
Stratification of Hepatocellular Carcinoma Using N6-Methyladenosine


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Acquisition of Data
2.2. Human Tissue Samples and Tissue Microarrays
2.3. Consensus Clustering of m6A Regulators
2.4. Identification of Differentially Expressed Genes
2.5. Construction of a Risk Score for Predicting the Overall Survival of Hepatocellular Carcinoma
2.6. Exploration of the Genomic Features and Stemness Characteristics of the Prognostic Signature
2.7. Validation of the Prognostic Signature and Clinical Correlation Analysis
2.8. Single-Cell RNA Sequencing Analysis
2.9. Cell Culture and Transfection
2.10. Quantitative Reverse Transcriptase PCR, Western Blot Analysis, and Immunohistochemistry Assay
2.11. Cell Proliferation Assays
2.12. Cell Migration Assays
2.13. Statistical Analysis
3. Results
3.1. Consensus Clustering of m6A Genes in Two Clusters with Different Clinical Outcomes of HCC
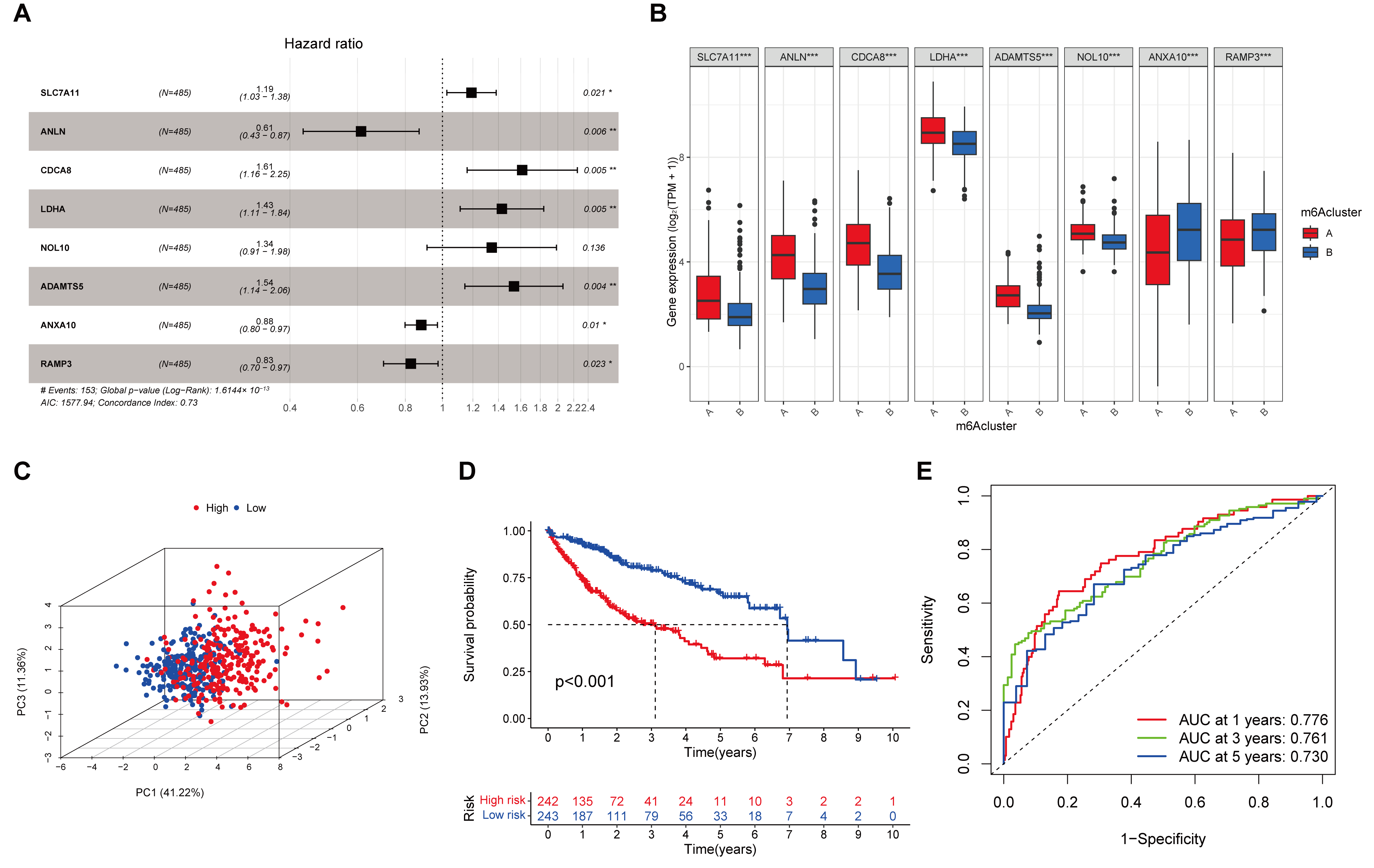
3.2. An 8-Gene Prognostic Signature Established in the Training Cohort
3.3. Evaluation and Validation of the Prognostic Signature
3.4. The Benefit of ICIs Therapy in Different Risk Subgroups
3.5. Exploration of m6A-Related Risk Genes in the Single-Cell Level
3.6. Association of Risk Score with Gene Mutation, Tumor Mutation Burden, and mRNA Stemness Index
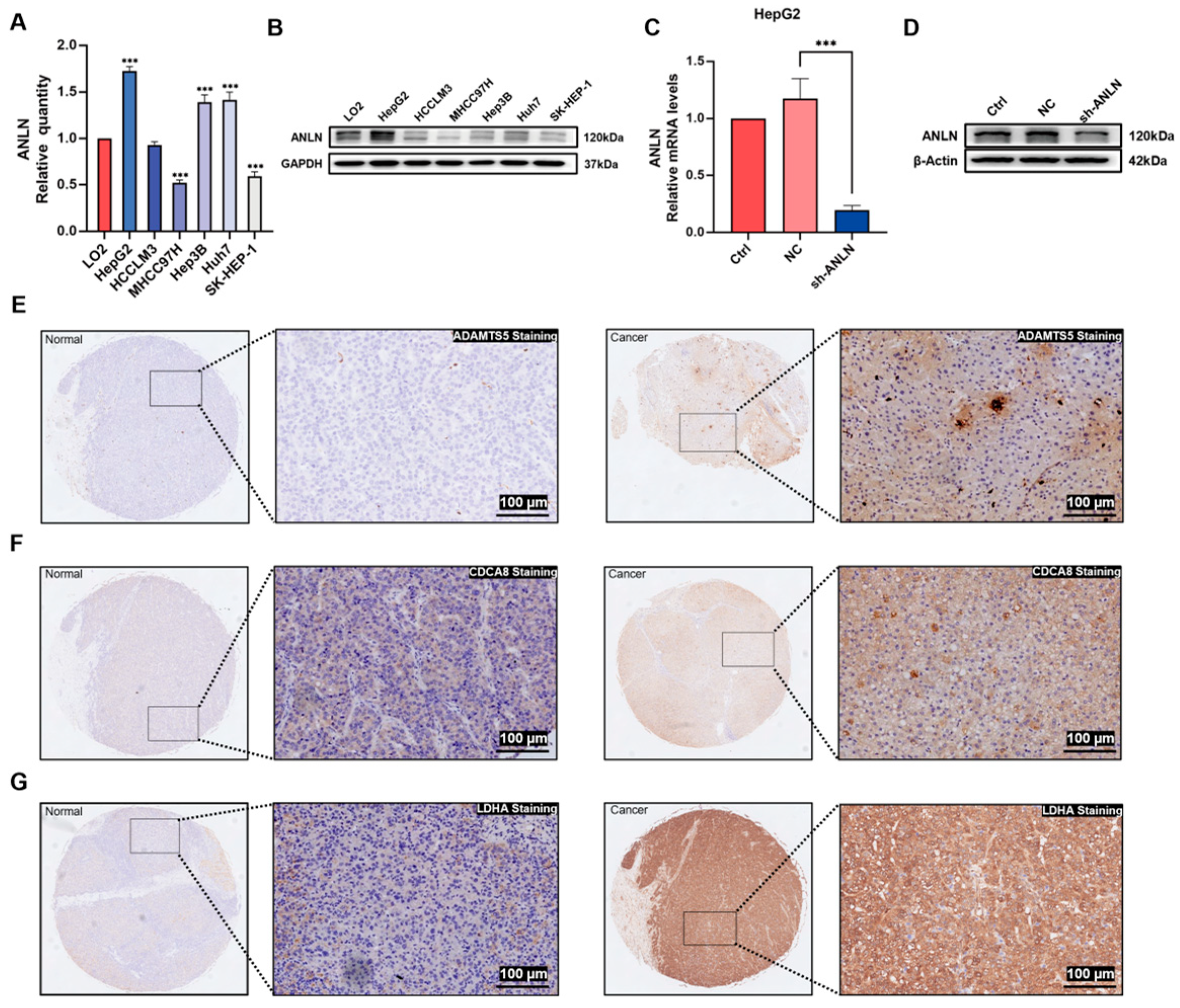
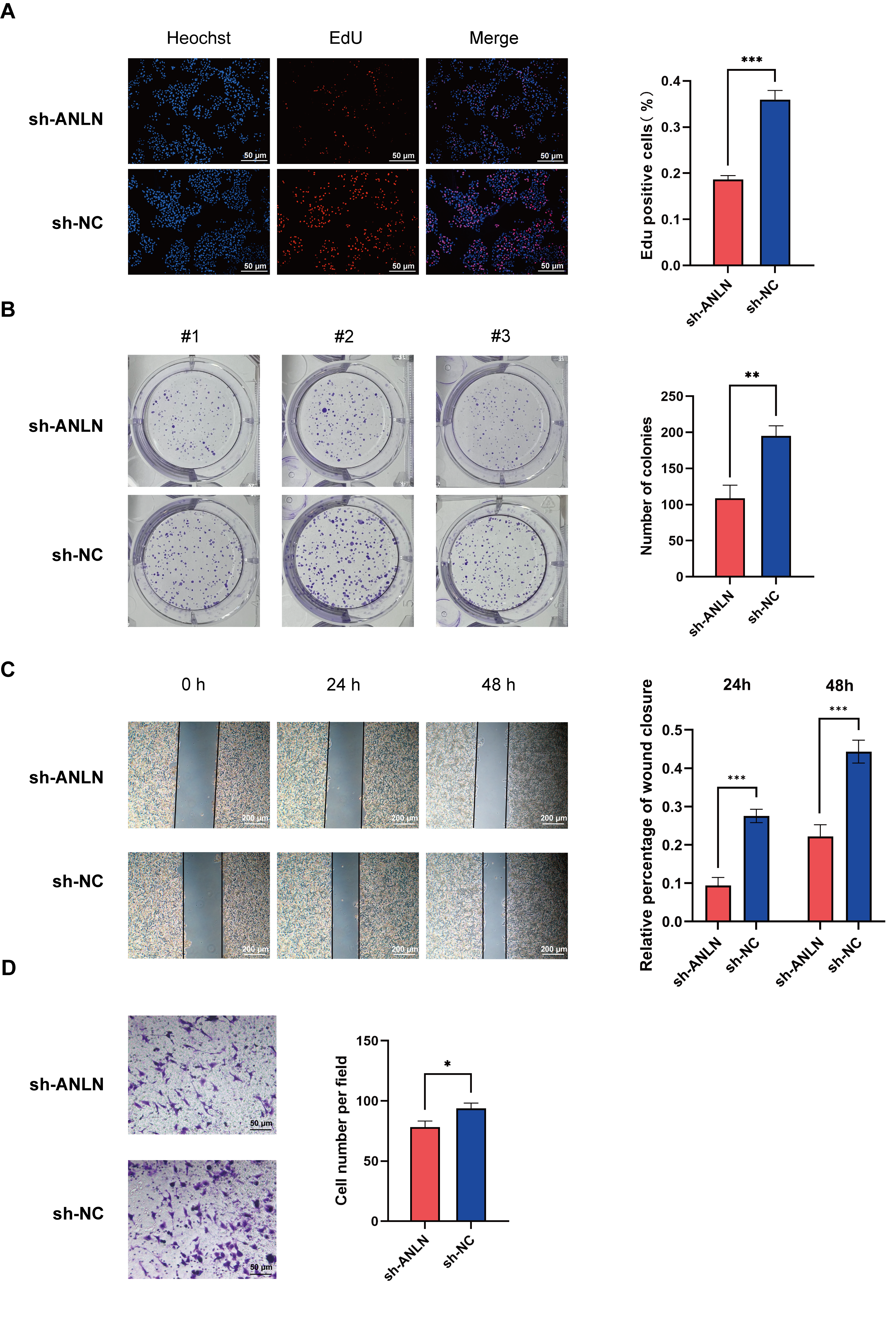
3.7. ANLN Inhibits HCC Cell Proliferation and Migration In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANLN | Actin-binding protein |
| ALKBH5 | AlkB homolog 5 |
