

Advancing the Landscape of Clinical Actionability in Von Hippel–Lindau Syndrome: An Evidence-Based Framework from the INT2GRATE Oncology Consortium
 , , , , , , ,
, , , , , , ,  , , and
, , and  add
Show full author list
add
Show full author list
Simple Summary
Abstract
1. Introduction
2. Methods
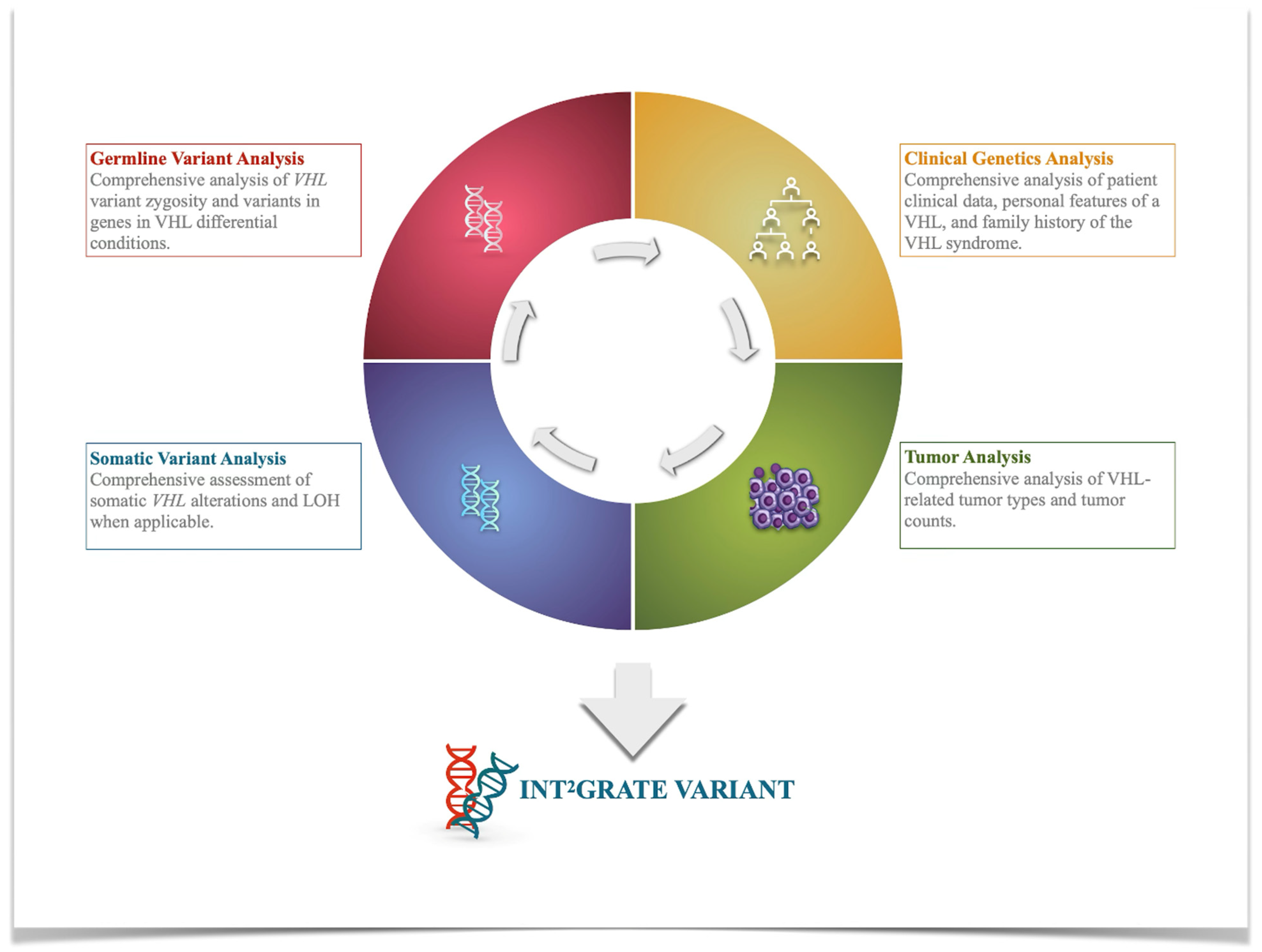
2.1. Development of the INT2GRATE|VHL Platform
INT2GRATE|VHL Variant Evidence Framework (VEF)
2.2. INT2GRATE Data Processing and Analysis
2.2.1. Development of the Digital INT2GRATE VEF
2.2.2. INT2GRATE Encoder
2.2.3. INT2GRATE Pattern Quantifier
2.2.4. INT2GRATE|Data Portal and Data Sharing
2.3. Patient Data Query and Cohorts
2.4. Germline Genetic Laboratory Testing
2.5. Tumor Genetic Laboratory Testing
2.5.1. Single-Nucleotide Variant (SNV)/Indel Analysis
2.5.2. OncoPanel Copy Number Analysis
2.5.3. OncoPanel Structural Variant Analysis
3. Results
3.1. Development of INT2GRATE|Variant Evidence Framework (VEF) for VHL
3.1.1. Germline Variants and Rationale
3.1.2. Clinical Genetics Criteria and Rationale
3.1.3. Tumor-Derived Information and Rationale
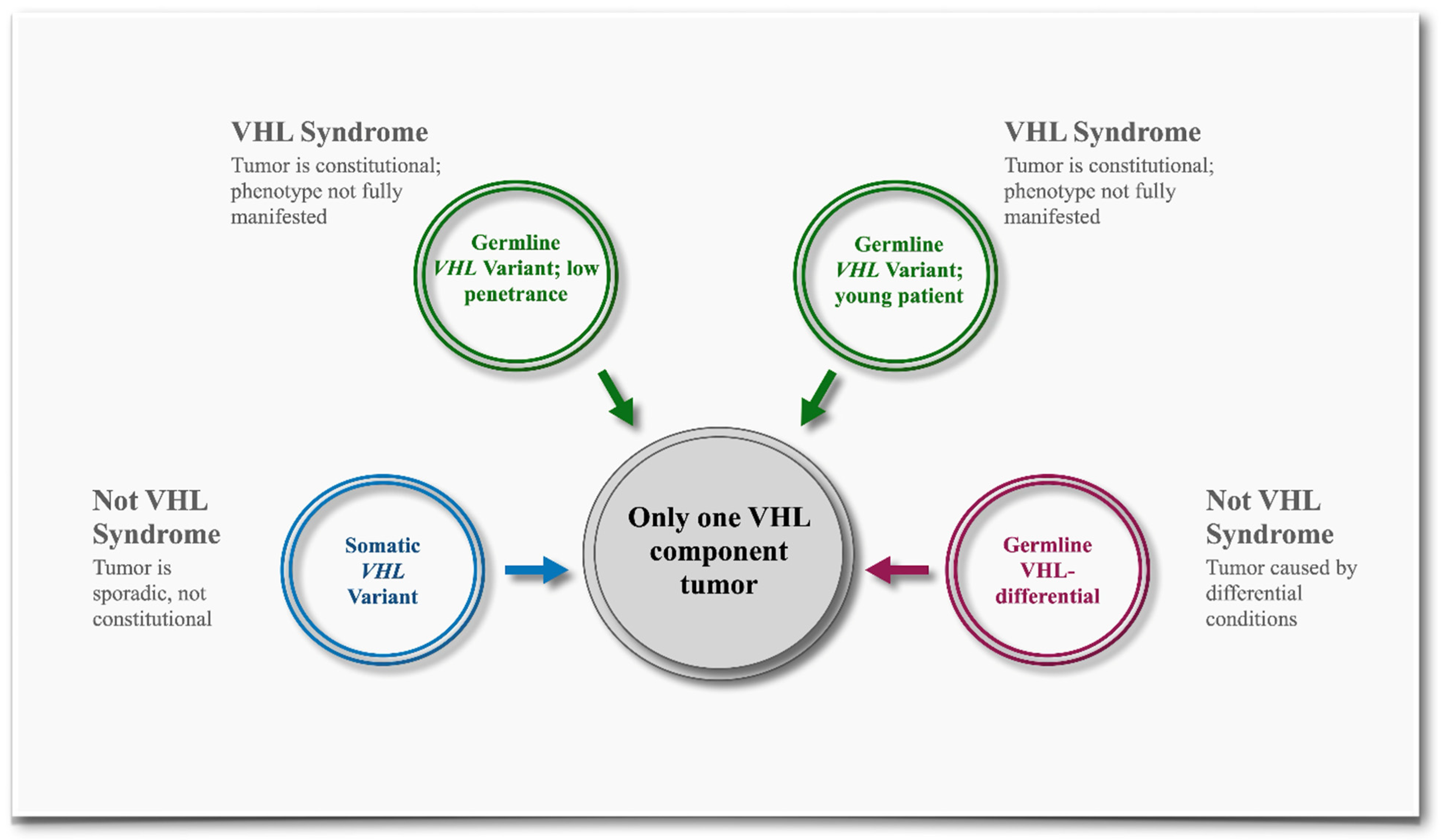
Only One VHL Component Tumor: Renal Cell Carcinoma
Only One VHL Component Tumor: Except Renal Cell Carcinoma
Two or More VHL Component Tumors
Non-VHL Tumor(s)
3.1.4. Somatic Variants and Rationale
3.2. Assignment of INT2GRATE Categories
3.3. Patient Cohorts and Clinical Presentations
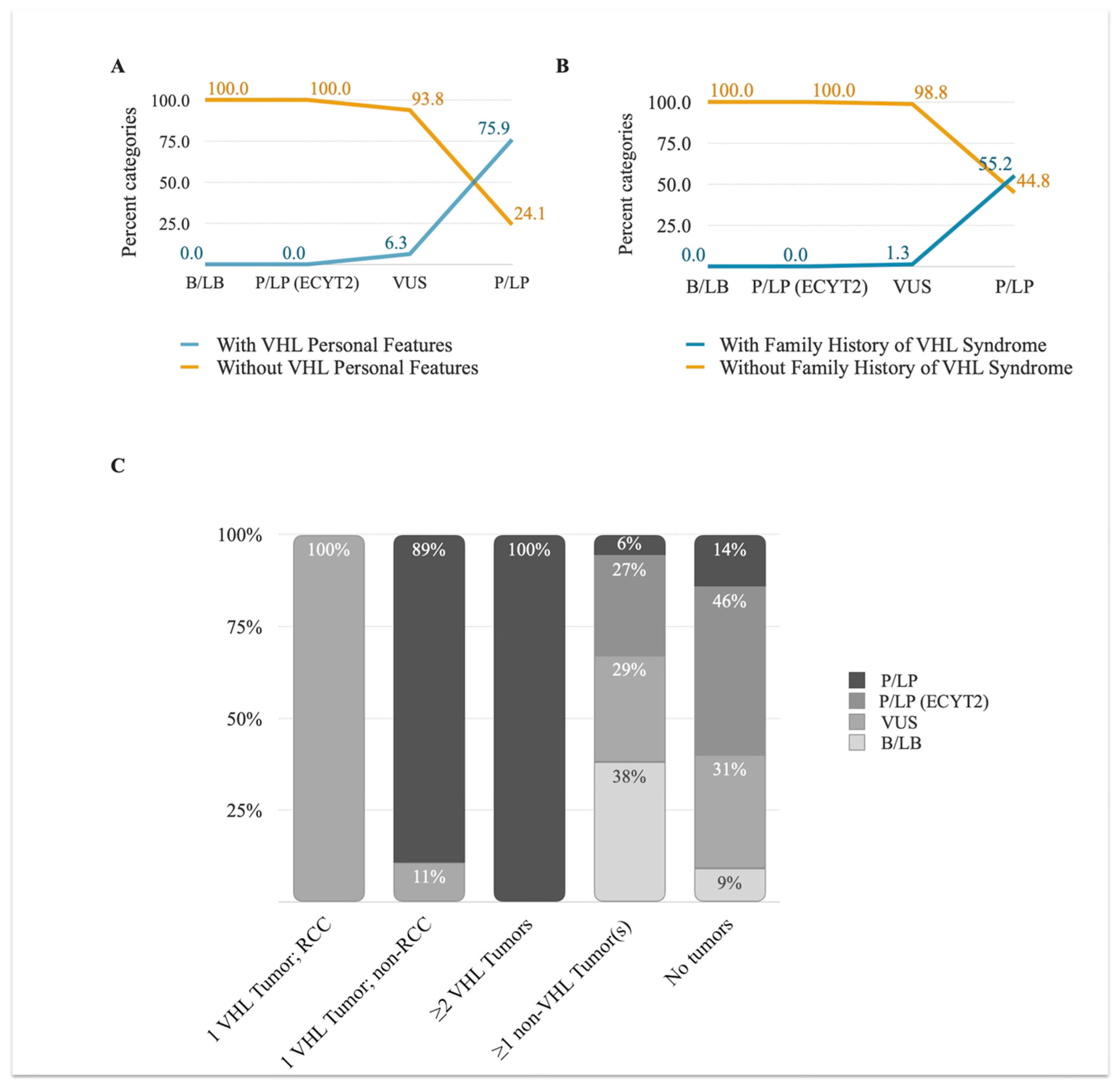
3.3.1. VHL Personal and Family History
3.3.2. VHL Tumor Information
3.4. INT2GRATE|VHL Variant Analysis
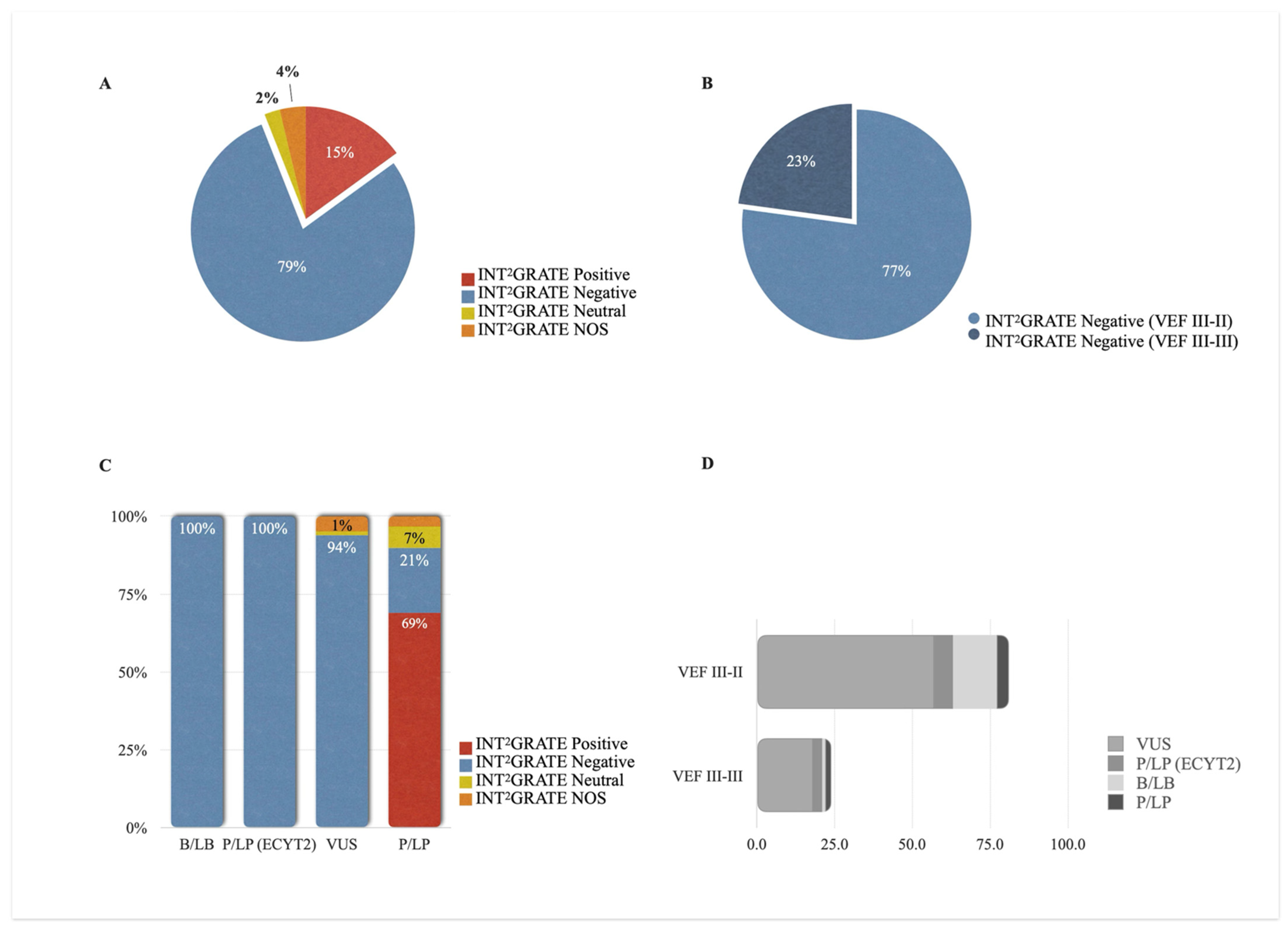
3.5. INT2GRATE Variants and Clinical Actionability
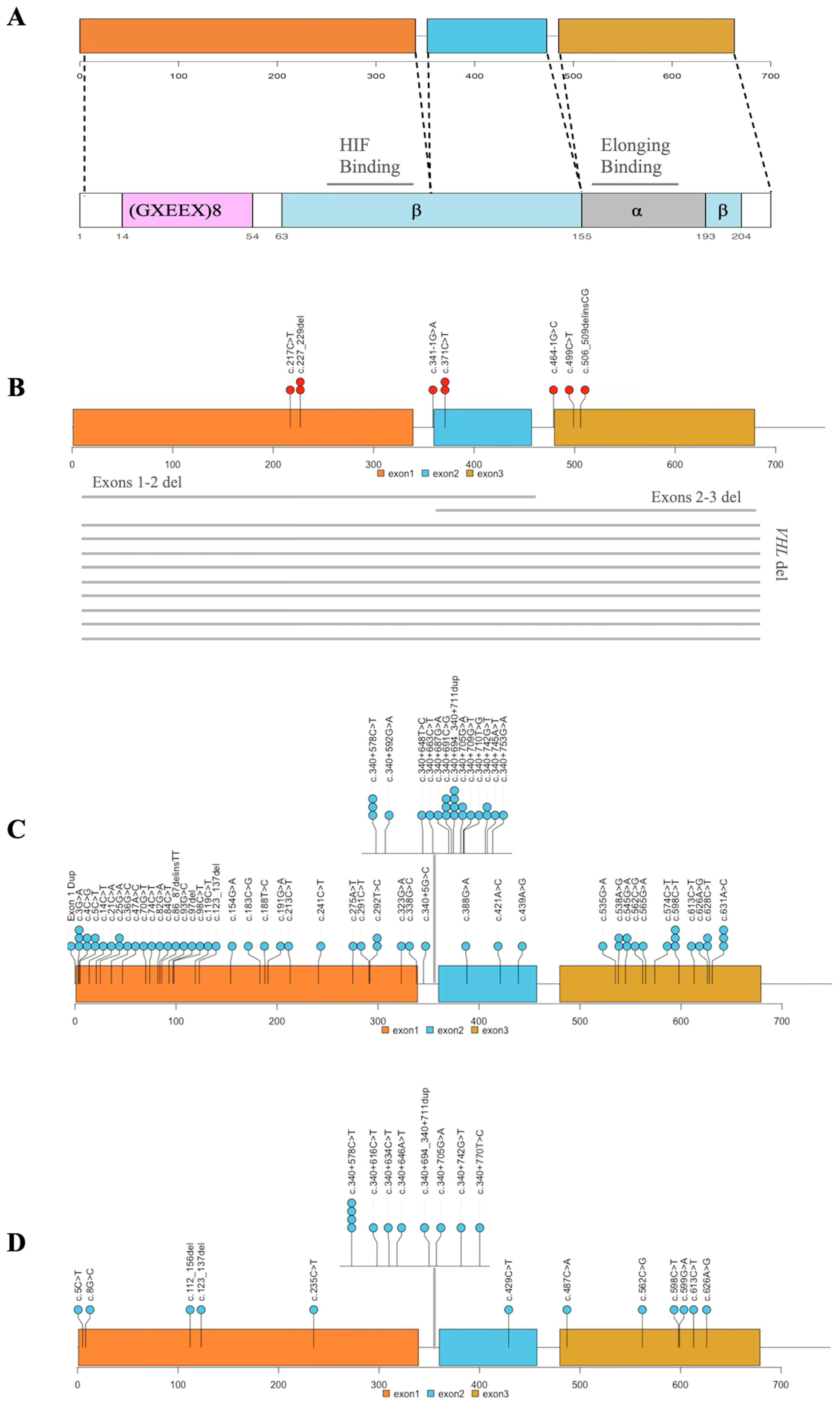
