
Pilot Transcriptomic Profiling of Canine Oral Melanoma Reveals Conserved Oncogenic Pathways and Uncharacterized Molecular Signatures

 ,
,  ,
,  , ,
, ,
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection, Processing, and Bioinformatics Analysis
- Read alignment and gene-level quantification: Reads were aligned using Bowtie2 and the Rsubread package, and gene counts were obtained using the summarizeOverlaps function from the GenomicAlignments package. Differential expression analysis was conducted using DESeq2. Transcript-level assembly and quantification: Reads were aligned using HISAT, assembled into transcripts with StringTie, and quantified using Ballgown. The results were then contrasted using DESeq2 to assess differential expression.
- The rationale for using these two approaches was to ensure a comprehensive analysis of gene expression. The Bowtie2 pipeline provides a traditional read count-based approach, which is efficient for gene-level expression analysis. In contrast, the HISAT/StringTie/Ballgown pipeline allows for transcript reconstruction, facilitating the detection of novel isoforms and alternative splicing events. By integrating both a gene-level and a transcript-level approach, we aimed to enhance the robustness of our analysis, ensuring the detection of both differentially expressed genes and novel isoforms.
2.2. Gene Set Enrichment Analysis and Pathways Analysis (KEGG)
2.3. RT-qPCR
3. Results
3.1. Main Patient Characteristics
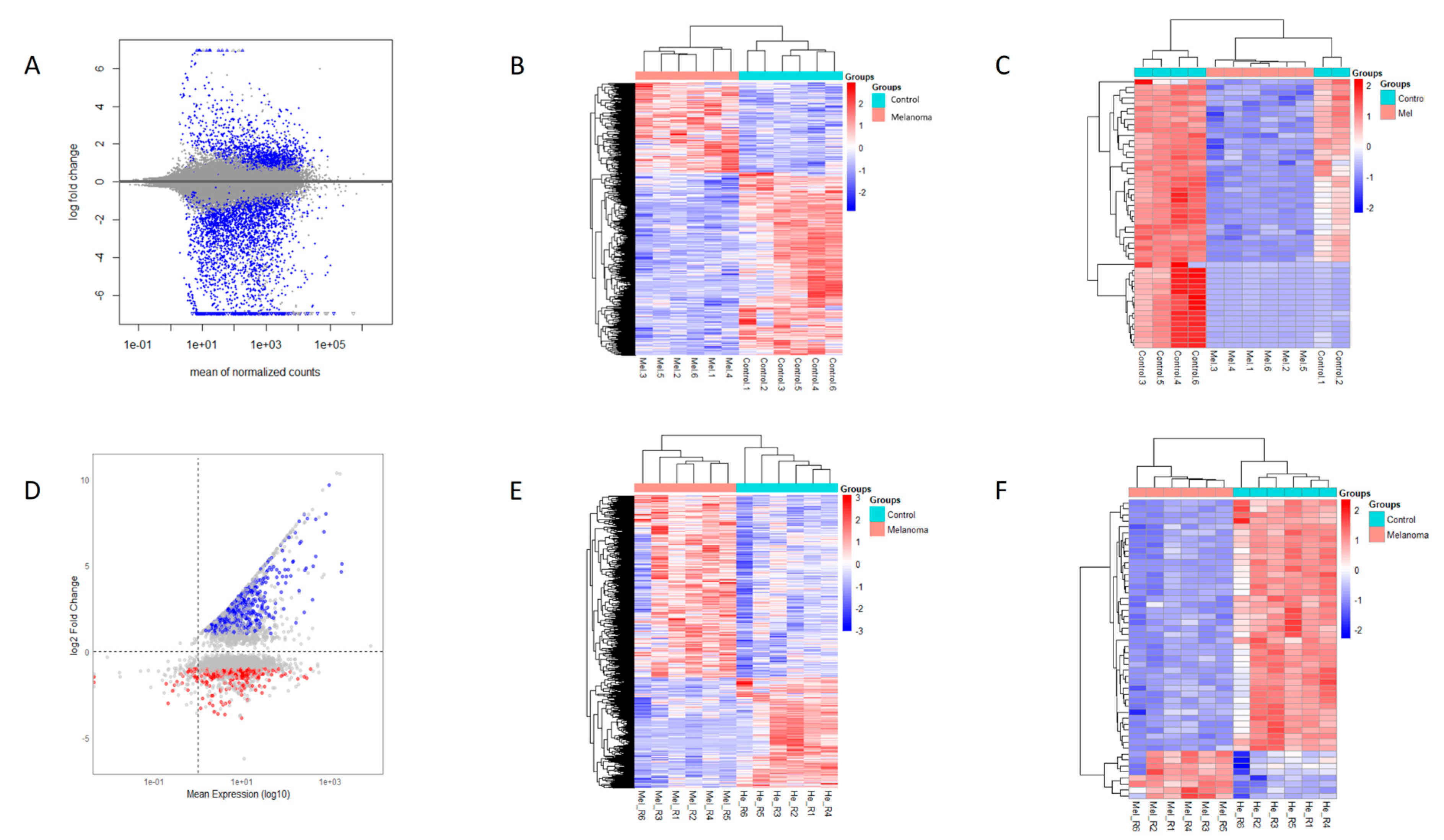
3.2. RNA-Seq Analysis
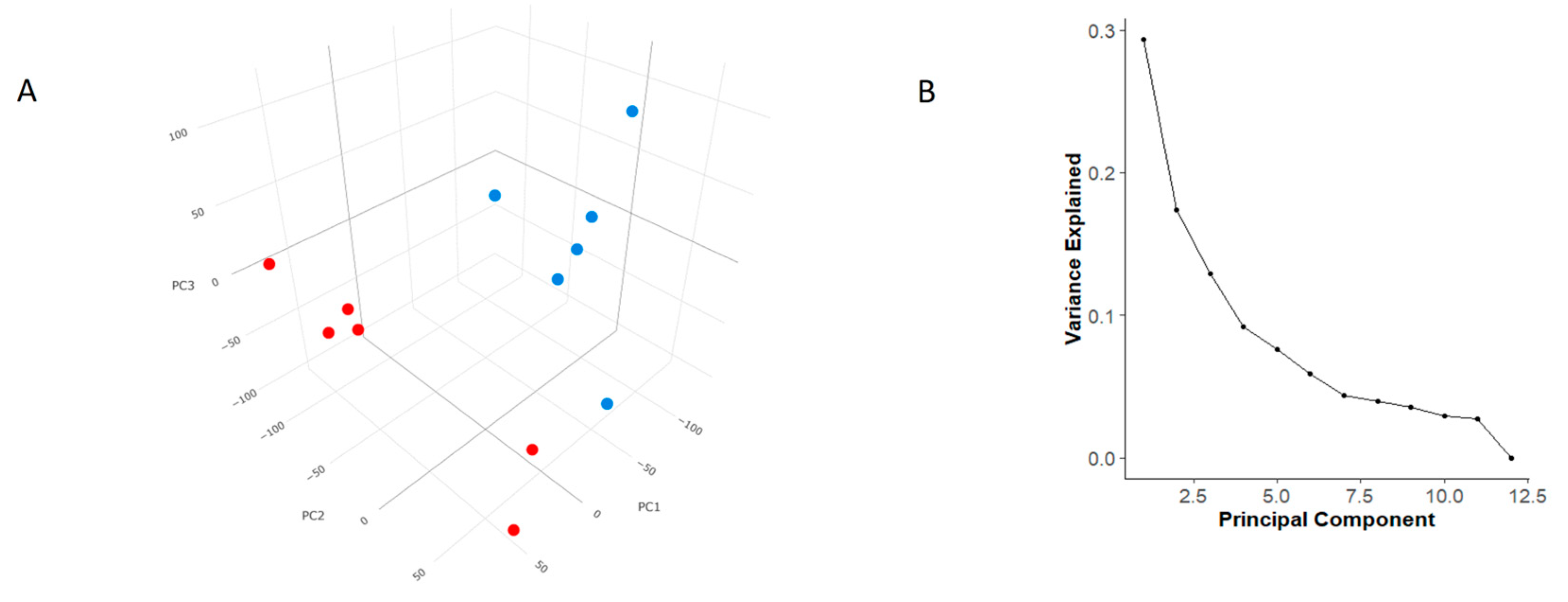
3.3. Principal Component Analysis (PCA)
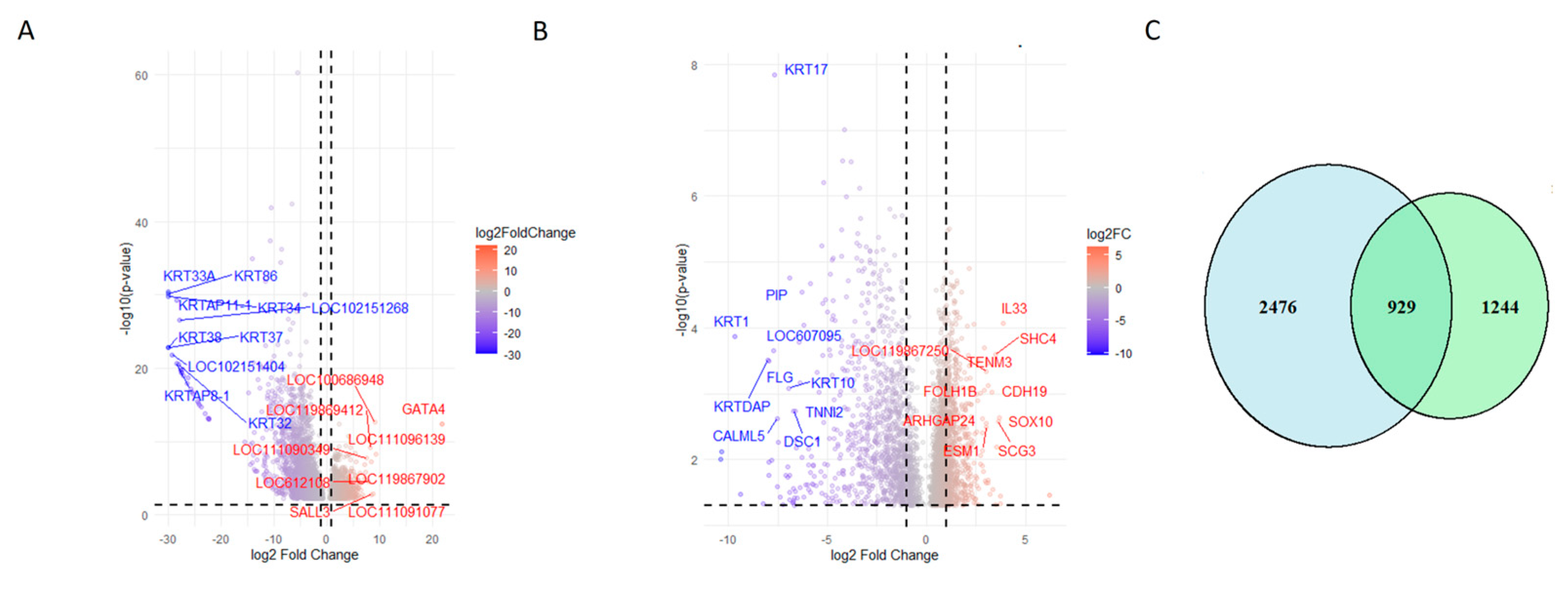
3.4. Differentially Expressed Genes (DEGs)
3.5. Comparison with HISAT-StringTie-Ballgown Pipeline
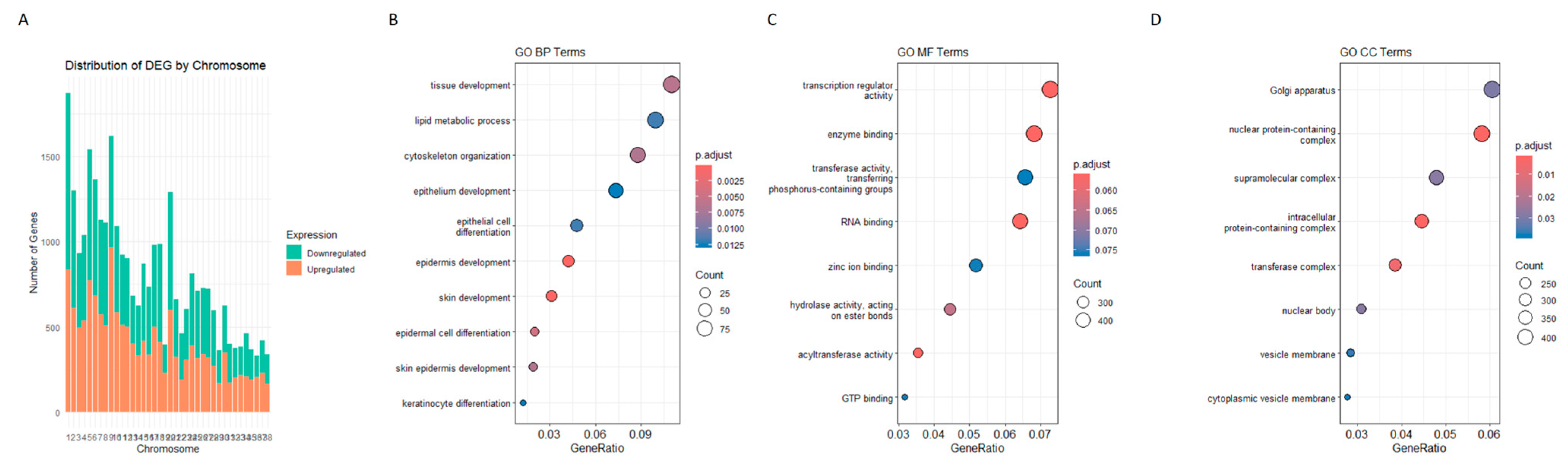
3.6. Chromosomal Distribution of DEGs
3.7. Human Oncogenes in the Dataset
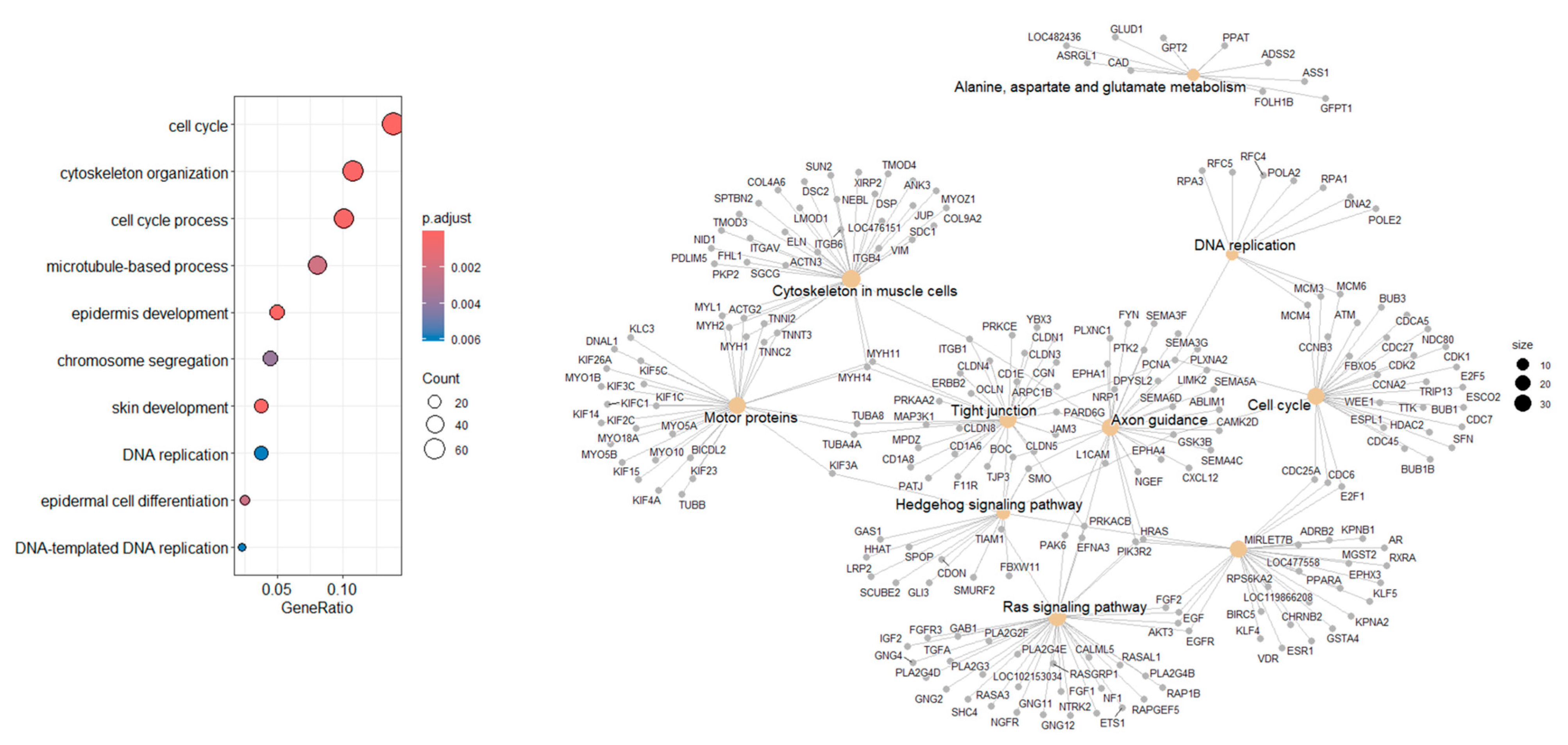
3.8. Go and KEGG Analysis
3.9. Isoform Analysis and DEGs Overlaps
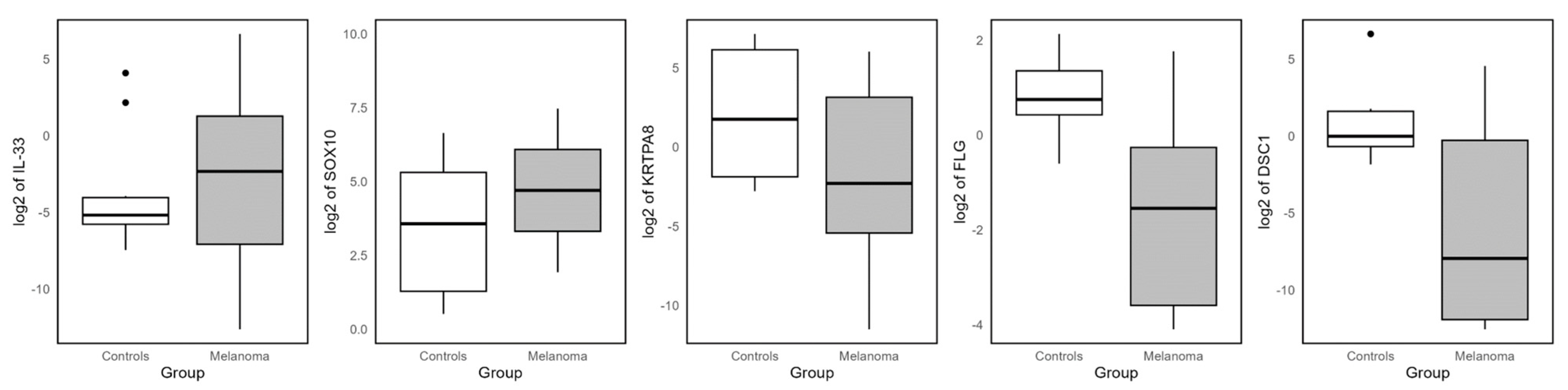
3.10. RT-PCR Determinations
4. Discussion
4.1. Uncharacterized Genes and Contextual Complexity
4.2. Upregulated Genes
4.3. Downregulated Genes
4.4. Ballgown Analysis Results
4.5. Comparison Between Methods
4.6. Comparison with Previous Studies
4.7. Chromosomal Distribution and Oncogenes
4.8. Gene Ontology and Pathway Analysis