| AUC | Area under the ROC curve |
| PD-L1 | Programmed death-ligand 1 |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DEGs | Differentially expressed genes |
| ECL | Enhanced chemiluminescence |
| EdU | Ethynyl-2′-deoxyuridine |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| FTO | Fat mass and obesity-associated protein |
| GAL-9 | Galectin-9 |
| GEO | Gene Expression Omnibus |
| HCC | Hepatocellular carcinoma |
| HNRNPC | Heterogeneous nuclear ribonucleoprotein C |
| HR | Hazard ratio |
| ICGC | International Cancer Genomics Consortium |
| ICIs | Immune checkpoint inhibitors |
| IPS | Immunophenotype score |
| LAG-3 | lymphocyte-activating 3 |
| LASSO | Least absolute shrinkage and selection operator |
| m6A | N6-methyladenosine |
| METTL | Methyltransferase-like |
| mRNAsi | mRNA stemness index |
| NK cells | Natural killer cells |
| OS | Overall survival |
| PCA | Principal component analysis |
| PDCD1 | Programmed cell death protein 1 |
| RBM15 | RNA-binding motif protein 15 |
| ROC | Receiver operating characteristic |
| shRNA | Short hairpin RNA |
| TCGA | The Cancer Genome Atlas |
| TIDE | Tumor immune dysfunction and exclusion |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TIM-3 | T cell immunoglobulin and mucin-domain containing-3 |
| TMB | Tumor mutation burden |
| TPM | Transcripts per million kilobase |
| UMAP | Uniform Manifold Approximation and Projection |
| VIRMA | Vir-Like m6A Methyltransferase Associated protein |
| WTAP | Wilms tumor 1-associating protein |
| YTH | YT521-B homology |
| ZC3H13 | Zinc finger CCCH-type containing 13 |