3.6. INT2GRATE Variants and Distribution Along VHL Exons
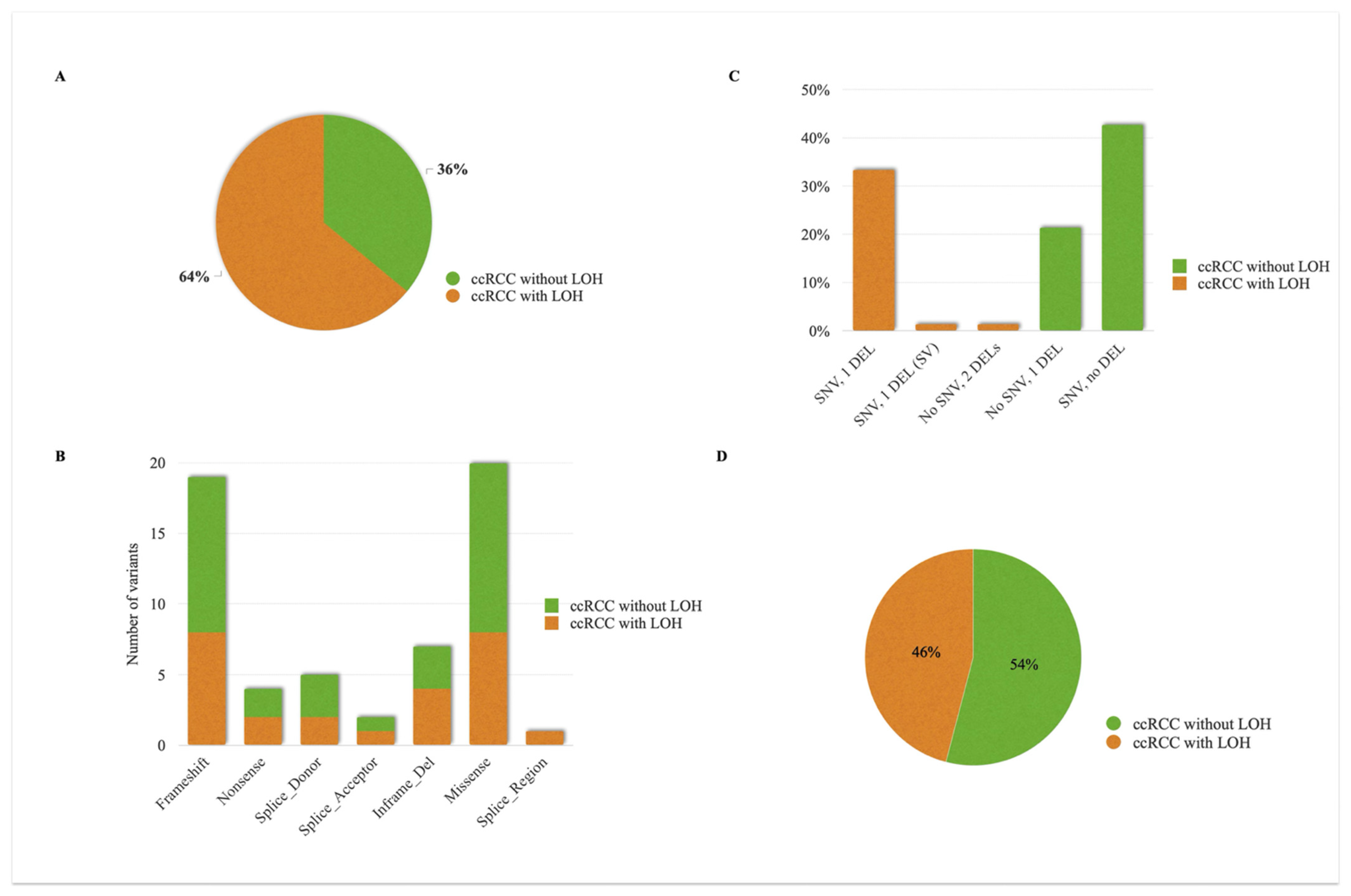
3.7. Analysis of Somatic VHL Allelic Tumors
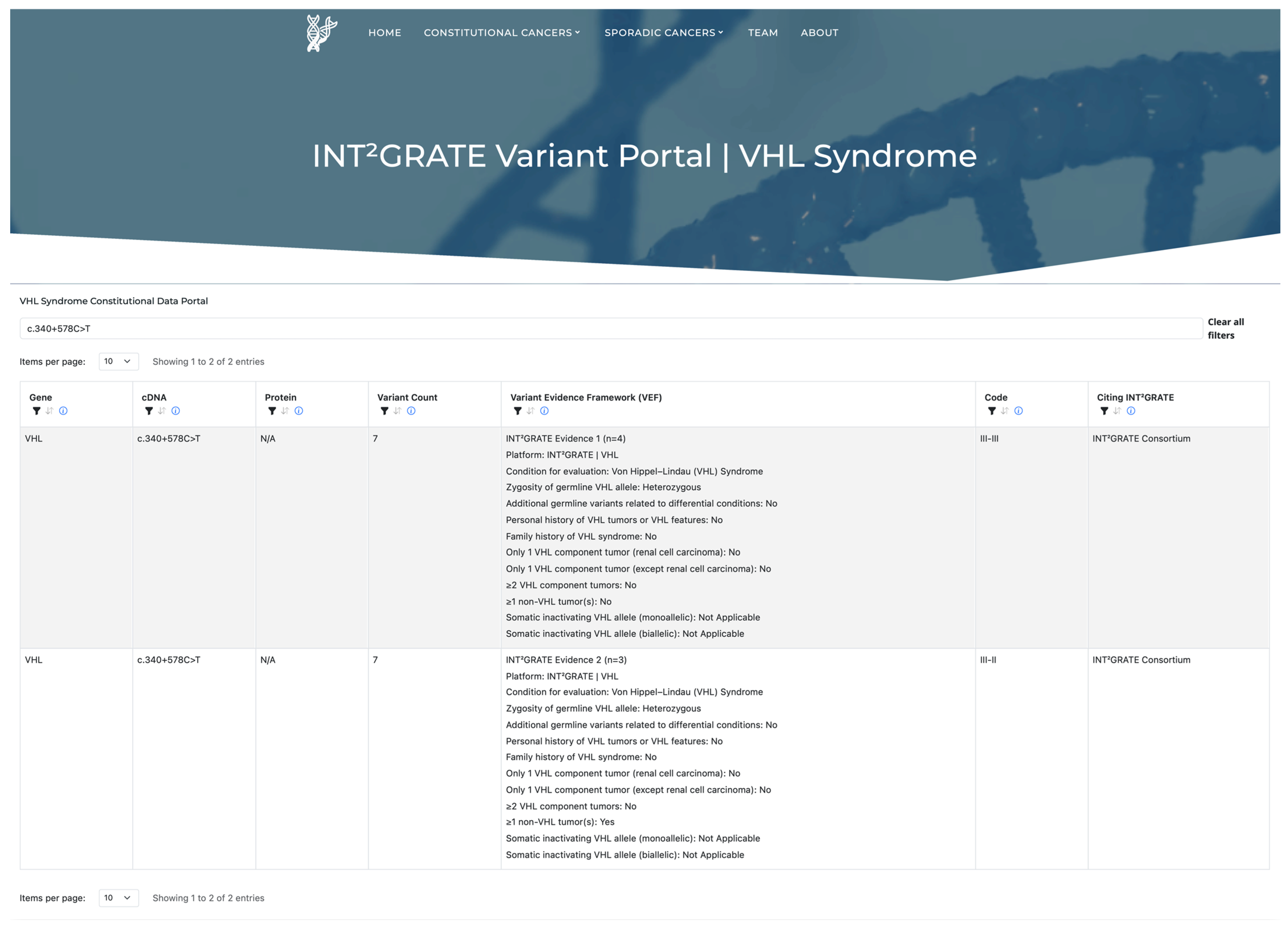
3.8. INT2GRATE|Variant Portal and Data Sharing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maher, E.R.; Iselius, L.; Yates, J.R.; Littler, M.; Benjamin, C.; Harris, R.; Sampson, J.; Williams, A.; Ferguson-Smith, M.A.; Morton, N. Von Hippel-Lindau disease: A genetic study. J. Med. Genet. 1991, 28, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A Review of Von Hippel-Lindau Syndrome. J. Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e68–e75. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Zhang, K.; Qiu, J.; Yang, W.; Ma, K.; Li, L.; Xie, H.; Xu, Y.; Gong, Y.; Zhou, J.; Cai, L.; et al. Clinical characteristics and risk factors for survival in affected offspring of von Hippel-Lindau disease patients. J. Med. Genet. 2022, 59, 951–956. [Google Scholar] [CrossRef]
- Rana, H.Q.; Koeller, D.R.; Schwartz, A.; Manning, D.K.; Schneider, K.A.; Krajewski, K.M.; Choueiri, T.K.; Lindeman, N.I.; Garber, J.E.; Ghazani, A.A. Pathogenicity of VHL variants in families with non-syndromic von Hippel-Lindau phenotypes: An integrated evaluation of germline and somatic genomic results. Eur. J. Med. Genet. 2021, 64, 104359. [Google Scholar] [CrossRef]
- Adam, M.P.; Feldman, J.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Amemiya, A. GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Rana, H.Q.; Koeller, D.R.; Walker, M.; Unal, B.; Levine, A.S.; Chittenden, A.; Isidro, R.A.; Hayes, C.P.; Manam, M.D.; Buehler, R.M.; et al. Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes. Cancers 2024, 16, 947. [Google Scholar] [CrossRef]
