
Role of Vascular Liver Diseases in Hepatocellular Carcinoma Development

 ,
,  ,
,
Simple Summary
Abstract

1. Introduction
2. Budd–Chiari Syndrome and Hepatocellular Carcinoma
3. Fontan-Associated Liver Disease and Hepatocellular Carcinoma
4. Congenital Porto-Systemic Shunts and Hepatocellular Carcinoma
5. Cavernous Transformation of the Portal Vein and Hepatocellular Carcinoma
6. Porto-Sinusoidal Vascular Disorder and Hepatocellular Carcinoma
7. Treatment Strategy of Hepatocellular Carcinoma in Vascular Liver Disease
8. Discussion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sangro, B.; Argemi, J.; Ronot, M.; Paradis, V.; Meyer, T.; Mazzaferro, V.; Jepsen, P.; Golfieri, R.; Galle, P.; Dawson, L.; et al. EASL Clinical Practice Guidelines on the Management of Hepatocellular Carcinoma. J. Hepatol. 2025, 82, 315–374. [Google Scholar] [CrossRef]
- Balogh, J.; Victor, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, M.; Monsour, H. Hepatocellular Carcinoma: A Review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef]
- Vilgrain, V.; Paradis, V.; Van Wettere, M.; Valla, D.; Ronot, M.; Rautou, P.-E. Benign and Malignant Hepatocellular Lesions in Patients with Vascular Liver Diseases. Abdom. Radiol. 2018, 43, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Sempoux, C.; Balabaud, C.; Paradis, V.; Bioulac-Sage, P. Hepatocellular Nodules in Vascular Liver Diseases. Virchows Arch. 2018, 473, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Vilgrain, V.; Rautou, P.-E.; Paradis, V.; Ronot, M. Benign and Malignant Hepatocellular Lesions in Patients with Vascular Liver Disease: Hepatocellular Lesions in Vascular Liver Disease. Clin. Liver Dis. 2014, 3, 122–125. [Google Scholar] [CrossRef]
- Valla, D.; Garcia-Pagan, J.C.; De Gottardi, A.; Rautou, P.-E. (Eds.) Vascular Disorders of the Liver: VALDIG’s Guide to Management and Causes; Springer International Publishing: Cham, Switzerland, 2022; ISBN 978-3-030-82987-2. [Google Scholar]
- EASL Clinical Practice Guidelines: Vascular Diseases of the Liver. J. Hepatol. 2016, 64, 179–202. [CrossRef]
- Valla, D.-C. Budd–Chiari Syndrome/Hepatic Venous Outflow Tract Obstruction. Hepatol. Int. 2018, 12, 168–180. [Google Scholar] [CrossRef]
- Prasad, D.; Nguyen, M.H. Epidemiology, Pathogenesis, Diagnosis, Surveillance, and Management of Hepatocellular Carcinoma Associated with Vascular Liver Disease. Kaohsiung J. Med. Sci. 2021, 37, 355–360. [Google Scholar] [CrossRef]
- Panvini, N.; Dioguardi Burgio, M.; Sartoris, R.; Maino, C.; Van Wettere, M.; Plessier, A.; Payancé, A.; Rautou, P.-E.; Ladouceur, M.; Vilgrain, V.; et al. MR Imaging Features and Long-Term Evolution of Benign Focal Liver Lesions in Budd-Chiari Syndrome and Fontan-Associated Liver Disease. Diagn. Interv. Imaging 2022, 103, 111–120. [Google Scholar] [CrossRef]
- Tanaka, M.; Wanless, I.R. Pathology of the Liver in Budd-Chiari Syndrome: Portal Vein Thrombosis and the Histogenesis of Veno-Centric Cirrhosis, Veno-Portal Cirrhosis, and Large Regenerative Nodules. Hepatology 1998, 27, 488–496. [Google Scholar] [CrossRef]
- Rizzetto, F.; Rutanni, D.; Carbonaro, L.A.; Vanzulli, A. Focal Liver Lesions in Budd-Chiari Syndrome: Spectrum of Imaging Findings. Diagnostics 2023, 13, 2346. [Google Scholar] [CrossRef]
- Paul, S.B.; Shalimar; Sreenivas, V.; Gamanagatti, S.R.; Sharma, H.; Dhamija, E.; Acharya, S.K. Incidence and Risk Factors of Hepatocellular Carcinoma in Patients with Hepatic Venous Outflow Tract Obstruction. Aliment. Pharmacol. Ther. 2015, 41, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhang, Q.; Xu, H.; Zu, M.; Gu, Y.; Ma, H.; Kang, W.; Ni, C. Clinical Characteristics and Risk Factors of Hepatocellular Carcinoma Development in Budd-Chiari Syndrome Patients after Endovascular Treatment. Dig. Liver Dis. 2025, 57, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Wester, A.; Åberg, F.; Rajani, R.; Hagström, H. Minimal Risk of Hepatocellular Carcinoma in Noncirrhotic Budd-Chiari Syndrome: A Three-Decade Population-Based Study. Clin. Gastroenterol. Hepatol. 2023, 21, 2689–2691.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-S.; Guo, S.; Chen, Y.-X.; Zhang, Z.-L. Budd-Chiari Syndrome and Its Associated Hepatocellular Carcinoma: Clinical Risk Factors and Potential Immunotherapeutic Benefit Analysis. Front. Oncol. 2022, 12, 1075685. [Google Scholar] [CrossRef]
