yang, F.; Ansari, D.; Patel, S.; Ji, H.; et al. Development of a human glioblastoma model using humanized DRAG mice for immunotherapy. Antibody Ther. 2023, 6, 253–264. [Google Scholar] [CrossRef]
- Verma, B.; Wesa, A. Establishment of Humanized Mice from Peripheral Blood Mononuclear Cells or Cord Blood CD34+ Hematopoietic Stem Cells for Immune-Oncology Studies Evaluating New Therapeutic Agents. Curr. Protoc. Pharmacol. 2020, 89, e77. [Google Scholar] [CrossRef]
- Valujskikh, A.; Baldwin, W.M., 3rd; Fairchild, R.L. Recent progress and new perspectives in studying T cell responses to allografts. Am. J. Transplant. 2010, 10, 1117–1125. [Google Scholar] [CrossRef]
- Lan, X.; Kedziorek, D.A.; Chu, C.; Jablonska, A.; Li, S.; Kai, M.; Liang, Y.; Janowski, M.; Walczak, P. Modeling human pediatric and adult gliomas in immunocompetent mice through costimulatory blockade. Oncoimmunology 2020, 9, 1776577. [Google Scholar] [CrossRef]
- Neff, E.P. Cancer modeling thinks big with the pig. Lab Anim. 2019, 48, 75–78. [Google Scholar] [CrossRef]
- Schook, L.B.; Collares, T.V.; Darfour-Oduro, K.A.; De, A.K.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K. Unraveling the swine genome: Implications for human health. Annu. Rev. Anim. Biosci. 2015, 3, 219–244. [Google Scholar] [CrossRef]
- Candolfi, M.; Kroeger, K.M.; Pluhar, G.E.; Bergeron, J.; Puntel, M.; Curtin, J.F.; McNiel, E.A.; Freese, A.B.; Ohlfest, J.R.; Moore, P.; et al. Adenoviral-mediated gene transfer into the canine brain in vivo. Neurosurgery 2007, 60, 167–177, discussion 178. [Google Scholar] [CrossRef]
- Whelan, H.T.; Clanton, J.A.; Wilson, R.E.; Tulipan, N.B. Comparison of CT and MRI brain tumor imaging using a canine glioma model. Pediatr. Neurol. 1988, 4, 279–283. [Google Scholar] [CrossRef]
- Rainov, N.G.; Koch, S.; Sena-Esteves, M.; Berens, M.E. Characterization of a canine glioma cell line as related to established experimental brain tumor models. J. Neuropathol. Exp. Neurol. 2000, 59, 607–613. [Google Scholar] [CrossRef]
- Hicks, J.; Platt, S.; Kent, M.; Haley, A. Canine brain tumours: A model for the human disease? Vet. Comp. Oncol. 2017, 15, 252–272. [Google Scholar] [CrossRef]
- Qiao, N.; Ma, L.; Zhang, Y.; Wang, L. Update on Nonhuman Primate Models of Brain Disease and Related Research Tools. Biomedicines 2023, 11, 2516. [Google Scholar] [CrossRef]
- Han, L.; Wei, X.; Liu, C.; Volpe, G.; Zhuang, Z.; Zou, X.; Wang, Z.; Pan, T.; Yuan, Y.; Zhang, X.; et al. Cell transcriptomic atlas of the non-human primate Macaca fascicularis. Nature 2022, 604, 723–731. [Google Scholar] [CrossRef]
- Langin, M.; Mayr, T.; Reichart, B.; Michel, S.; Buchholz, S.; Guethoff, S.; Dashkevich, A.; Baehr, A.; Egerer, S.; Bauer, A.; et al. Consistent success in life-supporting porcine cardiac xenotransplantation. Nature 2018, 564, 430–433. [Google Scholar] [CrossRef]
- Tang-Schomer, M.D.; Chandok, H.; Wu, W.B.; Lau, C.C.; Bookland, M.J.; George, J. 3D patient-derived tumor models to recapitulate pediatric brain tumors In Vitro. Transl. Oncol. 2022, 20, 101407. [Google Scholar] [CrossRef]
- Quinn, C.H.; Beierle, A.M.; Julson, J.R.; Erwin, M.E.; Alrefai, H.; Markert, H.R.; Stewart, J.E.; Hutchins, S.C.; Bownes, L.V.; Aye, J.M.; et al. Using 3D-bioprinted models to study pediatric neural crest-derived tumors. Int. J. Bioprint. 2023, 9, 723. [Google Scholar] [CrossRef]