- Isidro, R.A.; Chittenden, A.; Walker, M.; Schwartz, A.; Koeller, D.R.; Hayes, C.P.; Unal, B.; Manam, M.D.; Buehler, R.M.; Manning, D.K.; et al. Development and evaluation of INT. Front. Oncol. 2023, 13, 1284690. [Google Scholar] [CrossRef]
- Manning, D.K.; Shivdasani, P.; Koeller, D.R.; Schwartz, A.; Rana, H.Q.; Garber, J.E.; Lindeman, N.I.; Ghazani, A.A. Assessment of genomic alterations in non-syndromic von Hippel-Lindau: Insight from integrating somatic and germline next generation sequencing genomic data. Data Brief 2021, 39, 107653. [Google Scholar] [CrossRef] [PubMed]
- Koeller, D.R.; Manning, D.K.; Schwartz, A.; Chittenden, A.; Hayes, C.P.; Abraamyan, F.; Rana, H.Q.; Lindeman, N.I.; Garber, J.E.; Ghazani, A.A. An optimized protocol for evaluating pathogenicity of. MethodsX 2022, 9, 101761. [Google Scholar] [CrossRef] [PubMed]
- Abo, R.P.; Ducar, M.; Garcia, E.P.; Thorner, A.R.; Rojas-Rudilla, V.; Lin, L.; Sholl, L.M.; Hahn, W.C.; Meyerson, M.; Lindeman, N.I.; et al. BreaKmer: Detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015, 43, e19. [Google Scholar] [CrossRef]
- Louise, M.B.M.; Smerdel, M.; Borgwadt, L.; Beck Nielsen, S.S.; Madsen, M.G.; Moller, H.U.; Kiilgaard, J.F.; Friis-Hansen, L.; Harbud, V.; Cortnum, S.; et al. Von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur. J. Med. Genet. 2022, 65, 104538. [Google Scholar] [CrossRef]
- Wolters, W.P.G.; Dreijerink, K.M.A.; Giles, R.H.; van der Horst-Schrivers, A.N.A.; van Nesselrooij, B.; Zandee, W.T.; Timmers, H.; Seute, T.; de Herder, W.W.; Verrijn Stuart, A.A.; et al. Multidisciplinary integrated care pathway for von Hippel-Lindau disease. Cancer 2022, 128, 2871–2879. [Google Scholar] [CrossRef]