4.9. Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palma, S.D.; McConnell, A.; Verganti, S.; Starkey, M. Review on Canine Oral Melanoma: An Undervalued Authentic Genetic Model of Human Oral Melanoma? Vet. Pathol. 2021, 58, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Polton, G.; Borrego, J.F.; Clemente-Vicario, F.; Clifford, C.A.; Jagielski, D.; Kessler, M.; Kobayashi, T.; Lanore, D.; Queiroga, F.L.; Rowe, A.T.; et al. Melanoma of the Dog and Cat: Consensus and Guidelines. Front. Vet. Sci. 2024, 11, 1359426. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.; Massoco, C.; Felizzola, C.; Perlmann, E.; Batschinski, K.; Tedardi, M.; Garcia, J.; Mendonça, P.; Teixeira, T.; Zaidan Dagli, M. Comparative Aspects of Canine Melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef]
- Williams, L.E.; Packer, R.A. Association between Lymph Node Size and Metastasis in Dogs with Oral Malignant Melanoma: 100 Cases (1987–2001). J. Am. Vet. Med. Assoc. 2003, 222, 1234–1236. [Google Scholar] [CrossRef]
- Shearin, A.L.; Ostrander, E.A. Leading the Way: Canine Models of Genomics and Disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.-H.; Zhang, X.-Q.; et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes 2021, 12, 847. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome Sequence, Comparative Analysis and Haplotype Structure of the Domestic Dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Yan, G.-R.; He, Q.-Y. DOSE: An R/Bioconductor Package for Disease Ontology Semantic and Enrichment Analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of Mutations in BRAF, NRAS, and KIT in Different Populations and Histological Subtypes of Melanoma: A Systemic Review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef]
- Cronin, J.C.; Wunderlich, J.; Loftus, S.K.; Prickett, T.D.; Wei, X.; Ridd, K.; Vemula, S.; Burrell, A.S.; Agrawal, N.S.; Lin, J.C.; et al. Frequent Mutations in the MITF Pathway in Melanoma. Pigment Cell Melanoma Res. 2009, 22, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Prouteau, A.; Mottier, S.; Primot, A.; Cadieu, E.; Bachelot, L.; Botherel, N.; Cabillic, F.; Houel, A.; Cornevin, L.; Kergal, C.; et al. Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets. Cancers 2022, 14, 276. [Google Scholar] [CrossRef] [PubMed]
- Tawa, G.J.; Braisted, J.; Gerhold, D.; Grewal, G.; Mazcko, C.; Breen, M.; Sittampalam, G.; LeBlanc, A.K. Transcriptomic Profiling in Canines and Humans Reveals Cancer Specific Gene Modules and Biological Mechanisms Common to Both Species. PLoS Comput. Biol. 2021, 17, e1009450. [Google Scholar] [CrossRef]