- Xu, J.; Erdreich-Epstein, A.; Gonzalez-Gomez, I.; Melendez, E.Y.; Smbatyan, G.; Moats, R.A.; Rosol, M.; Biegel, J.A.; Reynolds, C.P. Novel cell lines established from pediatric brain tumors. J. Neurooncol. 2012, 107, 269–280. [Google Scholar] [CrossRef]
- Orcheston-Findlay, L.; Bax, S.; Utama, R.; Engel, M.; Govender, D.; O’Neill, G. Advanced Spheroid, Tumouroid and 3D Bioprinted In-Vitro Models of Adult and Paediatric Glioblastoma. Int. J. Mol. Sci. 2021, 22, 2962. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, D.; Liu, A.; Wu, K. Tumor organoids: Applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 2022, 15, 58. [Google Scholar] [CrossRef]
- Kook, M.G.; Lee, S.E.; Shin, N.; Kong, D.; Kim, D.H.; Kim, M.S.; Kang, H.K.; Choi, S.W.; Kang, K.S. Generation of Cortical Brain Organoid with Vascularization by Assembling with Vascular Spheroid. Int. J. Stem Cells 2022, 15, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Antonica, F.; Aiello, G.; Soldano, A.; Abballe, L.; Miele, E.; Tiberi, L. Modeling Brain Tumors: A Perspective Overview of in vivo and Organoid Models. Front. Mol. Neurosci. 2022, 15, 818696. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Liu, F.; Cheng, Q.; Weygant, N.; Liang, X.; Fan, F.; Li, C.; Zhang, L.; Liu, Z. Applications of organoid technology to brain tumors. CNS Neurosci. Ther. 2023, 29, 2725–2743. [Google Scholar] [CrossRef]
- Haag, D.; Mack, N.; Benites Goncalves da Silva, P.; Statz, B.; Clark, J.; Tanabe, K.; Sharma, T.; Jager, N.; Jones, D.T.W.; Kawauchi, D.; et al. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell 2021, 39, 407–422e413. [Google Scholar] [CrossRef]
- Anastasaki, C.; Chatterjee, J.; Cobb, O.; Sanapala, S.; Scheaffer, S.M.; De Andrade Costa, A.; Wilson, A.F.; Kernan, C.M.; Zafar, A.H.; Ge, X.; et al. Human induced pluripotent stem cell engineering establishes a humanized mouse platform for pediatric low-grade glioma modeling. Acta Neuropathol. Commun. 2022, 10, 120. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Zhang, D.Y.; Ming, G.L.; Song, H. Glioblastoma modeling with 3D organoids: Progress and challenges. Oxf. Open Neurosci. 2023, 2, kvad008. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Wang, E.; Xiang, K.; Zhang, Y.; Wang, X.F. Patient-derived organoids (PDOs) and PDO-derived xenografts (PDOXs): New opportunities in establishing faithful pre-clinical cancer models. J. Natl. Cancer Cent. 2022, 2, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.D.; Sangster, K.; Sajid, R.S.; Djuric, U.; Diamandis, P. Cerebral organoids: Emerging ex vivo humanoid models of glioblastoma. Acta Neuropathol. Commun. 2020, 8, 209. [Google Scholar] [CrossRef]
- Sarnow, K.; Majercak, E.; Qurbonov, Q.; Cruzeiro, G.A.V.; Jeong, D.; Harque, I.A.; Khalil, A.; Baird, L.C.; Filbin, M.G.; Tang, X. Neuroimmune-competent human brain organoid model of Diffuse Midline Glioma. Neuro Oncol. 2025, 27, 369–382. [Google Scholar] [CrossRef]