- Shuch, B.; Vourganti, S.; Ricketts, C.J.; Middleton, L.; Peterson, J.; Merino, M.J.; Metwalli, A.R.; Srinivasan, R.; Linehan, W.M. Defining early-onset kidney cancer: Implications for germline and somatic mutation testing and clinical management. J. Clin. Oncol. 2014, 32, 431–437. [Google Scholar] [CrossRef]
- Boedeker, C.C.; Erlic, Z.; Richard, S.; Kontny, U.; Gimenez-Roqueplo, A.P.; Cascon, A.; Robledo, M.; de Campos, J.M.; van Nederveen, F.H.; de Krijger, R.R.; et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 2009, 94, 1938–1944. [Google Scholar] [CrossRef]
- Bender, B.U.; Eng, C.; Olschewski, M.; Berger, D.P.; Laubenberger, J.; Altehöfer, C.; Kirste, G.; Orszagh, M.; van Velthoven, V.; Miosczka, H.; et al. VHL c.505 T>C mutation confers a high age related penetrance but no increased overall mortality. J. Med. Genet. 2001, 38, 508–514. [Google Scholar] [CrossRef]
- Favier, J.; Brière, J.J.; Burnichon, N.; Rivière, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reyniès, A.; Bertherat, J.; Badoual, C.; et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef]
- Pastore, Y.; Jedlickova, K.; Guan, Y.; Liu, E.; Fahner, J.; Hasle, H.; Prchal, J.F.; Prchal, J.T. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am. J. Hum. Genet. 2003, 73, 412–419. [Google Scholar] [CrossRef]
- Bento, M.C.; Chang, K.T.; Guan, Y.; Liu, E.; Caldas, G.; Gatti, R.A.; Prchal, J.T. Congenital polycythemia with homozygous and heterozygous mutations of von Hippel-Lindau gene: Five new Caucasian patients. Haematologica 2005, 90, 128–129. [Google Scholar] [PubMed]
