
Activation of Unfolded Protein Response Pathway in Malignancies: Interplay with Extracellular Matrix and Targeting Perspectives

 and
and
Simple Summary
Abstract
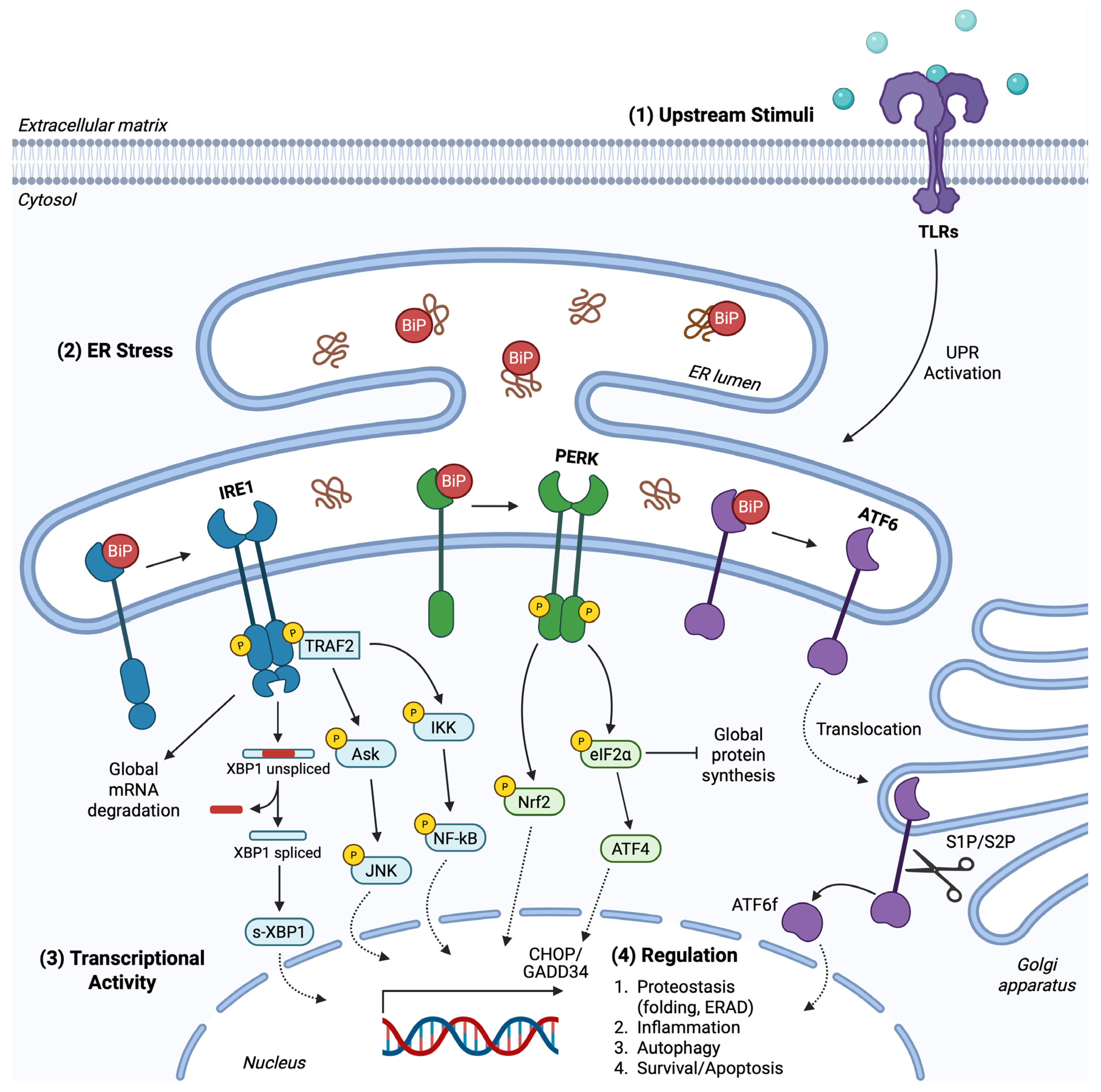
1. Endoplasmic Reticulum Stress
2. The Unfolded Protein Response Pathway
2.1. PERK Branch
2.2. IRE1 Branch
2.3. ATF6 Branch
2.4. UPR Crosstalk
3. UPR in Brain and Blood Malignancies
3.1. Glioblastoma
3.1.1. Integrated UPR and Tumorigenicity
3.1.2. Terminal UPR in Glioblastoma
3.2. Multiple Myeloma
3.2.1. Determining Cell Fate: The UPR Dichotomy
3.2.2. Clinical Relevance of Targeting UPR/UPS Equilibrium in Myeloma
4. Therapeutic Approaches
5. UPR Synergy with Extracellular Matrix in Malignancies
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADAM7 | A disintegrin and metalloprotease 7 |
| ARE | Antioxidant response element |
| ASK1 | Apoptosis-signal-regulating kinase 1 |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATF6f | ATF6 fragment |
| ATGs | Autophagy-related proteins |
| ATM/CHK2 | Ataxia Telangiectasia Mutated/Checkpoint kinase 2 |
| BAD | Bcl-2 antagonist of death |
| BAK | Bcl-2 antagonist/killer protein |
| BAX | Bcl-2-associated X protein |
| BBB | Blood-brain barrier |
| Bcl-2 | B-cell leukemia/lymphoma 2 protein |
| BIM | Bcl-2 interacting mediator of cell death |
| BiP | Binding immunoglobulin protein |
| Blos1 | Biogenesis of lysosome-related organelles 1 subunit 1 |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BTSCs | Brain tumor stem cells |
| BTZ | Bortezomib |
| b-ZIP | Basic leucine zipper |
| CAFs | Cancer-associated fibroblasts |
| CCR1 | C chemokine receptor type 1 |
| CDK12 | Cyclin-dependent kinase 12 |
| CHOP | C/EBP homologous protein |
| COPII | Coat protein complex II |
| CREB | cAMP-response element binding protein |
| CREBH | CREB hepatocyte |
| CSCs | Cancer stem-like cells |
| CSPGs | Chondroitin sulfate proteoglycans |
| DR5 | Death receptor 5 |
| DTT | Dithiothreitol |
| DUBs | Deubiquitinases |
| 2-DG | 2- deoxy-D-glucose |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| eIF2α | Eukaryotic translation initiation factor 2α |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | Extracellular signal-regulated kinases |
| FAK | Focal adhesion kinases |
| FLNA | Filamin A |
| FOXF1 | Forkhead box transcription factor 1 |
| 5-FU | 5-Fluorouracil |
| GADD34 | DNA damage-inducible 34 |
| GAGs | Glycosaminoglycans |
| GBM | Glioblastoma multiforme |
| GF | Growth factor |
| GFAP | Glial filament acidic protein |
| GPT | N-acetylglucosamine-1-phosphate transferase |
| GSCs | Glioma stem cells |
| GSH | Glutathione |
| HA | Hyaluronan |
| HDR1 | Hydroxymethyllutaryl reductase degradation protein 1 |
| HIF | Hypoxia-inducible factor |
| HK2 | Hexokinase 2 |
| HO-1 | Heme oxygenase-1 |
| HSP70 | Heat shock protein 70 |
| HUVECs | Human umbilical vein endothelial cells |
| IFN-γ | Interferon-γ |
| IKK | Inhibitor of nuclear factor-kB kinase |
| ILs | Interleukins |