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Kato, D.; Nakagawa, T.; Arif, M.; Miura, N. Hypoxia-Related Y RNA Fragments as a Novel Potential Biomarker for Distinguishing Metastatic Oral Melanoma from Non-Metastatic Oral Melanoma in Dogs. Vet. Q. 2024, 44, 1–8. [Google Scholar] [CrossRef]
- Husna, A.A.; Rahman, M.M.; Lai, Y.; Chen, H.; Hasan, M.N.; Nakagawa, T.; Miura, N. Identification of Melanoma-specific Exosomal MiRNAs as the Potential Biomarker for Canine Oral Melanoma. Pigment Cell Melanoma Res. 2021, 34, 1062–1073. [Google Scholar] [CrossRef]
- Hino, Y.; Rahman, M.M.; Lai, Y.-C.; Husna, A.A.; Chen, H.; Hasan, M.N.; Nakagawa, T.; Miura, N. Hypoxic MiRNAs Expression Are Different between Primary and Metastatic Melanoma Cells. Gene 2021, 782, 145552. [Google Scholar] [CrossRef]
- Husna, A.A.; Rahman, M.M.; Chen, H.; Hasan, M.N.; Nakagawa, T.; Miura, N. Long non-coding RNA and Transfer RNA-derived Small Fragments in Exosomes Are Potential Biomarkers for Canine Oral Melanoma. Vet. Comp. Oncol. 2022, 20, 653–663. [Google Scholar] [CrossRef]
- Ishizaki, T.; Yamazaki, J.; Jelinek, J.; Aoshima, K.; Kimura, T. Genome-Wide DNA Methylation Analysis Identifies Promoter Hypermethylation in Canine Malignant Melanoma. Res. Vet. Sci. 2020, 132, 521–526. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lai, Y.; Husna, A.; Chen, H.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Transcriptome Analysis of Dog Oral Melanoma and Its Oncogenic Analogy with Human Melanoma. Oncol. Rep. 2019, 42, 55–70. [Google Scholar] [CrossRef]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential Role of GATA-4 in Cell Survival and Drug-Induced Cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, C.; Quiroz, A.; Benítez-Riquelme, D.; Riffo, E.; Castro, A.F.; Pincheira, R. SALL Proteins; Common and Antagonistic Roles in Cancer. Cancers 2021, 13, 6292. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Brodesser, D.; Reifinger, M.; Forte, S.; Semp, P.; Eberspächer-Schweda, M.C.; Wolschek, M.; Brandt, S.; Kleiter, M.; Pratscher, B. Canine Oral Primary Melanoma Cells Exhibit Shift to Mesenchymal Phenotype and Phagocytic Behaviour. Vet. Comp. Oncol. 2019, 17, 211–220. [Google Scholar] [CrossRef]
- Takan, I.; Karakülah, G.; Louka, A.; Pavlopoulou, A. “In the Light of Evolution:” Keratins as Exceptional Tumor Biomarkers. PeerJ 2023, 11, e15099. [Google Scholar] [CrossRef]