- Lorenzo, F.R.; Yang, C.; Lanikova, L.; Butros, L.; Zhuang, Z.; Prchal, J.T. Novel compound VHL heterozygosity (VHL T124A/L188V) associated with congenital polycythaemia. Br. J. Haematol. 2013, 162, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Erlic, Z.; Hoffmann, M.M.; Sullivan, M.; Franke, G.; Peczkowska, M.; Harsch, I.; Schott, M.; Gabbert, H.E.; Valimäki, M.; Preuss, S.F.; et al. Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couvé, S.; et al. Identification of a new. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef]
- Capodimonti, S.; Teofili, L.; Martini, M.; Cenci, T.; Iachininoto, M.G.; Nuzzolo, E.R.; Bianchi, M.; Murdolo, M.; Leone, G.; Larocca, L.M. Von hippel-lindau disease and erythrocytosis. J. Clin. Oncol. 2012, 30, e137–e139. [Google Scholar] [CrossRef]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef]
- Maher, E.R.; Neumann, H.P.; Richard, S. Von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef]
- Arnon, L.; Halperin, R.; Tirosh, A. Impact of Pancreatic Neuroendocrine Tumor on Mortality in Patients with von Hippel-Lindau Disease. Endocr. Pract. 2021, 27, 1040–1045. [Google Scholar] [CrossRef]
- Webster, A.R.; Maher, E.R.; Moore, A.T. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch. Ophthalmol. 1999, 117, 371–378. [Google Scholar] [CrossRef]
- Kreusel, K.M.; Bechrakis, N.E.; Krause, L.; Neumann, H.P.; Foerster, M.H. Retinal angiomatosis in von Hippel-Lindau disease: A longitudinal ophthalmologic study. Ophthalmology 2006, 113, 1418–1424. [Google Scholar] [CrossRef]
- Dollfus, H.; Massin, P.; Taupin, P.; Nemeth, C.; Amara, S.; Giraud, S.; Béroud, C.; Dureau, P.; Gaudric, A.; Landais, P.; et al. Retinal hemangioblastoma in von Hippel-Lindau disease: A clinical and molecular study. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3067–3074. [Google Scholar]