| IRE1 | Inositol requiring enzyme 1 |
| JNK | c-Jun N-terminal kinase |
| KDELRs | Lys-Asp-Glu-Leu receptors |
| KPNB1 | Nuclear import receptor karyopherin β1 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LMW-HA | Low-molecular-weight hyaluronan |
| MAPK | Mitogen-activated protein kinases |
| MECs | Mammary epithelial cells |
| MEFs | Mouse embryonic fibroblasts |
| MEK | Mitogen-activated protein |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MM | Multiple myeloma |
| MMPs | Matrix metalloproteases |
| MPG | N-methylpurine DNA glycosylase |
| MSCs | Mesenchymal stem cells |
| mTOR | Mammalian target of rapamycin |
| NFATc1 | Nuclear factor of activated T-cells cytoplasmic 1 |
| NF-kB | Nuclear factor-kB |
| NK | Natural killer |
| NMR | Naked mole rats |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein 1 |
| NOX2/4 | NADPH oxidases 2/4 |
| Nrf-2 | Nuclear factor erythroid 2-related factor 2 |
| OASIS | Old astrocyte specifically induced substance |
| OPCs | Oligodendrocyte progenitor cells |
| PERK | Double-stranded RNA-activated protein kinase (PKR)–like ER kinase |
| PGs | Proteoglycans |
| PLC | Phospholipase C |
| PLK2 | Polo-like kinase 2 |
| PUMA | p53-upregulated modulator of apoptosis |
| Rad51 | DNA repair protein RAD51 homolog 1 |
| RCAN1 | Regulator of calcineurin 1 |
| RDV | Remdesivir |
| RER | Rough ER |
| RIDD | IRE1-dependent mRNA decay |
| RIP | Regulated intermembrane proteolysis |
| RNase | Ribonuclease |
| ROS | Reactive oxygen species |
| SER | Smooth ER |
| SLM | Salinomycin |
| SPARC | Secreted protein acidic and rich in cysteine |
| SPHK1/2 | Sphingosine kinase 1/2 |
| SRGN | Serglycin |
| S1P/S2P | Site-1 protease/Site-2 protease |
| TBI | Traumatic brain injury |
| TEVs | Tumor extracellular vesicles |
| TFs | Transcription factors |
| TGFβ | Transforming growth factor |
| Tg | Thapsigargin |
| TIMPs | Tissue inhibitors of matrix metalloproteases |
| TIS | Therapy-induced senescence |
| TLRs | Toll-like receptors |
| TM | Tunicamycin |
| TME | Tumor microenvironment |
| TMEM2 | Transmembrane protein 2 |
| TMZ | Temozolomide |
| TNBC | Triple negative breast cancer |
| TNF | Tumor necrosis factor |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TNFRSF6 | Tumor necrosis factor receptor superfamily 6 |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| TRAIL | Tumor necrosis factor-Related Apoptosis Inducing Ligand |
| TRIB3 | Tribbles homolog 3 |
| TRIM44 | Tripartite motif family member |
| UBL5 | Ubiquitin-like protein 5 |
| uPA | Urokinase plasminogen activator |
| uPAR | Urokinase plasminogen activator receptor |
| UPR | Unfolded protein response |
| UPS | Ubiquitin-proteasome system |
| UPS19 | Ubiquitin specific protease 19 |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| WARS1 | Tryptophanyl-tRNA synthetase 1 |
| XBP1 | X-box binding protein 1 |
| s-XBP1 | Spliced-XBP1 |
| u-XBP1 | Unspliced-XBP1 |
| XPOT | Exportin-T |
References