References
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef]
- Tabrizian, P.; Jibara, G.; Shrager, B.; Schwartz, M.; Roayaie, S. Recurrence of hepatocellular cancer after resection: Patterns, treatments, and prognosis. Ann. Surg. 2015, 261, 947–955. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Liu, Q.; Gregory, R.I. RNAmod: An integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019, 47, W548–W555. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef]
- Du, Y.; Ma, Y.; Zhu, Q.; Liu, T.; Jiao, Y.; Yuan, P.; Wang, X. An m6A-Related Prognostic Biomarker Associated with the Hepatocellular Carcinoma Immune Microenvironment. Front. Pharmacol. 2021, 12, 707930. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kuzmanović, A.; Wang, N.; Liao, L.; Ernst, S.; Penners, C.; Jans, A.; Hammoor, T.; Stach, P.B.; Peltzer, M.; et al. Exceptional Uptake, Limited Protein Expression: Liver Macrophages Lost in Translation of Synthetic mRNA. Adv. Sci. 2025, 12, e2409729. [Google Scholar] [CrossRef]
- Wang, N.; Hao, F.; Ren, J.; Fei, X.; Chen, Y.; Xu, W.; Wang, J. Positive feedback loop of AKR1B10P1/miR-138/SOX4 promotes cell growth in hepatocellular carcinoma cells. Am. J. Transl. Res. 2020, 12, 5465–5480. [Google Scholar]
- Long, M.T.; Noureddin, M.; Lim, J.K. AGA Clinical Practice Update: Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Lean Individuals: Expert Review. Gastroenterology 2022, 163, 764–774e761. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, S.; Zhuang, H.; Ruan, S.; Zhou, Z.; Huang, K.; Ji, F.; Ma, Z.; Hou, B.; He, X. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene 2020, 39, 4507–4518. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [PubMed]