- Huntoon, K.; Shepard, M.J.; Lukas, R.V.; McCutcheon, I.E.; Daniels, A.B.; Asthagiri, A.R. Hemangioblastoma diagnosis and surveillance in von Hippel-Lindau disease: A consensus statement. J. Neurosurg. 2022, 136, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. Von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Lefebvre, F.; Poon, B.P.; Bousard, A.; Fan, X.; Lathrop, M.; Tost, J.; Kim, W.Y.; Riazalhosseini, Y.; Ohh, M. Consequences of VHL Loss on Global DNA Methylome. Sci. Rep. 2018, 8, 3313. [Google Scholar] [CrossRef]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef]
- Li, L.; Bao, H.; Xu, Y.; Yang, W.; Zhang, Z.; Ma, K.; Zhang, K.; Zhou, J.; Gong, Y.; Ci, W.; et al. Preliminary Study of Whole-Genome Bisulfite Sequencing and Transcriptome Sequencing in VHL Disease-Associated ccRCC. Mol. Diagn. Ther. 2023, 27, 741–752. [Google Scholar] [CrossRef]
- Singh, R.B.; Amare Kadam, P.S. Investigation of tumor suppressor genes apart from VHL on 3p by deletion mapping in sporadic clear cell renal cell carcinoma (cRCC). Urol. Oncol. 2013, 31, 1333–1342. [Google Scholar] [CrossRef]
- Chen, M.Y.; Chew, E.Y.; Reynolds, J.C.; Chao, D.L.; Oldfield, E.H. Metastatic brainstem pheochromocytoma in a patient with von Hippel-Lindau disease. Case illustration. J. Neurosurg. 2001, 94, 138. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Cohorts Based on Availability of Data Type | Numbers |
|---|---|
| Total patients in the study | 2672 |
| Breakdown of Cohort 1 | |
| Patients had somatic VHL variants and detailed tumor data, evaluated in genetics, and had germline VHL variants. | 11 |
| Patients had no somatic sequencing data, had detailed tumor data, were evaluated in genetics, and had germline VHL variants. | 122 |