- Di Stefano, J.; Di Marco, F.; Cicalini, I.; FitzGerald, U.; Pieragostino, D.; Verhoye, M.; Ponsaerts, P.; Van Breedam, E. Generation, interrogation, and future applications of microglia-containing brain organoids. Neural Regen. Res. 2025, 20, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Lago, C.; Gianesello, M.; Santomaso, L.; Leva, G.; Ballabio, C.; Anderle, M.; Antonica, F.; Tiberi, L. Medulloblastoma and high-grade glioma organoids for drug screening, lineage tracing, co-culture and in vivo assay. Nat. Protoc. 2023, 18, 2143–2180. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, C.; Gianesello, M.; Lago, C.; Okonechnikov, K.; Anderle, M.; Aiello, G.; Antonica, F.; Zhang, T.; Gianno, F.; Giangaspero, F.; et al. Notch1 switches progenitor competence in inducing medulloblastoma. Sci. Adv. 2021, 7, eabd2781. [Google Scholar] [CrossRef]
- Pagliaro, A.; Finger, R.; Zoutendijk, I.; Bunschuh, S.; Clevers, H.; Hendriks, D.; Artegiani, B. Temporal morphogen gradient-driven neural induction shapes single expanded neuroepithelium brain organoids with enhanced cortical identity. Nat. Commun. 2023, 14, 7361. [Google Scholar] [CrossRef]
- Jacob, F.; Schnoll, J.G.; Song, H.; Ming, G.L. Building the brain from scratch: Engineering region-specific brain organoids from human stem cells to study neural development and disease. Curr. Top. Dev. Biol. 2021, 142, 477–530. [Google Scholar] [CrossRef]
- Antonica, F.; Santomaso, L.; Pernici, D.; Petrucci, L.; Aiello, G.; Cutarelli, A.; Conti, L.; Romanel, A.; Miele, E.; Tebaldi, T.; et al. A slow-cycling/quiescent cells subpopulation is involved in glioma invasiveness. Nat. Commun. 2022, 13, 4767. [Google Scholar] [CrossRef]
- Yip, S.; Wang, N.; Sugimura, R. Give Them Vasculature and Immune Cells: How to Fill the Gap of Organoids. Cells Tissues Organs 2023, 212, 369–382. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Comba, A.; Varela, M.L.; Faisal, S.M.; Abel, C.C., 2nd; Argento, A.E.; Al-Holou, W.N.; Hollon, T.C.; Perelman, J.D.; Dunn, P.J.; Motsch, S.; et al. Generation of 3D ex vivo mouse- and patient-derived glioma explant slice model for integration of confocal time-lapse imaging and spatial analysis. STAR Protoc. 2023, 4, 102174. [Google Scholar] [CrossRef]
- Ravi, V.M.; Joseph, K.; Wurm, J.; Behringer, S.; Garrelfs, N.; d’Errico, P.; Naseri, Y.; Franco, P.; Meyer-Luehmann, M.; Sankowski, R.; et al. Human organotypic brain slice culture: A novel framework for environmental research in neuro-oncology. Life Sci. Alliance 2019, 2, e201900305. [Google Scholar] [CrossRef] [PubMed]
- Neises, L.; Delbrouck, C.; Schuster, A.; Rezaipour, M.; Eiden, K.; Oudin, A.; Fabian, C.; Niclou, S.P.; Golebiewska, A.; Meiser, J. Protocol using ex vivo mouse brain slice culture mimicking in vivo conditions to study tumor growth and cell motility of glioblastoma cells. STAR Protoc. 2024, 5, 103401. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, X.; Ming, G.L.; Song, H. Brain tumors on slice: A novel platform for personalized therapeutic screening. Cell Rep. Med. 2023, 4, 101059. [Google Scholar] [CrossRef]
- Sawlani, V.; Patel, M.D.; Davies, N.; Flintham, R.; Wesolowski, R.; Ughratdar, I.; Pohl, U.; Nagaraju, S.; Petrik, V.; Kay, A.; et al. Multiparametric MRI: Practical approach and pictorial review of a useful tool in the evaluation of brain tumours and tumour-like lesions. Insights Imaging 2020, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Qian, J.; Yang, L.; Sun, Z.; Hu, C.; Wang, X.; Hu, S.; Xie, Y. Multiparametric MRI radiomics for the differentiation of brain glial cell hyperplasia from low-grade glioma. BMC Med. Imaging 2023, 23, 116. [Google Scholar] [CrossRef]