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER Structure and Function. Curr. Opin. Cell Biol. 2013, 25, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural Organization of the Endoplasmic Reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The Endoplasmic Reticulum: A Multifunctional Signaling Organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Kettel, P.; Karagöz, G.E. Endoplasmic Reticulum: Monitoring and Maintaining Protein and Membrane Homeostasis in the Endoplasmic Reticulum by the Unfolded Protein Response. Int. J. Biochem. Cell Biol. 2024, 172, 106598. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Parodi, A.J. Quality Control and Protein Folding in the Secretory Pathway. Annu. Rev. Cell Dev. Biol. 2003, 19, 649–676. [Google Scholar] [CrossRef]
- Farhan, H.; Rabouille, C. Signalling to and from the Secretory Pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form Follows Function: The Importance of Endoplasmic Reticulum Shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef]
- Johnson, S. The Ins and Outs of Calreticulin: From the ER Lumen to the Extracellular Space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Breitling, J.; Aebi, M. N-Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef]
- Hirata, T.; Kizuka, Y. N-Glycosylation. In The Role of Glycosylation in Health and Disease; Lauc, G., Trbojević-Akmačić, I., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; Volume 1325, pp. 3–24. ISBN 978-3-030-70114-7. [Google Scholar]
- Bieberich, E. Synthesis, Processing, and Function of N-Glycans in N-Glycoproteins. In Glycobiology of the Nervous System; Yu, R.K., Schengrund, C.-L., Eds.; Advances in Neurobiology; Springer: New York, NY, USA, 2014; Volume 9, pp. 47–70. ISBN 978-1-4939-1153-0. [Google Scholar]
- Desai, M.; Singh, A.; Pham, D.; Chowdhury, S.R.; Sun, B. Discovery and Visualization of the Hidden Relationships among N-Glycosylation, Disulfide Bonds, and Membrane Topology. Int. J. Mol. Sci. 2023, 24, 16182. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Recent Advances in Understanding Catalysis of Protein Folding by Molecular Chaperones. FEBS Lett. 2020, 594, 2770–2781. [Google Scholar] [CrossRef]
- Adams, B.M.; Canniff, N.P.; Guay, K.P.; Hebert, D.N. The Role of Endoplasmic Reticulum Chaperones in Protein Folding and Quality Control. In Cellular Biology of the Endoplasmic Reticulum; Agellon, L.B., Michalak, M., Eds.; Progress in Molecular and Subcellular Biology; Springer International Publishing: Cham, Switzerland, 2021; Volume 59, pp. 27–50. ISBN 978-3-030-67695-7. [Google Scholar]
- Yoshida, H. ER Stress and Diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The Endoplasmic Reticulum (ER) Chaperone BiP Is a Master Regulator of ER Functions: Getting by with a Little Help from ERdj Friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef]
- Griesemer, M.; Young, C.; Robinson, A.S.; Petzold, L. BiP Clustering Facilitates Protein Folding in the Endoplasmic Reticulum. PLoS Comput. Biol. 2014, 10, e1003675. [Google Scholar] [CrossRef]