- Agarwal, A.; Biswas, S.; Swaroop, S.; Aggarwal, A.; Agarwal, A.; Jain, G.; Elhence, A.; Vaidya, A.; Gupte, A.; Mohanka, R.; et al. Clinical Profile and Outcomes of Hepatocellular Carcinoma in Primary Budd-Chiari Syndrome. World J. Gastrointest. Oncol. 2024, 16, 699–715. [Google Scholar] [CrossRef]
- Moucari, R.; Rautou, P.-E.; Cazals-Hatem, D.; Geara, A.; Bureau, C.; Consigny, Y.; Francoz, C.; Denninger, M.-H.; Vilgrain, V.; Belghiti, J.; et al. Hepatocellular Carcinoma in Budd-Chiari Syndrome: Characteristics and Risk Factors. Gut 2008, 57, 828–835. [Google Scholar] [CrossRef]
- Park, H. Hepatocellular Carcinoma in Budd-Chiari Syndrome: A Single Center Experience with Long-Term Follow-up in South Korea. World J. Gastroenterol. 2012, 18, 1946. [Google Scholar] [CrossRef]
- Simonetto, D.A.; Yang, H.; Yin, M.; De Assuncao, T.M.; Kwon, J.H.; Hilscher, M.; Pan, S.; Yang, L.; Bi, Y.; Beyder, A.; et al. Chronic Passive Venous Congestion Drives Hepatic Fibrogenesis via Sinusoidal Thrombosis and Mechanical Forces. Hepatology 2015, 61, 648–659. [Google Scholar] [CrossRef]
- Cazals-Hatem, D.; Vilgrain, V.; Genin, P.; Denninger, M.-H.; Durand, F.; Belghiti, J.; Valla, D.; Degott, C. Arterial and Portal Circulation and Parenchymal Changes in Budd–Chiari Syndrome: A Study in 17 Explanted Livers. Hepatology 2003, 37, 510–519. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, H.; Zhang, X.; Xu, Z.; Jiang, Q.; Shen, Q.; Yu, M.; Li, K.; Jia, M. Clinical and Pathological Features and Surgical Treatment of Budd-Chiari Syndrome-Associated Hepatocellular Carcinoma. Chin. Med. J. (Engl.) 2013, 126, 3632–3638. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Chung, Y.-H.; Suh, D.D.; Shin, J.W.; Jang, M.K.; Ryu, S.H.; Park, N.H.; Lee, H.C.; Lee, Y.S.; Suh, D.J. Characteristic Clinical Features of Hepatocellular Carcinoma Associated with Budd???Chiari Syndrome: Evidence of Different Carcinogenic Process from Hepatitis B Virus-Associated Hepatocellular Carcinoma. Eur. J. Gastroenterol. Hepatol. 2004, 16, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Elsayes, K.M.; Kielar, A.Z.; Elmohr, M.M.; Chernyak, V.; Masch, W.R.; Furlan, A.; Marks, R.M.; Cruite, I.; Fowler, K.J.; Tang, A.; et al. White Paper of the Society of Abdominal Radiology Hepatocellular Carcinoma Diagnosis Disease-Focused Panel on LI-RADS V2018 for CT and MRI. Abdom. Radiol. 2018, 43, 2625–2642. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Walker, T.T.; Bove, K.; Veldtman, G. Fontan-Associated Liver Disease: A Review. J. Cardiol. 2019, 74, 223–232. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Kamath, P.S. Fontan-Associated Liver Disease. Clin. Liver Dis. 2023, 22, 130–133. [Google Scholar] [CrossRef]
- Kiesewetter, C.H.; Sheron, N.; Vettukattill, J.J.; Hacking, N.; Stedman, B.; Millward-Sadler, H.; Haw, M.; Cope, R.; Salmon, A.P.; Sivaprakasam, M.C.; et al. Hepatic Changes in the Failing Fontan Circulation. Heart 2007, 93, 579–584. [Google Scholar] [CrossRef]
- Navaratnam, D.; Fitzsimmons, S.; Grocott, M.; Rossiter, H.B.; Emmanuel, Y.; Diller, G.-P.; Gordon-Walker, T.; Jack, S.; Sheron, N.; Pappachan, J.; et al. Exercise-Induced Systemic Venous Hypertension in the Fontan Circulation. Am. J. Cardiol. 2016, 117, 1667–1671. [Google Scholar] [CrossRef]
- Lightsey, J.M.; Rockey, D.C. Current Concepts in Ischemic Hepatitis. Curr. Opin. Gastroenterol. 2017, 33, 158–163. [Google Scholar] [CrossRef]
- Seeto, R.K.; Fenn, B.; Rockey, D.C. Ischemic Hepatitis: Clinical Presentation and Pathogenesis. Am. J. Med. 2000, 109, 109–113. [Google Scholar] [CrossRef]