- Hu, R.; Hoch, M.J. Application of Diffusion Weighted Imaging and Diffusion Tensor Imaging in the Pretreatment and Post-treatment of Brain Tumor. Radiol. Clin. N. Am. 2021, 59, 335–347. [Google Scholar] [CrossRef]
- Henderson, F.; Abdullah, K.G.; Verma, R.; Brem, S. Tractography and the connectome in neurosurgical treatment of gliomas: The premise, the progress, and the potential. Neurosurg. Focus 2020, 48, E6. [Google Scholar] [CrossRef]
- Jabehdar Maralani, P.; Chan, R.W.; Lam, W.W.; Oakden, W.; Oglesby, R.; Lau, A.; Mehrabian, H.; Heyn, C.; Chan, A.K.M.; Soliman, H.; et al. Chemical Exchange Saturation Transfer MRI: What Neuro-Oncology Clinicians Need To Know. Technol. Cancer Res. Treat. 2023, 22, 15330338231208613. [Google Scholar] [CrossRef]
- Grosse, F.; Wedel, F.; Thomale, U.W.; Steffen, I.; Koch, A.; Brenner, W.; Plotkin, M.; Driever, P.H. Benefit of Static FET PET in Pretreated Pediatric Brain Tumor Patients with Equivocal Conventional MRI Results. Klin. Padiatr. 2021, 233, 127–134. [Google Scholar] [CrossRef]
- Ladefoged, C.N.; Marner, L.; Hindsholm, A.; Law, I.; Hojgaard, L.; Andersen, F.L. Deep Learning Based Attenuation Correction of PET/MRI in Pediatric Brain Tumor Patients: Evaluation in a Clinical Setting. Front. Neurosci. 2018, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Najjar, A.M.; Johnson, J.M.; Schellingerhout, D. The Emerging Role of Amino Acid PET in Neuro-Oncology. Bioengineering 2018, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.W.; Park, I.; Kline, C.; Chen, H.Y.; Gordon, J.W.; Raber, S.; Hoffman, C.; Kim, Y.; Okamoto, K.; Vigneron, D.B.; et al. Pilot Study of Hyperpolarized (13)C Metabolic Imaging in Pediatric Patients with Diffuse Intrinsic Pontine Glioma and Other CNS Cancers. AJNR Am. J. Neuroradiol. 2021, 42, 178–184. [Google Scholar] [CrossRef]
- Albalkhi, I.; Bhatia, A.; Losch, N.; Goetti, R.; Mankad, K. Current state of radiomics in pediatric neuro-oncology practice: A systematic review. Pediatr. Radiol. 2023, 53, 2079–2091. [Google Scholar] [CrossRef]
- Madhogarhia, R.; Haldar, D.; Bagheri, S.; Familiar, A.; Anderson, H.; Arif, S.; Vossough, A.; Storm, P.; Resnick, A.; Davatzikos, C.; et al. Radiomics and radiogenomics in pediatric neuro-oncology: A review. Neurooncol. Adv. 2022, 4, vdac083. [Google Scholar] [CrossRef]
- Sheikh, S.R.; Patel, N.J.; Recinos, V.M.R. Safety and Technical Efficacy of Pediatric Brainstem Biopsies: An Updated Meta-Analysis of 1000+ Children. World Neurosurg. 2024, 189, 428–438e422. [Google Scholar] [CrossRef]
- Cantor, E.; Wierzbicki, K.; Tarapore, R.S.; Ravi, K.; Thomas, C.; Cartaxo, R.; Nand Yadav, V.; Ravindran, R.; Bruzek, A.K.; Wadden, J.; et al. Serial H3K27M cell-free tumor DNA (cf-tDNA) tracking predicts ONC201 treatment response and progression in diffuse midline glioma. Neuro Oncol. 2022, 24, 1366–1374. [Google Scholar] [CrossRef]