- Duran-Romaña, R.; Houben, B.; De Vleeschouwer, M.; Louros, N.; Wilson, M.P.; Matthijs, G.; Schymkowitz, J.; Rousseau, F. N-Glycosylation as a Eukaryotic Protective Mechanism against Protein Aggregation. Sci. Adv. 2024, 10, eadk8173. [Google Scholar] [CrossRef]
- Gregersen, N.; Bross, P.; Andresen, B.S.; Pedersen, C.B.; Corydon, T.J.; Bolund, L. The Role of Chaperone-assisted Folding and Quality Control in Inborn Errors of Metabolism: Protein Folding Disorders. J. Inherit. Metab. Dis. 2001, 24, 189–212. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Parodi, A.J. Getting In and Out from Calnexin/Calreticulin Cycles. J. Biol. Chem. 2008, 283, 10221–10225. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond Lectins: The Calnexin/Calreticulin Chaperone System of the Endoplasmic Reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef]
- Bernasconi, R.; Molinari, M. ERAD and ERAD Tuning: Disposal of Cargo and of ERAD Regulators from the Mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. The Protective and Destructive Roles Played by Molecular Chaperones during ERAD (Endoplasmic-Reticulum-Associated Degradation). Biochem. J. 2007, 404, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Qi, L. ER-Associated Degradation in Health and Disease—From Substrate to Organism. J. Cell Sci. 2019, 132, jcs232850. [Google Scholar] [CrossRef]
- Schröder, M. Endoplasmic Reticulum Stress Responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Shen, Y.; Ballar, P.; Apostolou, A.; Doong, H.; Fang, S. ER Stress Differentially Regulates the Stabilities of ERAD Ubiquitin Ligases and Their Substrates. Biochem. Biophys. Res. Commun. 2007, 352, 919–924. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Bánhegyi, G.; Baumeister, P.; Benedetti, A.; Dong, D.; Fu, Y.; Lee, A.S.; Li, J.; Mao, C.; Margittai, É.; Ni, M.; et al. Endoplasmic Reticulum Stress. Ann. N. Y. Acad. Sci. 2007, 1113, 58–71. [Google Scholar] [CrossRef]
- Wu, J.; Kaufman, R.J. From Acute ER Stress to Physiological Roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Niessner, H.; Hüsch, A.; Kosnopfel, C.; Meinhardt, M.; Westphal, D.; Meier, F.; Schilling, B.; Sinnberg, T. Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma. Cancers 2023, 15, 5521. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic Reticulum Stress Signals in the Tumour and Its Microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Chng, W.-J.; Zhou, J. Crosstalk between Endoplasmic Reticulum Stress and Oxidative Stress: A Dynamic Duo in Multiple Myeloma. Cell. Mol. Life Sci. 2021, 78, 3883–3906. [Google Scholar] [CrossRef] [PubMed]
- Ri, M. Endoplasmic-Reticulum Stress Pathway-Associated Mechanisms of Action of Proteasome Inhibitors in Multiple Myeloma. Int. J. Hematol. 2016, 104, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Namazi, A.; Reger, R.; Levy, E.; Berg, M.; St. Hilaire, C.; Childs, R.W. Bortezomib Sensitizes Multiple Myeloma to NK Cells via ER-Stress-Induced Suppression of HLA-E and Upregulation of DR5. OncoImmunology 2019, 8, e1534664. [Google Scholar] [CrossRef]