- Dmello, C.; Srivastava, S.S.; Tiwari, R.; Chaudhari, P.R.; Sawant, S.; Vaidya, M.M. Multifaceted Role of Keratins in Epithelial Cell Differentiation and Transformation. J. Biosci. 2019, 44, 33. [Google Scholar] [CrossRef]
- Han, W.; Hu, C.; Fan, Z.-J.; Shen, G.-L. Transcript Levels of Keratin 1/5/6/14/15/16/17 as Potential Prognostic Indicators in Melanoma Patients. Sci. Rep. 2021, 11, 1023. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in Health and Disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.; Wickström, S.L.; Gao, J.; He, X.; Jing, X.; Wu, J.; Du, Q.; Yang, M.; Chen, Y.; et al. Interleukin-33 Is a Novel Immunosuppressor That Protects Cancer Cells from TIL Killing by a Macrophage-Mediated Shedding Mechanism. Adv. Sci. 2021, 8. [Google Scholar] [CrossRef]
- Peng, L.; Sun, W.; Wei, F.; Chen, L.; Wen, W. Interleukin-33 Modulates Immune Responses in Cutaneous Melanoma in a Context-Specific Way. Aging 2021, 13, 6740–6751. [Google Scholar] [CrossRef]
- Hatzioannou, A.; Banos, A.; Sakelaropoulos, T.; Fedonidis, C.; Vidali, M.-S.; Köhne, M.; Händler, K.; Boon, L.; Henriques, A.; Koliaraki, V.; et al. An Intrinsic Role of IL-33 in Treg Cell–Mediated Tumor Immunoevasion. Nat. Immunol. 2020, 21, 75–85. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Thiravialingam, A.; Sriganeshan, K.; Gonzalez, J.; Alvarez, V.; Ocejo, S.; Abreu, A.R.; Avellan, R.; Arzola, A.H.; Hachem, S.; et al. Clinical Significance of SOX10 Expression in Human Pathology. Curr. Issues Mol. Biol. 2023, 45, 10131–10158. [Google Scholar] [CrossRef] [PubMed]
- Elgash, M.; Young, J.; White, K.; Leitenberger, J.; Bar, A. An Update and Review of Clinical Outcomes Using Immunohistochemical Stains in Mohs Micrographic Surgery for Melanoma. Dermatol. Surg. 2024, 50, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.L.; Baxter, L.L.; Loftus, S.K.; Pavan, W.J. Sox Proteins in Melanocyte Development and Melanoma. Pigment Cell Melanoma Res. 2010, 23, 496–513. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Wong, K.K.; Rostomily, R.; Wong, S.T.C. Prognostic Gene Discovery in Glioblastoma Patients Using Deep Learning. Cancers 2019, 11, 53. [Google Scholar] [CrossRef]
- Sun, M.; Sun, T.; He, Z.; Xiong, B. Identification of Two Novel Biomarkers of Rectal Carcinoma Progression and Prognosis via Co-Expression Network Analysis. Oncotarget 2017, 8, 69594–69609. [Google Scholar] [CrossRef]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12. [Google Scholar] [CrossRef]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK Pathway in Melanoma: Why Some Approaches Succeed and Other Fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef]