- Wanless, I.R.; Liu, J.J.; Butany, J. Role of Thrombosis in the Pathogenesis of Congestive Hepatic Fibrosis (Cardiac Cirrhosis). Hepatology 1995, 21, 1232–1237. [Google Scholar]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Severino, B.; Fiorentina, R.; Baldoni, M.; Caliendo, G.; Santagada, V.; Morelli, A.; Cirino, G. PAR1 Antagonism Protects against Experimental Liver Fibrosis. Role of Proteinase Receptors in Stellate Cell Activation. Hepatology 2004, 39, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Airola, C.; Pallozzi, M.; Cerrito, L.; Santopaolo, F.; Stella, L.; Gasbarrini, A.; Ponziani, F.R. Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications. Cells 2023, 12, 1712. [Google Scholar] [CrossRef] [PubMed]
- Wallihan, D.B.; Podberesky, D.J. Hepatic Pathology after Fontan Palliation: Spectrum of Imaging Findings. Pediatr. Radiol. 2013, 43, 330–338. [Google Scholar] [CrossRef]
- Bryant, T.; Ahmad, Z.; Millward-Sadler, H.; Burney, K.; Stedman, B.; Kendall, T.; Vettukattil, J.; Haw, M.; Salmon, A.P.; Cope, R.; et al. Arterialised Hepatic Nodules in the Fontan Circulation: Hepatico-Cardiac Interactions. Int. J. Cardiol. 2011, 151, 268–272. [Google Scholar] [CrossRef]
- Brancatelli, G.; Federle, M.P.; Grazioli, L.; Golfieri, R.; Lencioni, R. Benign Regenerative Nodules in Budd-Chiari Syndrome and Other Vascular Disorders of the Liver: Radiologic-Pathologic and Clinical Correlation. RadioGraphics 2002, 22, 847–862. [Google Scholar] [CrossRef]
- Téllez, L.; Payancé, A.; Tjwa, E.; Del Cerro, M.J.; Idorn, L.; Ovroutski, S.; De Bruyne, R.; Verkade, H.J.; De Rita, F.; De Lange, C.; et al. EASL-ERN Position Paper on Liver Involvement in Patients with Fontan-Type Circulation. J. Hepatol. 2023, 79, 1270–1301. [Google Scholar] [CrossRef]
- Sagawa, T.; Kogiso, T.; Sugiyama, H.; Hashimoto, E.; Yamamoto, M.; Tokushige, K. Characteristics of Hepatocellular Carcinoma Arising from Fontan-associated Liver Disease. Hepatol. Res. 2020, 50, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.G.; Iyengar, A.J.; Hardikar, W.; Sood, S.; d’Udekem, Y. Prevalence of Hepatocellular Carcinoma in the Entire Fontan Population of Australia and New Zealand. JTCVS Tech. 2020, 2, 128–130. [Google Scholar] [CrossRef]
- Hansen, S.; Gilroy, R.; Lindsay, I.; Doty, J.R.; Butschek, R.A.; Danford, C.J. A Meta-Analysis of Cumulative Incidence of Hepatocellular Carcinoma After the Fontan Operation. Dig. Dis. Sci. 2024, 69, 4467–4475. [Google Scholar] [CrossRef]
- Ghaferi, A.A.; Hutchins, G.M. Progression of Liver Pathology in Patients Undergoing the Fontan Procedure: Chronic Passive Congestion, Cardiac Cirrhosis, Hepatic Adenoma, and Hepatocellular Carcinoma. J. Thorac. Cardiovasc. Surg. 2005, 129, 1348–1352. [Google Scholar] [CrossRef]
- Yoon, J.S.; Lee, D.H.; Cho, E.J.; Song, M.K.; Choi, Y.H.; Kim, G.B.; Lee, Y.B.; Lee, J.-H.; Yu, S.J.; Kim, H.; et al. Risk of Liver Cirrhosis and Hepatocellular Carcinoma after Fontan Operation: A Need for Surveillance. Cancers 2020, 12, 1805. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez De Santiago, E.; Téllez, L.; Guerrero, A.; Albillos, A. Hepatocellular Carcinoma after Fontan Surgery: A Systematic Review. Hepatol. Res. 2021, 51, 116–134. [Google Scholar] [CrossRef]
- Axley, P.; Ahmed, Z.; Ravi, S.; Singal, A.K. Hepatitis C Virus and Hepatocellular Carcinoma: A Narrative Review. J. Clin. Transl. Hepatol. 2018, 6, 79–84. [Google Scholar] [CrossRef]
- Inuzuka, R.; Nii, M.; Inai, K.; Shimada, E.; Shinohara, T.; Kogiso, T.; Ono, H.; Otsuki, S.; Kurita, Y.; Takeda, A.; et al. Predictors of Liver Cirrhosis and Hepatocellular Carcinoma among Perioperative Survivors of the Fontan Operation. Heart 2023, 109, 276–282. [Google Scholar] [CrossRef]