| Breakdown of Cohort 2 | |
| Patients had somatic VHL variants and detailed tumor data, were evaluated in genetics, and had no germline VHL variants. | 638 |
| Patients had somatic VHL variants and detailed tumor data, were not evaluated in genetics for any hereditary cancer, and had no germline sequencing data *. | 1901 |
| Sex and Age Distribution of Patients with the Germline VHL Variant | Numbers (%) |
|---|---|
| Female | 98 (74%) |
| Male | 35 (26%) |
| Median Age | 54 |
| Patient with VHL Component Tumor | 27 (20%) |
| Patient with no VHL Component Tumor | 106 (80%) |
| Patient with No Tumor or Cancer Diagnosis | 24 (18%) |
| Patient with Positive family History of VHL Diagnosis | 17 (13%) |
| Patient with Negative Family History of VHL Diagnosis | 114 (86%) |
| Patient with Unavailable Family History of VHL Diagnosis | 2 (2%) |
| VHL Tumor Type | VHL Tumors (%) | Females (%) | Males (%) | Mean Age | Patients with Family History of VHL Diagnosis |
|---|---|---|---|---|---|
| Hemangioblastomas | |||||
| CNS hemangioblastoma | 16 (12%) | 15 (12%) | 1 (1%) | 36.9 | 11 (9%) |
| Retinal hemangioblastoma | 11 (9%) | 9 (7%) | 2 (2%) | 32.3 | 8 (6%) |
| Renal lesions | |||||
| Multiple renal cysts | 13 (10%) | 11 (9%) | 2 (2%) | 32.9 | 10 (8%) |
| Renal cell carcinoma | 9 (7%) | 4 (3%) | 5 (4%) | 44.3 | 5 (4%) |
| Pheochromocytoma | 4 (3%) | 2 (2%) | 2 (2%) | 30.5 | 3 (2%) |
| Paraganglioma | 3 (2%) | 2 (2%) | 1 (1%) | 31 | 1 (1%) |
| Pancreatic lesions | |||||
| Pancreatic cysts | 14 (11%) | 11 (9%) | 3 (2%) | 30.6 | 10 |
| Neuroendocrine tumors of the pancreas | 1 (1%) | 1 (1%) | 0 | 54 | 0 |
| Endolymphatic sac tumors | 1 (1%) | 1 (1%) | 0 | 30 | 0 |