- Pages, M.; Rotem, D.; Gydush, G.; Reed, S.; Rhoades, J.; Ha, G.; Lo, C.; Fleharty, M.; Duran, M.; Jones, R.; et al. Liquid biopsy detection of genomic alterations in pediatric brain tumors from cell-free DNA in peripheral blood, CSF, and urine. Neuro Oncol. 2022, 24, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Aittaleb, R.; Doherty, R.; Gera, A.; Lau, B.; Messinger, D.; Wadden, J.; Franson, A.; Saratsis, A.; Koschmann, C. Liquid biopsy in H3K27M diffuse midline glioma. Neuro Oncol. 2024, 26, S101–S109. [Google Scholar] [CrossRef]
- Tripathy, A.; John, V.; Wadden, J.; Kong, S.; Sharba, S.; Koschmann, C. Liquid biopsy in pediatric brain tumors. Front. Genet. 2022, 13, 1114762. [Google Scholar] [CrossRef]
- Bakker, A.; Ixkes, A.E.; Venugopal, H.; Ries, M.G.; Lak, N.S.M.; de Vos, F.; van Vuurden, D.G.; Snijders, T.J. Focused Ultrasound-Enhanced Liquid Biopsy: A Promising Diagnostic Tool for Brain Tumor Patients. Cancers 2024, 16, 1576. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Pediatric High-Grade Gliomas (pHGGs) | |||||
| Subtype | Genetic Alterations | Age Groups | Prevalence | Prognosis | 5-Year OS |
| Diffuse midline gliomas (DMGs) | TP53; H3.1/3K27M; NF1; ACVR1; PIK3CA; FGFR; PDGFRA | Younger children (H3.1) (age range: <3–7 years) and teens (H3.3) | 40% | Worst | <1% |
| Diffuse hemispheric gliomas (DHGs) | TP53; H3.3G34R/V; ATRX; PDGFRA | Older children (age range: 6–15 years) and young adults | 10% | Poor | <5% |
| Infantile receptor tyrosine kinase (RTK) fusion glioma | ALK; ROS1; MET; NTRK fusions | Infants (<3 years) | 15–20% | Intermediate | ~42.9–53.8% |
| H3-wt/IDH-wt | TP53; MYCN; PDGFRA; EGFR | Children (3–10 years) and young adults | 50% | Intermediate; poor | ≤5% |
| Pediatric Low-Grade Gliomas (pLGGs) | |||||
| IDH-mutant glioma | IDH1/IDH2 mutant | Adolescents and young adults (15–30 years) | <5% | Better | 30–40% |
| Pilocytic astrocytoma (PA) | BRAF-KIAA1549 fusion, NF1 loss | 5–15 years | 30–40% | Excellent | >95% |
| Diffuse astrocytoma (MYB/MYBL1) | MYB/MYBL1 rearrangements | 10–20 years | ~5% | Good | ~85–95% |
| Ganglioglioma | BRAF V600E mutation | 5–20 years | ~10% | Favorable | ~80–95% |
| Pleomorphic xanthoastrocytoma | BRAF V600E mutation | 10–25 years | 1–3% | Favorable | ~70–80% |
| Subependymal giant cell astrocytoma | TSC1/TSC2 mutations | Infants and young children (0–10 years) | ~1–2% | Excellent (mTOR inhibitors) | >95% |
| Dysembryoplastic neuroepithelial tumor | FGFR1 alterations | 5–20 years | <1–2% | Excellent | >95% |
| Pilomyxoid astrocytoma | BRAF alterations | Infants and young children (0–5 years) | ~1–2% | Worse than PA | ~80% |
| Ependymoma | |||||
| Hemispheric supratentorial-RELA (ST-RELA) | ZFTA-RELA; ZFTA-YAP1; ZFTA-MAML2 | Children (3–10 years) | 18% | Poor | <70% |
| Hemispheric supratentorial-YAP1 (ST-YAP1) | YAP1-MAMLD1; YAP1-FAM118B | Infants (<3 years) | 3% | Good | >90% |
| Cerebellar posterior fossa-A (PF-A) | EZHIP; MAP3K20; TGA6; Chr 1q gain or 6q loss | Children (3–10 years) | 48% | Poor | 70–85% |
| Cerebellar posterior fossa-B (PF-B) | Chromosomal instability; H3K27M | Teens (age range: 13–19 years) and adults | 10% | Good | >90% |
| Spinal cord (SP-EPN: spinal ependymoma) | MYCN; NF2; chromosomal instability; Chr 22q loss | Adults | 4% | Good | >70% |
| Medulloblastoma (MB) | |||||