- Sakamori, R.; Yamada, R.; Tahata, Y.; Kodama, T.; Hikita, H.; Tatsumi, T.; Yamada, T.; Takehara, T. The Absence of Warfarin Treatment and Situs Inversus Are Associated with the Occurrence of Hepatocellular Carcinoma after Fontan Surgery. J. Gastroenterol. 2022, 57, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Yang, H.K.; Jang, H.-J.; Yoo, S.-J.; Khalili, K.; Kim, T.K. Abdominal Imaging Findings in Adult Patients with Fontan Circulation. Insights Imaging 2018, 9, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Lee, K.H.; Khalili, K.; Jang, H.-J. Hepatocellular Nodules in Liver Cirrhosis: Contrast-Enhanced Ultrasound. Abdom. Imaging 2011, 36, 244–263. [Google Scholar] [CrossRef]
- Kim, T.K. Contrast-Enhanced Ultrasound in the Diagnosis of Nodules in Liver Cirrhosis. World J. Gastroenterol. 2014, 20, 3590. [Google Scholar] [CrossRef]
- Thrane, K.J.; Müller, L.S.O.; Suther, K.R.; Thomassen, K.S.; Holmström, H.; Thaulow, E.; Almaas, R.; Möller, T.; De Lange, C. Spectrum of Fontan-Associated Liver Disease Assessed by MRI and US in Young Adolescents. Abdom. Radiol. 2021, 46, 3205–3216. [Google Scholar] [CrossRef]
- Dillman, J.R.; Trout, A.T.; Alsaied, T.; Gupta, A.; Lubert, A.M. Imaging of Fontan-Associated Liver Disease. Pediatr. Radiol. 2020, 50, 1528–1541. [Google Scholar] [CrossRef]
- Zentner, D.; Celermajer, D.S.; Gentles, T.; d’Udekem, Y.; Ayer, J.; Blue, G.M.; Bridgman, C.; Burchill, L.; Cheung, M.; Cordina, R.; et al. Management of People With a Fontan Circulation: A Cardiac Society of Australia and New Zealand Position Statement. Heart Lung Circ. 2020, 29, 5–39. [Google Scholar] [CrossRef] [PubMed]
- Possner, M.; Gordon-Walker, T.; Egbe, A.C.; Poterucha, J.T.; Warnes, C.A.; Connolly, H.M.; Ginde, S.; Clift, P.; Kogon, B.; Book, W.M.; et al. Hepatocellular Carcinoma and the Fontan Circulation: Clinical Presentation and Outcomes. Int. J. Cardiol. 2021, 322, 142–148. [Google Scholar] [CrossRef]
- Sharma, R.; Suddle, A.; Quaglia, A.; Peddu, P.; Karani, J.; Satyadas, T.; Heaton, N. Congenital Extrahepatic Portosystemic Shunt Complicated by the Development of Hepatocellular Carcinoma. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Papamichail, M.; Pizanias, M.; Heaton, N. Congenital Portosystemic Venous Shunt. Eur. J. Pediatr. 2018, 177, 285–294. [Google Scholar] [CrossRef] [PubMed]
- McLin, V.A.; Franchi-Abella, S.; Brütsch, T.; Bahadori, A.; Casotti, V.; De Ville De Goyet, J.; Dumery, G.; Gonzales, E.; Guérin, F.; Hascoet, S.; et al. Expert Management of Congenital Portosystemic Shunts and Their Complications. JHEP Rep. 2024, 6, 100933. [Google Scholar] [CrossRef]
- Sokollik, C.; Bandsma, R.H.J.; Gana, J.C.; Van Den Heuvel, M.; Ling, S.C. Congenital Portosystemic Shunt: Characterization of a Multisystem Disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 675–681. [Google Scholar] [CrossRef]
- Franchi-Abella, S.; Branchereau, S.; Lambert, V.; Fabre, M.; Steimberg, C.; Losay, J.; Riou, J.; Pariente, D.; Gauthier, F.; Jacquemin, E.; et al. Complications of Congenital Portosystemic Shunts in Children: Therapeutic Options and Outcomes. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 322–330. [Google Scholar] [CrossRef]
- Tyraskis, A.; Deganello, A.; Sellars, M.; De Vito, C.; Thompson, R.; Quaglia, A.; Heaton, N.; Davenport, M. Portal Venous Deprivation in Patients with Portosystemic Shunts and Its Effect on Liver Tumors. J. Pediatr. Surg. 2020, 55, 651–654. [Google Scholar] [CrossRef]
- Tyraskis, A.; Davenport, M.; Deganello, A.; Sellars, M.; De Vito, C.; Kane, P.; Thompson, R.J.; Quaglia, A.; Heaton, N. Complications of Congenital Portosystemic Shunts: Liver Tumors Are Affected by Shunt Severity, but Pulmonary and Neurocognitive Associations Are Not. Hepatol. Int. 2022, 16, 918–925. [Google Scholar] [CrossRef]
- Lautz, T.B.; Shah, S.A.; Superina, R.A. Hepatoblastoma in Children With Congenital Portosystemic Shunts. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 542–545. [Google Scholar] [CrossRef]