| Epididymal and broad ligament cystadenomas | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koeller, D.R.; Walker, M.; Unal, B.; Chittenden, A.; Levine, A.S.; Hayes, C.P.; Oramasionwu, P.C.; Manam, M.D.; Buehler, R.M.; Gomy, I.; et al. Advancing the Landscape of Clinical Actionability in Von Hippel–Lindau Syndrome: An Evidence-Based Framework from the INT2GRATE Oncology Consortium. Cancers 2025, 17, 2173. https://doi.org/10.3390/cancers17132173
Koeller DR, Walker M, Unal B, Chittenden A, Levine AS, Hayes CP, Oramasionwu PC, Manam MD, Buehler RM, Gomy I, et al. Advancing the Landscape of Clinical Actionability in Von Hippel–Lindau Syndrome: An Evidence-Based Framework from the INT2GRATE Oncology Consortium. Cancers. 2025; 17(13):2173. https://doi.org/10.3390/cancers17132173
Chicago/Turabian StyleKoeller, Diane R., McKenzie Walker, Busra Unal, Anu Chittenden, Alison Schwartz Levine, Connor P. Hayes, Paul C. Oramasionwu, Monica D. Manam, Ryan M. Buehler, Israel Gomy, and et al. 2025. "Advancing the Landscape of Clinical Actionability in Von Hippel–Lindau Syndrome: An Evidence-Based Framework from the INT2GRATE Oncology Consortium" Cancers 17, no. 13: 2173. https://doi.org/10.3390/cancers17132173
APA StyleKoeller, D. R., Walker, M., Unal, B., Chittenden, A., Levine, A. S., Hayes, C. P., Oramasionwu, P. C., Manam, M. D., Buehler, R. M., Gomy, I., Silva, W. A., Jr., Lerner-Ellis, J., Casalino, S., Mahajan, R., Watkins, N., Agaoglu, N. B., Manning, D. K., Barletta, J. A., Hornick, J. L., ... Ghazani, A. A. (2025). Advancing the Landscape of Clinical Actionability in Von Hippel–Lindau Syndrome: An Evidence-Based Framework from the INT2GRATE Oncology Consortium. Cancers, 17(13), 2173. https://doi.org/10.3390/cancers17132173