| WNT-activated (WNT-MB) | TP53; CTNNB1; SMARCA4; DDX3X | Older children (age range: 6–15 years) and adults | 10% | Best | 95% |
| Sonic hedgehog-activated (SHH-MB) | TP53; PTCH1; SMO; MYCN; GLI1; GLI2; SUFU; MLL2 | Infants (<3 years), children (3–10 years), and adults | 30% | Intermediate; poor | 75% |
| Group-3 MB | MYC/MYCN; OTX2; MLL2; CHD7 | Infants (<3 years) and younger children (age range: <3–7 years) | 25% | Very poor | 50% |
| Group-4 MB | Group-3 MB + CDK6; KDM6A; UTX; PRDM6; CBFA complex; DDX31; GFI1/GFI1B; KMT2C; MLL3 | Infants (<3 years), older children (age range: 6–15 years), and adults | 35% | Intermediate, poor | 75% |
| Preclinical Pediatric Brain Tumor Model | Strengths/Advantages | Limitations/Disadvantages |
|---|---|---|
| In vitro 2D cell culture | Cost-effective, rapid drug screening, enables study of specific molecular mechanisms. | Does not represent tumor heterogeneity, lacks microenvironmental interactions and hypoxic regions. |
| Neurosphere cultures | Maintains tumor heterogeneity, retains tumor genotype, preserves stem-like properties. | Requires enriched medium, limited scalability, stem-like cells grow disproportionately. |
| Patient-derived xenografts (PDXs) | Maintains tumor histological features, versatile for drug screening and toxicity studies. | Engraftment rate is variable, lacks immune system contribution in immunodeficient models. |
| Genetically engineered mouse models (GEMMs) | Replicates tumor initiation in vivo, includes immune interactions, supports blood–brain barrier integrity and tumor microenvironment (TME). | Species differences limit translational relevance, costly, lacks complete tumor heterogeneity. |
| In utero electroporation (IUE) GEMMs | Allows precise genetic manipulation, recapitulates human-like tumor features, syngeneic models for immune-competent studies. IUE–PiggyBac system able to carry significantly larger cargo, typically up to 100 kb, making it highly suitable for transducing larger oncogene constructs or multiple genes simultaneously. | Although offering large cargo capacity, PiggyBac often considered more complex in terms of the delivery process and optimization. |
| Sleeping Beauty transposon (SB-GEMMs) | Stable and controlled gene integration, ideal for studying specific genetic mutations in gliomas. | Risk of off-target effects, labor-intensive validation. SB system typically carrying cargo of up to ~10–15 kb, which is relatively smaller compared with PiggyBac. |
| Syngeneic allograft models | Supports immune-competent studies, replicates tumor histology and immune response. | Limited molecular alignment with human tumors, challenging standardization protocols. |
| Humanized mouse models | Facilitates study of human tumor–immune interactions, suitable for testing preclinical immunotherapies. | Expensive, risk of graft-versus-host disease (GVHD), incomplete central nervous system (CNS)-specific immune response. |
| Zebrafish brain tumor models | Cost-effective and high-throughput screening for drug discovery and testing, transparent embryos allow real-time imaging of tumor growth, invasion, angiogenesis. Rapid tumor development and shorter experimental timelines compared to mammalian models. | Limited physiological and anatomical similarity to the human brain, lack of a mature adaptive immune system in early developmental stages, affecting immunotherapy studies. Smaller brain size restricting the ability to model complex tumor behaviors. Ethical and technical considerations for scaling to advanced therapeutic interventions. |