- Miñoza, J.M.A.; Rico, J.A.; Zamora, P.R.F.; Bacolod, M.; Laubenbacher, R.; Dumancas, G.G.; de Castro, R. Biomarker Discovery for Meta-Classification of Melanoma Metastatic Progression Using Transfer Learning. Genes 2022, 13, 2303. [Google Scholar] [CrossRef]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the Frontline: Role in Skin Barrier Function and Disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Andersen, Y.M.F.; Balslev, E.; Szecsi, P.B.; Stender, S.; Kaae, J.; Linneberg, A.; Skov, L. Loss-of-function Mutations in Filaggrin Gene and Malignant Melanoma: A Case–Control Study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A Census of Human Transcription Factors: Function, Expression and Evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Geng, H.; Ji, L.; Ma, X.; Yin, Q.; Xiong, H. ESM-1: A Novel Tumor Biomaker and Its Research Advances. Anticancer Agents Med. Chem. 2019, 19, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.S.; Denton, C.L.; Gustafson, D.L. Comparative Analysis of MAPK and PI3K/AKT Pathway Activation and Inhibition in Human and Canine Melanoma. Vet. Comp. Oncol. 2015, 13, 288–304. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Pisamai, S.; Edwards, S.W.; Cheng, C.W.; Chaivichit, P.; Sooksiri, M.; Yanakam, S.; Maneewong, S.; Suriyaphol, G. Tissue Transcriptome Profiling and Pathway Analyses Revealed Novel Potential Biomarkers in the Tumor Progression of Canine Oral Melanoma. Res. Vet. Sci. 2023, 165. [Google Scholar] [CrossRef]
- Ploypetch, S.; Roytrakul, S.; Jaresitthikunchai, J.; Phaonakrop, N.; Teewasutrakul, P.; Rungsipipat, A.; Suriyaphol, G. Salivary Proteomics in Monitoring the Therapeutic Response of Canine Oral Melanoma. PLoS ONE 2021, 16, e0256167. [Google Scholar] [CrossRef]
- Kawakami, A.; Fisher, D.E. The Master Role of Microphthalmia-Associated Transcription Factor in Melanocyte and Melanoma Biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef]
- Brocca, G.; Ferraresso, S.; Zamboni, C.; Martinez-Merlo, E.M.; Ferro, S.; Goldschmidt, M.H.; Castagnaro, M. Array Comparative Genomic Hybridization Analysis Reveals Significantly Enriched Pathways in Canine Oral Melanoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Ploypetch, S.; Luo, X.; Zhao, S.; Roytrakul, S.; Li, L.; Suriyaphol, G. Salivary Metabolomic Identification of Biomarker Candidates for Oral Melanoma and Oral Squamous Cell Carcinoma in Dogs. J. Vet. Intern. Med. 2024, 38, 2293–2304. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age | Breed | Tumor Location | Size | TNM |