- Guirguis, A.A.; Ofir-Rosenfeld, Y.; Knezevic, K.; Blackaby, W.; Hardick, D.; Chan, Y.C.; Motazedian, A.; Gillespie, A.; Vassiliadis, D.; Lam, E.Y.N.; et al. Inhibition of METTL3 Results in a Cell-Intrinsic Interferon Response That Enhances Antitumor Immunity. Cancer Discov. 2023, 13, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; He, K.; Wang, Y.; Sun, J.; Chen, Y.; Yang, Z. Prognostic Signature of Hepatocellular Carcinoma and Analysis of Immune Infiltration Based on m6A-Related lncRNAs. Front. Oncol. 2021, 11, 691372. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, T.; Chen, H.; Ding, X.; Chen, H.; Liang, Z.; Zhao, Y.; Ma, S.; Liu, X. METTL3 inhibition promotes radiosensitivity in hepatocellular carcinoma through regulation of SLC7A11 expression. Cell Death Dis. 2025, 16, 9. [Google Scholar] [CrossRef]
- Shi, S.; Wang, C.; Cai, Q.; Yang, R.; Peng, M.; Liang, H.; Qian, B.; Jiang, Y.; Xiao, B.; Wang, L.; et al. RBM15 drives the progression of lung adenocarcinoma by regulating N6-methyladenosine-mediated LDHA mRNA stability. Life Sci. 2024, 358, 123146. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, Z.J.; Liang, B.; Wang, Z.G.; Tao, Y.P.; Huang, S.Y.; Ni, J.S.; Li, H.F.; Yang, L.; Yuan, S.X.; et al. N6-Methyladenosine Modification of ANLN Enhances Hepatocellular Carcinoma Bone Metastasis. Int. J. Biol. Sci. 2023, 19, 1009–1023. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA-LIHC | GSE76427 | LIRI-JP | |