- Kawano, S.; Hasegawa, S.; Urushihara, N.; Okazaki, T.; Yoshida, A.; Kusafuka, J.; Mimaya, J.; Horikoshi, Y.; Aoki, K.; Hamazaki, M. Hepatoblastoma with Congenital Absence of the Portal Vein—A Case Report. Eur. J. Pediatr. Surg. 2007, 17, 292–294. [Google Scholar] [CrossRef]
- Sanada, Y.; Mizuta, K.; Niki, T.; Tashiro, M.; Hirata, Y.; Okada, N.; Yamada, N.; Ihara, Y.; Urahashi, T.; Soejima, Y.; et al. Hepatocellular Nodules Resulting from Congenital Extrahepatic Portosystemic Shunts Can Differentiate into Potentially Malignant Hepatocellular Adenomas. J. Hepato-Biliary-Pancreat. Sci. 2015, 22, 746–756. [Google Scholar] [CrossRef]
- Lundstedt, C.; Lindell, G.; Tranberg, K.-G.; Svartholm, E. Congenital Absence of the Intrahepatic Portion of the Portal Vein in an Adult Male Resected for Hepatocellular Carcinoma. Eur. Radiol. 2001, 11, 2228–2231. [Google Scholar] [CrossRef]
- Pupulim, L.F.; Vullierme, M.-P.; Paradis, V.; Valla, D.; Terraz, S.; Vilgrain, V. Congenital Portosystemic Shunts Associated with Liver Tumours. Clin. Radiol. 2013, 68, e362–e369. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, L.; Alberti, D.; Olivetti, L.; Rigamonti, W.; Codazzi, F.; Matricardi, L.; Fugazzola, C.; Chiesa, A. Congenital Absence of Portal Vein with Nodular Regenerative Hyperplasia of the Liver. Eur. Radiol. 2000, 10, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Tajima, T.; Taguchi, K.; Kuroiwa, T.; Yoshimitsu, K.; Irie, H.; Aibe, H.; Shinozaki, K.; Shimada, M.; Masuda, K.; et al. Recent Developments in Imaging Diagnostics for HCC: CT Arteriography and CT Arterioportography Evaluation of Vascular Changes in Premalignant and Malignant Hepatic Nodules. J. Hepatobiliary. Pancreat. Surg. 2000, 7, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, U.; Foltys, D.; Otto, G. Focal Nodular Hyperplasia Proceeds Hepatocellular Carcinoma in an Adult with Congenital Absence of the Portal Vein: Letter to the Editors. Transpl. Int. 2012, 25, e67–e68. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype Correlation of CTNNB1 Mutations Reveals Different SS-catenin Activity Associated with Liver Tumor Progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef]
- Coilly, A.; Potier, P.; Broué, P.; Kounis, I.; Valla, D.; Hillaire, S.; Lambert, V.; Dutheil, D.; Hernández-Gea, V.; Plessier, A.; et al. Budd-Chiari Syndrome. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 420–425. [Google Scholar] [CrossRef]
- Qi, X.; Han, G.; Yin, Z.; He, C.; Guo, W.; Niu, J.; Wu, K.; Fan, D. Cavernous Vessels around a Patent Portal Trunk in the Liver Hilum. Abdom. Imaging 2012, 37, 422–430. [Google Scholar] [CrossRef]
- Vilgrain, V.; Condat, B.; Bureau, C.; Hakimé, A.; Plessier, A.; Cazals-Hatem, D.; Valla, D.C. Atrophy-Hypertrophy Complex in Patients with Cavernous Transformation of the Portal Vein: CT Evaluation. Radiology 2006, 241, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Harmanci, O. How Can Portal Vein Cavernous Transformation Cause Chronic Incomplete Biliary Obstruction? World J. Gastroenterol. 2012, 18, 3375. [Google Scholar] [CrossRef]
- Klopfenstein, K.J.; Grossman, N.J.; Fishbein, M.; Ruymann, F.B. Cavernous Transformation of the Portal Vein: A Cause of Thrombocytopenia and Splenomegaly. Clin. Pediatr. Phila. 2000, 39, 727–730. [Google Scholar] [CrossRef]
- Vasilescu, C.; Stanciulea, O.; Popa, M.; Colita, A.; Arion, C. Subtotal Laparoscopic Splenectomy and Esophagogastric Devascularization for the Thrombocytopenia Because of Portal Cavernoma—Case Report. J. Pediatr. Surg. 2008, 43, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Huang, Z.; Tang, C. Optimal Treatment for Patients With Cavernous Transformation of the Portal Vein. Front. Med. 2022, 9, 853138. [Google Scholar] [CrossRef] [PubMed]
- Ribera Cano, A.; Debette-Gratien, M.; Descottes, B.; Languepin, J. Hyperplasie nodulaire multifocale compliquant un cavernome porte. Arch. Pédiatr. 2007, 14, 1315–1317. [Google Scholar] [CrossRef]