- He, Y.; Alejo, S.; Johnson, J.D.; Jayamohan, S.; Sareddy, G.R. Reticulocalbin 3 Is a Novel Mediator of Glioblastoma Progression. Cancers 2023, 15, 2008. [Google Scholar] [CrossRef]
- Shi, P.; Zhang, Z.; Xu, J.; Zhang, L.; Cui, H. Endoplasmic Reticulum Stress-induced Cell Death as a Potential Mechanism for Targeted Therapy in Glioblastoma (Review). Int. J. Oncol. 2021, 59, 60. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Papavassiliou, A.G.; Piperi, C. Targeting of Endoplasmic Reticulum (ER) Stress in Gliomas. Pharmacol. Res. 2020, 157, 104823. [Google Scholar] [CrossRef]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib Enhances Radiosensitivity of Hypoxic Glioblastoma Cells through Endoplasmic Reticulum Stress. Neuro-Oncol. 2013, 15, 1186–1199. [Google Scholar] [CrossRef]
- Dadey, D.; Kapoor, V.; Khudanyan, A.; Thotala, D.; Hallahan, D. Abstract 3323: Radiation-Induced ER Stress Contributes to Survival in Glioblastoma Cell Lines. Cancer Res. 2015, 75, 3323. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre dâ€TMHellencourt, C.; Ravanan, P. A Molecular Web: Endoplasmic Reticulum Stress, Inflammation, and Oxidative Stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross Talk Between ER Stress, Oxidative Stress, and Inflammation in Health and Disease. In Stress Responses; Oslowski, C.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1292, pp. 205–214. ISBN 978-1-4939-2521-6. [Google Scholar]
- Zhang, K.; Kaufman, R.J. From Endoplasmic-Reticulum Stress to the Inflammatory Response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.; McGuckin, M.A. The Interplay between Endoplasmic Reticulum Stress and Inflammation. Immunol. Cell Biol. 2012, 90, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 490, pp. 71–92. ISBN 978-0-12-385114-7. [Google Scholar]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER Stress in Vivo and in Vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef]
- Dufey, E.; Sepúlveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. 1. An Overview. Am. J. Physiol.-Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef]
- Dabsan, S.; Twito, G.; Biadsy, S.; Igbaria, A. Less Is Better: Various Means to Reduce Protein Load in the Endoplasmic Reticulum. FEBS J. 2025, 292, 976–989. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Lai, C.W.; Aronson, D.E.; Snapp, E.L. BiP Availability Distinguishes States of Homeostasis and Stress in the Endoplasmic Reticulum of Living Cells. Mol. Biol. Cell 2010, 21, 1909–1921. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical Binding of BiP ATPase Domain to Ire1 and Perk Is Dissociated by Unfolded Protein CH1 to Initiate ER Stress Signaling. eLife 2015, 4, e03522. [Google Scholar] [CrossRef]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR Proteins IRE1 and PERK Switch BiP from Chaperone to ER Stress Sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of Mammalian IRE1α upon ER Stress Depends on Dissociation of BiP Rather than on Direct Interaction with Unfolded Proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable Binding of ATF6 to BiP in the Endoplasmic Reticulum Stress Response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef]