|---|---|---|---|
| Age (%) | |||
| ≤65 | 141 (37.4%) | 65 (56.5%) | 98 (37.7%) |
| >65 | 235 (62.3%) | 50 (43.5%) | 162 (62.3%) |
| Unknown | 1 (0.3%) | NA | NA |
| Gender (%) | |||
| Female | 122 (32.4%) | 22 (19.1%) | 68 (26.2%) |
| Male | 255 (67.6%) | 93 (80.9%) | 192 (73.8%) |
| Stage (%) | |||
| I | 175 (46.4%) | 55 (47.8%) | 40 (15.4%) |
| II | 87 (23.1%) | 35 (30.4%) | 117 (45.0%) |
| III | 86 (22.8%) | 21 (18.3%) | 80 (30.8%) |
| IV | 5 (1.3%) | 4 (3.5%) | 23 (8.8%) |
| Unknown | 24 (6.4%) | NA | NA |
| Grade (%) | |||
| Grade 1 | 55 (14.6%) | NA | NA |
| Grade 2 | 180 (47.7%) | NA | NA |
| Grade 3 | 124 (32.9%) | NA | NA |
| Grade 4 | 13 (3.4%) | NA | NA |
| Unknown | 5 (1.3%) | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Shi, J.-X.; Bartneck, M.; Dahl, E.; Wang, J. Stratification of Hepatocellular Carcinoma Using N6-Methyladenosine. Cancers 2025, 17, 2220. https://doi.org/10.3390/cancers17132220
Wang N, Shi J-X, Bartneck M, Dahl E, Wang J. Stratification of Hepatocellular Carcinoma Using N6-Methyladenosine. Cancers. 2025; 17(13):2220. https://doi.org/10.3390/cancers17132220
Chicago/Turabian StyleWang, Nan, Jia-Xin Shi, Matthias Bartneck, Edgar Dahl, and Junqing Wang. 2025. "Stratification of Hepatocellular Carcinoma Using N6-Methyladenosine" Cancers 17, no. 13: 2220. https://doi.org/10.3390/cancers17132220
APA StyleWang, N., Shi, J.-X., Bartneck, M., Dahl, E., & Wang, J. (2025). Stratification of Hepatocellular Carcinoma Using N6-Methyladenosine. Cancers, 17(13), 2220. https://doi.org/10.3390/cancers17132220