- Bureau, C.; Péron, J.M.; Sirach, E.; Selves, J.; Otal, P.; Vinel, J.P. Liver Nodules Ressembling Focal Nodular Hyperplasia after Portal Vein Thrombosis. J. Hepatol. 2004, 41, 499–500. [Google Scholar] [CrossRef]
- Marin, D.; Galluzzo, A.; Plessier, A.; Brancatelli, G.; Valla, D.; Vilgrain, V. Focal Nodular Hyperplasia-like Lesions in Patients with Cavernous Transformation of the Portal Vein: Prevalence, MR Findings and Natural History. Eur. Radiol. 2011, 21, 2074–2082. [Google Scholar] [CrossRef]
- Amarapurkar, P.; Bhatt, N.; Patel, N.; Amarapurkar, D. Primary Extrahepatic Portal Vein Obstruction in Adults: A Single Center Experience. Indian J. Gastroenterol. 2014, 33, 19–22. [Google Scholar] [CrossRef]
- Attanasi, M.L.; Bou Daher, H.; Rockey, D.C. Natural History and Outcomes of Cavernous Transformation of the Portal Vein in Cirrhosis. Dig. Dis. Sci. 2023, 68, 3458–3466. [Google Scholar] [CrossRef]
- De Gottardi, A.; Rautou, P.-E.; Schouten, J.; Rubbia-Brandt, L.; Leebeek, F.; Trebicka, J.; Murad, S.D.; Vilgrain, V.; Hernandez-Gea, V.; Nery, F.; et al. Porto-Sinusoidal Vascular Disease: Proposal and Description of a Novel Entity. Lancet Gastroenterol. Hepatol. 2019, 4, 399–411. [Google Scholar] [CrossRef]
- De Gottardi, A.; Sempoux, C.; Berzigotti, A. Porto-Sinusoidal Vascular Disorder. J. Hepatol. 2022, 77, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Kage, M. Pathology of Idiopathic Non-Cirrhotic Portal Hypertension. Hepatol. Int. 2017, 11, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Giuli, L.; Pallozzi, M.; Venturini, G.; Gasbarrini, A.; Ponziani, F.R.; Santopaolo, F. Molecular Mechanisms Underlying Vascular Liver Diseases: Focus on Thrombosis. Int. J. Mol. Sci. 2023, 24, 12754. [Google Scholar] [CrossRef]
- Siramolpiwat, S.; Seijo, S.; Miquel, R.; Berzigotti, A.; Garcia-Criado, A.; Darnell, A.; Turon, F.; Hernandez-Gea, V.; Bosch, J.; Garcia-Pagán, J.C. Idiopathic Portal Hypertension: Natural History and Long-Term Outcome. Hepatology 2014, 59, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Glatard, A.-S.; Hillaire, S.; d’Assignies, G.; Cazals-Hatem, D.; Plessier, A.; Valla, D.C.; Vilgrain, V. Obliterative Portal Venopathy: Findings at CT Imaging. Radiology 2012, 263, 741–750. [Google Scholar] [CrossRef]
- Jin, S.J.; Choi, W.-M. Porto-Sinusoidal Vascular Disease: A Concise Updated Summary of Epidemiology, Pathophysiology, Imaging, Clinical Features, and Treatments. Korean J. Radiol. 2023, 24, 31. [Google Scholar] [CrossRef]
- Schouten, J.N.L.; Nevens, F.; Hansen, B.; Laleman, W.; Van Den Born, M.; Komuta, M.; Roskams, T.; Verheij, J.; Janssen, H.L.A. Idiopathic Noncirrhotic Portal Hypertension Is Associated with Poor Survival: Results of a Long-term Cohort Study. Aliment. Pharmacol. Ther. 2012, 35, 1424–1433. [Google Scholar] [CrossRef]
- Magaz, M.; Giudicelli-Lett, H.; Abraldes, J.G.; Nicoară-Farcău, O.; Turon, F.; Rajoriya, N.; Goel, A.; Raymenants, K.; Hillaire, S.; Téllez, L.; et al. Porto-Sinusoidal Vascular Liver Disorder with Portal Hypertension: Natural History and Long-Term Outcome. J. Hepatol. 2025, 82, 72–83. [Google Scholar] [CrossRef]
- Wöran, K.; Semmler, G.; Jachs, M.; Simbrunner, B.; Bauer, D.J.M.; Binter, T.; Pomej, K.; Stättermayer, A.F.; Schwabl, P.; Bucsics, T.; et al. Clinical Course of Porto-Sinusoidal Vascular Disease Is Distinct From Idiopathic Noncirrhotic Portal Hypertension. Clin. Gastroenterol. Hepatol. 2022, 20, e251–e266. [Google Scholar] [CrossRef]
- Isobe, Y.; Yamasaki, T.; Yokoyama, Y.; Kurokawa, F.; Hino, K.; Sakaida, I. Hepatocellular Carcinoma Developing Six and a Half Years after a Diagnosis of Idiopathic Portal Hypertension. J. Gastroenterol. 2007, 42, 407–409. [Google Scholar] [CrossRef] [PubMed]
- De Franchis, R.; Bosch, J.; Garcia-Tsao, G.; Reiberger, T.; Ripoll, C.; Abraldes, J.G.; Albillos, A.; Baiges, A.; Bajaj, J.; Bañares, R.; et al. Baveno VII—Renewing Consensus in Portal Hypertension. J. Hepatol. 2022, 76, 959–974. [Google Scholar] [CrossRef]