|---|---|---|---|---|---|---|
| Control_1 | Male | 6 | American Stafford | - | - | - |
| Control_2 | Female | 5 | Mix Stafford | - | - | - |
| Control_3 | Female | 9 | Pitbull | - | - | - |
| Control_4 | Female | 8 | Pitbull | - | - | - |
| Control_5 | Male | 11 | Mix Pitbull | - | - | - |
| Control_6 | Female | 5 | Mix Stafford | - | - | - |
| Melanoma_1 | Female | 10 | American Staffordshire Terrier | Lip | 2 cm | Stage II |
| Melanoma_2 | Female | 10 | Chihuahua | Mandible | 3.5 cm | Stage III |
| Melanoma_3 | Male | 8 | Canary Mastiff | Cheek | 7.7 cm | Stage III |
| Melanoma_4 | Male | 14 | Yorkshire Terrier | Lip | 2.1 cm | Stage II |
| Melanoma_5 | Female | 12 | Mixed | Lip | 1.5 cm | Stage I |
| Melanoma_6 | Male | 12 | Schnauzer | Maxilla | 3.8 cm | Stage III |
| Gene Name | Chromosome | Start Position | End Position | Mean Control | Mean Melanoma |
|---|---|---|---|---|---|
| ABCC11 | 2 | 67,365,969 | 67,434,568 | 1,206,093,988 | 513,458,128 |
| ATRNL1 | 28 | 26,352,303 | 27,138,636 | 8,453,868,178 | 107,012,498 |
| CASZ1 | 2 | 85,844,387 | 85,966,963 | 1,145,693,802 | 5,715,992,023 |
| CEP170 | 7 | 34,091,156 | 34,222,655 | 1,065,593,156 | 1,303,246,121 |
| CES1 | 2 | 60,780,757 | 60,850,933 | 1,199,658,343 | 4,492,333,331 |
| COL17A1 | 28 | 16,828,097 | 16,880,431 | 144,398,744 | 867,508,665 |
| DSC1 | 7 | 58,308,188 | 58,387,461 | 1,286,131,338 | 4,125,987,355 |
| ETV1 | 14 | 28,575,516 | 28,670,323 | 8,799,008,458 | 111,593,121 |
| FAT2 | 4 | 58,185,984 | 58,263,273 | 126,269,534 | 8,691,156,552 |
| FOLH1B | 21 | 10,581,865 | 10,647,452 | 8,524,754,322 | 1,171,190,646 |
| GATA4 | 25 | 26,280,510 | 26,337,654 | 3,825,626,408 | 4,758,170,722 |
| IL22RA1 | 2 | 76,014,607 | 76,034,628 | 951,117,109 | 4,381,898,873 |
| IL33 | 11 | 28,069,663 | 28,108,194 | 967,667,444 | 1,412,622,417 |
| KRT34 | 9 | 22,098,153 | 22,101,234 | 9,432,768,672 | 3,825,626,408 |
| KRT86 | 27 | 2,656,611 | 2,826,996 | 1,003,341,814 | 3,825,626,408 |
| KRTAP11-1 | 31 | 25,995,174 | 25,995,635 | 838,372,259 | 3,825,626,408 |
| MMP16 | 29 | 34,120,818 | 34,403,822 | 8,285,952,144 | 1,029,971,631 |
| MYEF2 | 30 | 14,532,118 | 14,564,995 | 9,328,405,867 | 1,145,666,304 |
| PCSK1N | X | 42,166,878 | 42,172,740 | 4,946,786,732 | 7,722,763,719 |