| Ex vivo models (brain slice cultures) | Retains native tissue architecture, enables study of tumor invasion and cellular interactions. | Short viability, lacks active blood flow, limited to small-scale studies. |
| Large animal models | Physiological similarity to humans, ideal for drug delivery and tumor infiltration studies. | High cost, ethical and logistical challenges. |
| CRISPR-Cas9-engineered models | Precise genetic editing for specific mutations, relevant for studying H3K27M and other targets. | Risk of off-target effects, requires advanced expertise. |
| Tumor organoids | Recapitulates tumor architecture, hypoxic gradients, useful for biomarker testing. | Absence of vasculature, host immune cells, limited cell diversity, technically demanding. |
| Cerebral organoids | Human-like brain microenvironment, retains heterogeneity and tumor invasiveness. | Lacks mature brain tissue, no immune compartment, resembles fetal brain structures. |
| Advanced organoids with vascularization | Incorporates endothelial and immune components, enhances physiological relevance. | Technically demanding, scalability issues, high cost. |
| Microfluidic devices | Mimics TME dynamics, compartmentalization increases reproducibility. | Complex and costly, not standardized, typically uses pre-differentiated cell types. |
| 3D bioprinting | Enables precise spatial control, supports co-culture of various cell types, mimics extracellular matrix interactions. | Requires high technical expertise, variability between bioinks, increased cost. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faisal, S.M.; Yadav, M.; Gibson, G.R.; Klinestiver, A.T.; Sorenson, R.M.; Cantor, E.; Ghishan, M.; Prensner, J.R.; Franson, A.T.; Ginn, K.F.; et al. Current Landscape of Preclinical Models for Pediatric Gliomas: Clinical Implications and Future Directions. Cancers 2025, 17, 2221. https://doi.org/10.3390/cancers17132221
Faisal SM, Yadav M, Gibson GR, Klinestiver AT, Sorenson RM, Cantor E, Ghishan M, Prensner JR, Franson AT, Ginn KF, et al. Current Landscape of Preclinical Models for Pediatric Gliomas: Clinical Implications and Future Directions. Cancers. 2025; 17(13):2221. https://doi.org/10.3390/cancers17132221
Chicago/Turabian StyleFaisal, Syed M., Monika Yadav, Garrett R. Gibson, Adora T. Klinestiver, Ryan M. Sorenson, Evan Cantor, Maria Ghishan, John R. Prensner, Andrea T. Franson, Kevin F. Ginn, and et al. 2025. "Current Landscape of Preclinical Models for Pediatric Gliomas: Clinical Implications and Future Directions" Cancers 17, no. 13: 2221. https://doi.org/10.3390/cancers17132221
APA StyleFaisal, S. M., Yadav, M., Gibson, G. R., Klinestiver, A. T., Sorenson, R. M., Cantor, E., Ghishan, M., Prensner, J. R., Franson, A. T., Ginn, K. F., Koschmann, C., & Yadav, V. N. (2025). Current Landscape of Preclinical Models for Pediatric Gliomas: Clinical Implications and Future Directions. Cancers, 17(13), 2221. https://doi.org/10.3390/cancers17132221