- Vitale, A.; Cabibbo, G.; Iavarone, M.; Viganò, L.; Pinato, D.J.; Ponziani, F.R.; Lai, Q.; Casadei-Gardini, A.; Celsa, C.; Galati, G.; et al. Personalised Management of Patients with Hepatocellular Carcinoma: A Multiparametric Therapeutic Hierarchy Concept. Lancet Oncol. 2023, 24, e312–e322. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-Y.; Wang, M.-Q.; Duan, F.; Fan, Q.-S.; Song, P.; Wang, Y. Hepatocellular Carcinoma Associated with Budd-Chiari Syndrome: Imaging Features and Transcatheter Arterial Chemoembolization. BMC Gastroenterol. 2013, 13, 105. [Google Scholar] [CrossRef]
- Asrani, S.K.; Warnes, C.A.; Kamath, P.S. Hepatocellular Carcinoma after the Fontan Procedure. N. Engl. J. Med. 2013, 368, 1756–1757. [Google Scholar] [CrossRef]
- Ben Khaled, N.; Möller, M.; Jochheim, L.S.; Leyh, C.; Ehmer, U.; Böttcher, K.; Pinter, M.; Balcar, L.; Scheiner, B.; Weich, A.; et al. Atezolizumab/Bevacizumab or Lenvatinib in Hepatocellular Carcinoma: Multicenter Real-World Study with Focus on Bleeding and Thromboembolic Events. JHEP Rep. 2024, 6, 101065. [Google Scholar] [CrossRef] [PubMed]
- Kinami, T.; Uchikawa, S.; Kawaoka, T.; Yamasaki, S.; Kosaka, M.; Johira, Y.; Yano, S.; Amioka, K.; Naruto, K.; Yamaoka, K.; et al. Efficacy and Safety of Atezolizumab plus Bevacizumab in Patients with Portal Hypertension for Unresectable Hepatocellular Carcinoma. Cancer Med. 2024, 13, e7025. [Google Scholar] [CrossRef]
- Niimura, T.; Goda, M.; Miyata, K.; Matsumoto, J.; Yoshioka, T.; Hamano, H.; Aizawa, F.; Yagi, K.; Izawa-Ishizawa, Y.; Zamami, Y.; et al. Evaluation of Cardiovascular Toxicity of the Atezolizumab and Bevacizumab Combination. Front. Drug Saf. Regul. 2023, 3, 1213771. [Google Scholar] [CrossRef]
- Itai, Y.; Matsui, O. Blood Flow and Liver Imaging. Radiology 1997, 202, 306–314. [Google Scholar] [CrossRef]
- Weinbren, K.; Washington, S.L.A. Hyperplastic Nodules after Portacaval Anastomosis in Rats. Nature 1976, 264, 440–442. [Google Scholar] [CrossRef]
- Rasenack, U. Changes in the liver and brain after portacaval and modified portacaval end-to-side anastomosis: Histology, autoradiography and clinical studies. Fortschr. Med. 1981, 99, 107–112. [Google Scholar] [PubMed]
- Gwon, D.; Ko, G.-Y.; Yoon, H.-K.; Sung, K.-B.; Kim, J.H.; Lee, S.S.; Lee, J.M.; Ohm, J.-Y.; Shin, J.H.; Song, H.-Y. Hepatocellular Carcinoma Associated with Membranous Obstruction of the Inferior Vena Cava: Incidence, Characteristics, and Risk Factors and Clinical Efficacy of TACE. Radiology 2010, 254, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Guérin, F.; Porras, J.; Fabre, M.; Guettier, C.; Pariente, D.; Bernard, O.; Gauthier, F. Liver Nodules after Portal Systemic Shunt Surgery for Extrahepatic Portal Vein Obstruction in Children. J. Pediatr. Surg. 2009, 44, 1337–1343. [Google Scholar] [CrossRef]
- Umetsu, S.E.; Joseph, N.M.; Cho, S.-J.; Morotti, R.; Deshpande, V.; Jain, D.; Kakar, S. Focal Nodular Hyperplasia–like Nodules Arising in the Setting of Hepatic Vascular Disorders with Portosystemic Shunting Show β-Catenin Activation. Hum. Pathol. 2023, 142, 20–26. [Google Scholar] [CrossRef]
- Kawai, H.; Osawa, Y.; Matsuda, M.; Tsunoda, T.; Yanagida, K.; Hishikawa, D.; Okawara, M.; Sakamoto, Y.; Shimagaki, T.; Tsutsui, Y.; et al. Sphingosine-1-phosphate Promotes Tumor Development and Liver Fibrosis in Mouse Model of Congestive Hepatopathy. Hepatology 2022, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, H.; Shan, W.; Zhou, J.; You, Y. Liver Sinusoidal Endothelial Cells in Hepatic Fibrosis: Opportunities for Future Strategies. Biochem. Biophys. Res. Commun. 2025, 766, 151881. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; DeLeve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef]