| POU2F3 | 5 | 13,632,917 | 13,707,369 | 9,638,707,384 | 4,926,011,945 |
| PRKACB | 6 | 64,155,572 | 64,269,867 | 1,075,963,493 | 1,234,943,757 |
| SBSN | 1 | 117,733,283 | 117,738,080 | 1,294,205,254 | 6,188,281,965 |
| SDSL | 26 | 10,990,071 | 11,015,079 | 4,681,131,408 | 7,338,404,409 |
| SHC4 | 30 | 15,185,863 | 15,322,777 | 8,292,860,068 | 1,262,090,089 |
| SKA3 | 25 | 17,118,211 | 17,138,178 | 7,694,448,604 | 9,481,288,937 |
| SLF1 | 3 | 14,670,381 | 14,744,679 | 9,059,277,209 | 1,07,288,861 |
| TENM3 | 16 | 49,667,658 | 50,270,179 | 929,343,987 | 1,298,114,629 |
| TP63 | 34 | 21,700,663 | 21,918,660 | 1,302,620,425 | 7,773,881,578 |
| Gene | Description | Log2 Fold Change | p-Value Adjusted |
|---|---|---|---|
| GATA4 | GATA binding protein 4 | 2,178,905,058 | 4.79 × 10−11 |
| LOC111096139 | Uncharacterized | 9,282,033,508 | 3.05 × 10−7 |
| LOC100686948 | Serine/arginine-rich splicing factor SR45-like | 9,089,771,615 | 3.11 × 10−11 |
| LOC111091077 | Uncharacterized | 8,810,707,393 | 0.016373206 |
| SALL3 | Spalt-like transcription factor 3 | 844,175,455 | 0.017528697 |
| LOC119867902 | Uncharacterized | 8,421,755,882 | 2.86 × 10−6 |
| LOC119869412 | Uncharacterized | 8,300,275,074 | 4.87 × 10−8 |
| LOC612108 | Uncharacterized | 7,933,603,842 | 0.000639468 |
| LOC111090349 | Uncharacterized | 7,525,602,976 | 9.04 × 10−7 |
| CDH18 | Cadherin 18 | 7,448,216,395 | 5.22 × 10−5 |
| KRT33A * | Keratin 33A | −30 | 1.07 × 10−27 |
| KRT86 * | Keratin 86 | −30 | 2.10 × 10−27 |
| KRT34 * | Keratin 34 | −30 | 3.76 × 10−27 |
| KRT37 * | Keratin 37 | −30 | 1.77 × 10−20 |
| KRT38 | Keratin 38 | −30 | 1.77 × 10−20 |
| LOC102151404 * | Keratin, high-sulfur matrix protein, B2A-like | −2,933,651,986 | 1.43 × 10−19 |
| KRTAP11-1 * | Keratin-associated protein 11-1 | −285,367,828 | 1.30 × 10−26 |
| KRTAP8-1 * | Keratin-associated protein 8-1 | −2,853,215,131 | 1.70 × 10−18 |
| KRT32 * | Keratin 32 | −2,809,598,496 | 2.72 × 10−18 |
| LOC102151268 * | Keratin-associated protein 1-3-like | −2,801,190,653 | 4.40 × 10−24 |
| Gene | Description | Log Fold Change | q-Value # |
|---|---|---|---|
| IL33 * | Interleukin 33 | 4.0 × 1014 | 0.008 |
| SOX10 | SRY-box transcription factor 10 | 4.0 × 1014 | 0.041 |
| SCG3 | Secretogranin III | 4.0 × 1014 | 0.044 |
| SHC4 | SHC adaptor protein 4 | 3.0 × 1014 | 0.015 |
| CDH19 | Cadherin 19 | 3.0 × 1014 | 0.027 |
| TENM3 | Teneurin transmembrane protein 3 | 3.0 × 1014 | 0.017 |
| FOLH1B | Folate hydrolase 1B | 3.0 × 1014 | 0.024 |