- Van Wettere, M.; Purcell, Y.; Bruno, O.; Payancé, A.; Plessier, A.; Rautou, P.-E.; Cazals-Hatem, D.; Valla, D.; Vilgrain, V.; Ronot, M. Low Specificity of Washout to Diagnose Hepatocellular Carcinoma in Nodules Showing Arterial Hyperenhancement in Patients with Budd-Chiari Syndrome. J. Hepatol. 2019, 70, 1123–1132. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Vascular Liver Disease | HCC Risk | Surveillance Recommendation | Guideline Availability |
|---|---|---|---|
| Budd–Chiari Syndrome | Moderate | Abdominal ultrasound every 6 months ± AFP | Practice guidelines [70] |
| Fontan-Associated Liver Disease | Moderate | Starting 10 years post-Fontan, CT/MRI at baseline, ultrasound and AFP every 6 months + CT/MRI every 1–2 years | Expert consensus [37] |
| Congenital Portosystemic Shunts | Low | Pre-shunt closure: imaging every 6 months Post-shunt closure: imaging every 3 to 6 months for 2 years, and yearly beyond that | Expert consensus [56] |
| Cavernous transformation of the portal vein | Very low | Consider ultrasound every 6 months in presence of liver disease | No specific guideline |
| Porto-Sinusoidal Vascular Disorder | Very low | Not currently recommended; consider ultrasound every 6 months for the risk of PVT development | No specific guideline |
| Vascular Liver Diseases | Definition | Types of Hepatic Lesions | HCC Frequency | Pathophysiological Mechanism |
|---|---|---|---|---|
BCS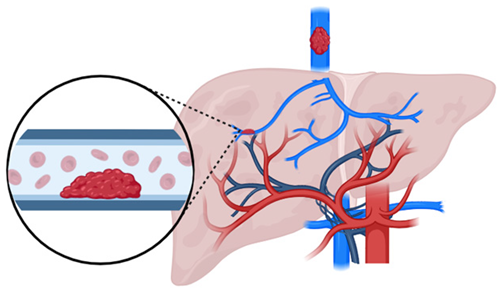 | Obstruction of hepatic venous outflow tract (between HVs and the junction IVC and the right atrium) |
| Frequent (cumulative incidence 0.3%, 4.7% and 7.7% after 1-, 3-, and 5-years [14]) | Hepatic congestion + centrilobular ischemia → ↑ compensatory arterial inflow + liver cirrhosis |
FALD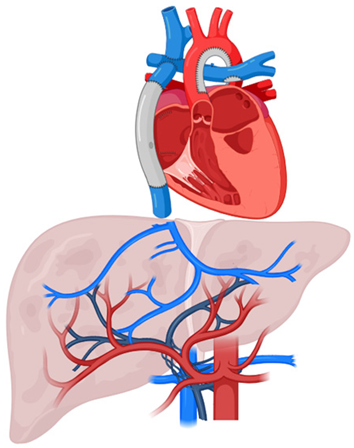 | Congestive hepatopathies caused by hemodynamic disturbances following Fontan surgery in patients with univentricular congenital heart disease |
| Moderately common (cumulative incidence 0%, 2% and 7% after 10-, 20- and 30 years, respectively, from surgery [40]; annual incidence 1.04% in pts with liver cirrhosis [42]) | Chronic increase in central venous pressure + ↓ hepatic drainage → hepatic congestion + hypoxia → ↑ compensatory arterial inflow + liver cirrhosis |
CPSSs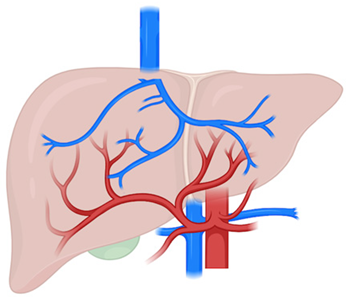 | Alterations in the communication between the PV and the systemic circulation |
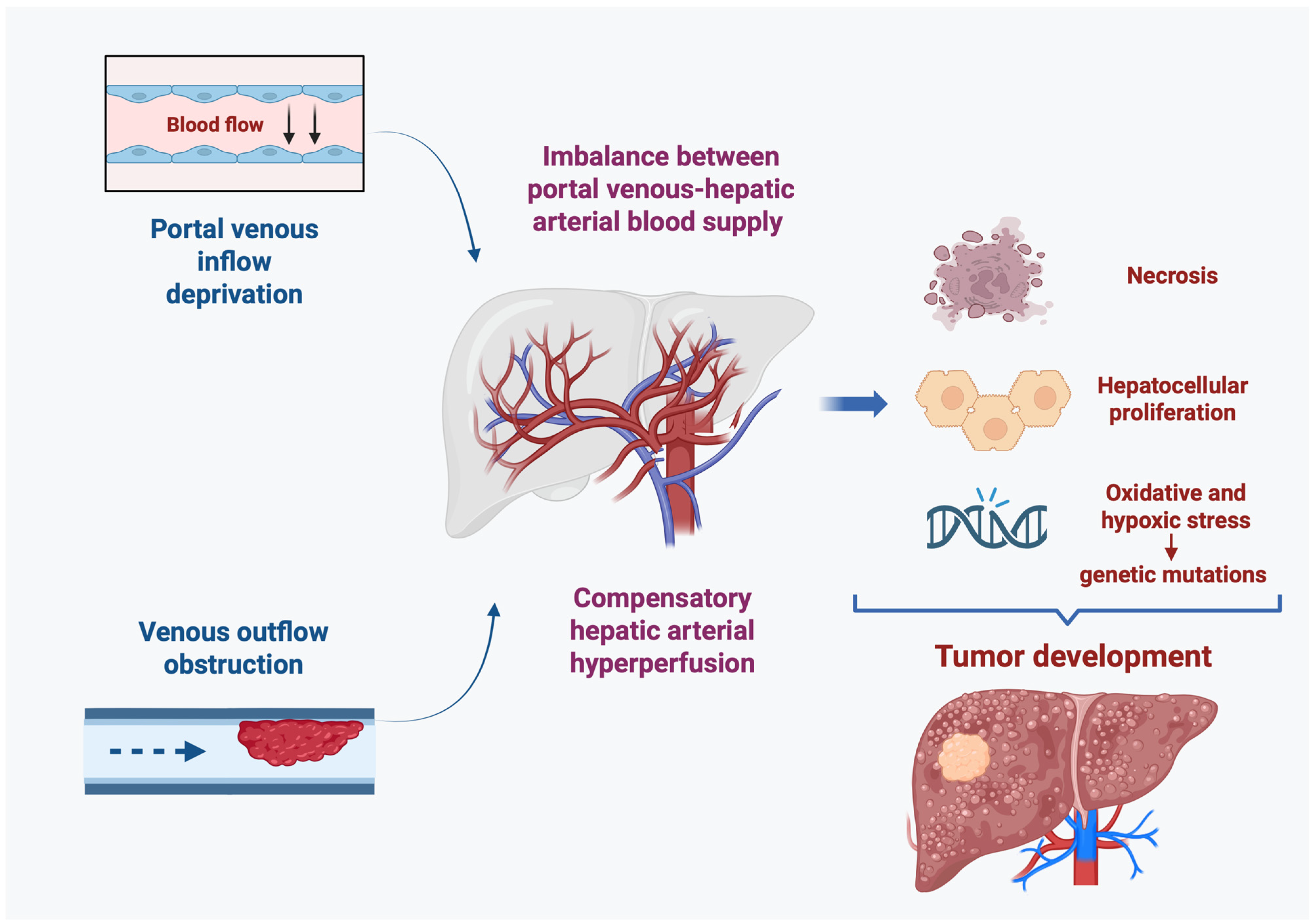
| Rare | Deprivation of portal inflow → arterial hyperperfusion → proliferative stimulus |
CTPV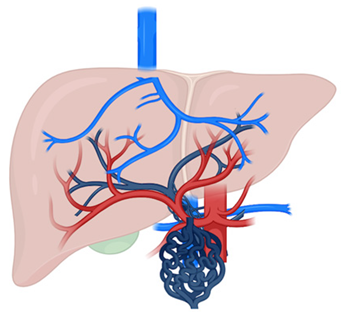 | Compensatory response to chronic PVT with collateral vessel formation |
| Very rare | Altered portal perfusion → arterial adaptation with increased hepatic arterial flow |
PSVD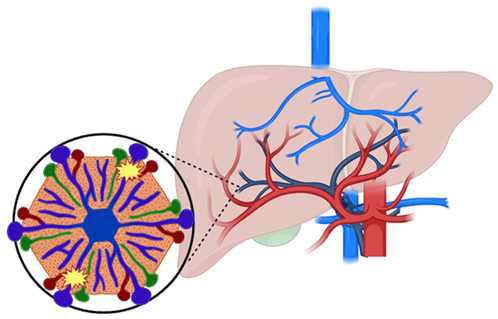 | Portal microvascular pathology involving the portal venules and sinusoids in the absence of cirrhosis. |
| Very rare | Portal venules and sinusoids disorder → reduced portal venous inflow + increased compensatory arterial inflow |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuli, L.; De Gaetano, V.; Venturini, G.; Arvonio, E.; Murgiano, M.; Gasbarrini, A.; Santopaolo, F.; Ponziani, F.R. Role of Vascular Liver Diseases in Hepatocellular Carcinoma Development. Cancers 2025, 17, 2060. https://doi.org/10.3390/cancers17132060
Giuli L, De Gaetano V, Venturini G, Arvonio E, Murgiano M, Gasbarrini A, Santopaolo F, Ponziani FR. Role of Vascular Liver Diseases in Hepatocellular Carcinoma Development. Cancers. 2025; 17(13):2060. https://doi.org/10.3390/cancers17132060
Chicago/Turabian StyleGiuli, Lucia, Valeria De Gaetano, Giulia Venturini, Ersilia Arvonio, Marco Murgiano, Antonio Gasbarrini, Francesco Santopaolo, and Francesca Romana Ponziani. 2025. "Role of Vascular Liver Diseases in Hepatocellular Carcinoma Development" Cancers 17, no. 13: 2060. https://doi.org/10.3390/cancers17132060
APA StyleGiuli, L., De Gaetano, V., Venturini, G., Arvonio, E., Murgiano, M., Gasbarrini, A., Santopaolo, F., & Ponziani, F. R. (2025). Role of Vascular Liver Diseases in Hepatocellular Carcinoma Development. Cancers, 17(13), 2060. https://doi.org/10.3390/cancers17132060