| ESM1 * | Endothelial cell-specific molecule 1 | 3.0 × 1014 | 0.048 |
| LOC119867250 | U6 spliceosomal RNA | 3.0 × 1014 | 0.019 |
| ARHGAP24 | Rho GTPase activating protein 24 | 3.0 × 1014 | 0.046 |
| KRT1 * | Keratin 1 | −9.680895 | 0.010 |
| KRTDAP * | Keratinocyte differentiation associated protein | −8.004239 | 0.016 |
| FLG * | Filaggrin | −7.937734 | 0.016 |
| LOC607095 * | Lysine-rich arabinogalactan protein 19-like | −7.715431 | 0.013 |
| KRT17 * | Keratin 17 | −7.653794 | 0.0001 |
| CALML5 * | Calmodulin-like 5 | −7.545614 | 0.041 |
| KRT10 * | Keratin 10 | −6.935247 | 0.024 |
| PIP * | Prolactin-induced protein | −6.912647 | 0.004 |
| DSC1 * | Desmocollin 1 | −6.702341 | 0.037 |
| TNNI2 * | Troponin I2, fast skeletal type | −6.613600 | 0.037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Santana, C.G.; Rodríguez-Esparragón, F.; Cazorla-Rivero, S.E.; Jiménez-Alonso, A.A.; Clavo, B.; González-Martín, J.M.; Cánovas-Molina, Á.; Bartolomé, C.; Estupiñán, L.; Rodríguez Grau-Bassas, E. Pilot Transcriptomic Profiling of Canine Oral Melanoma Reveals Conserved Oncogenic Pathways and Uncharacterized Molecular Signatures. Cancers 2025, 17, 2106. https://doi.org/10.3390/cancers17132106
Pérez-Santana CG, Rodríguez-Esparragón F, Cazorla-Rivero SE, Jiménez-Alonso AA, Clavo B, González-Martín JM, Cánovas-Molina Á, Bartolomé C, Estupiñán L, Rodríguez Grau-Bassas E. Pilot Transcriptomic Profiling of Canine Oral Melanoma Reveals Conserved Oncogenic Pathways and Uncharacterized Molecular Signatures. Cancers. 2025; 17(13):2106. https://doi.org/10.3390/cancers17132106
Chicago/Turabian StylePérez-Santana, Carmen G., Francisco Rodríguez-Esparragón, Sara E. Cazorla-Rivero, Ana A. Jiménez-Alonso, Bernardino Clavo, Jesús M. González-Martín, Ángeles Cánovas-Molina, Carmen Bartolomé, Lidia Estupiñán, and Enrique Rodríguez Grau-Bassas. 2025. "Pilot Transcriptomic Profiling of Canine Oral Melanoma Reveals Conserved Oncogenic Pathways and Uncharacterized Molecular Signatures" Cancers 17, no. 13: 2106. https://doi.org/10.3390/cancers17132106
APA StylePérez-Santana, C. G., Rodríguez-Esparragón, F., Cazorla-Rivero, S. E., Jiménez-Alonso, A. A., Clavo, B., González-Martín, J. M., Cánovas-Molina, Á., Bartolomé, C., Estupiñán, L., & Rodríguez Grau-Bassas, E. (2025). Pilot Transcriptomic Profiling of Canine Oral Melanoma Reveals Conserved Oncogenic Pathways and Uncharacterized Molecular Signatures. Cancers, 17(13), 2106. https://doi.org/10.3390/cancers17132106