- Shen, J.; Prywes, R. ER Stress Signaling by Regulated Proteolysis of ATF6. Methods 2005, 35, 382–389. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis Control by the Unfolded Protein Response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Kohno, K. Self-Association and BiP Dissociation Are Not Sufficient for Activation of the ER Stress Sensor Ire1. J. Cell Sci. 2007, 120, 1681–1688. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Wofford, J.A.; Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Wieman, H.L.; Day, A.E.; Ilkayeva, O.; Rathmell, J.C. Autophagy Provides Nutrients but Can Lead to Chop-Dependent Induction of Bim to Sensitize Growth Factor–Deprived Cells to Apoptosis. Mol. Biol. Cell 2009, 20, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Radanović, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in Endoplasmic Reticulum Stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating Transcription Factor 3 Is Integral to the Eukaryotic Initiation Factor 2 Kinase Stress Response. Mol. Cell. Biol. 2004, 24, 1365–1377. [Google Scholar] [CrossRef]
- Averous, J.; Bruhat, A.; Jousse, C.; Carraro, V.; Thiel, G.; Fafournoux, P. Induction of CHOP Expression by Amino Acid Limitation Requires Both ATF4 Expression and ATF2 Phosphorylation. J. Biol. Chem. 2004, 279, 5288–5297. [Google Scholar] [CrossRef]
- Márton, M.; Bánhegyi, G.; Gyöngyösi, N.; Kálmán, E.É.; Pettkó-Szandtner, A.; Káldi, K.; Kapuy, O. A Systems Biological Analysis of the ATF4-GADD34-CHOP Regulatory Triangle upon Endoplasmic Reticulum Stress. FEBS Open Bio 2022, 12, 2065–2082. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Lu, P.D.; Jousse, C.; Marciniak, S.J.; Zhang, Y.; Novoa, I.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Harding, H.P. Cytoprotection by Pre-Emptive Conditional Phosphorylation of Translation Initiation Factor 2. EMBO J. 2004, 23, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Gonen, N.; Sabath, N.; Burge, C.B.; Shalgi, R. Widespread PERK-Dependent Repression of ER Targets in Response to ER Stress. Sci. Rep. 2019, 9, 4330. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Scheuner, D.; Chen, J.-J.; Kaufman, R.J.; Ron, D. Ppp1r15 Gene Knockout Reveals an Essential Role for Translation Initiation Factor 2 Alpha (eIF2α) Dephosphorylation in Mammalian Development. Proc. Natl. Acad. Sci. USA 2009, 106, 1832–1837. [Google Scholar] [CrossRef]
- Bashir, S.; Banday, M.; Qadri, O.; Bashir, A.; Hilal, N.; Nida-i-Fatima; Rader, S.; Fazili, K.M. The Molecular Mechanism and Functional Diversity of UPR Signaling Sensor IRE1. Life Sci. 2021, 265, 118740. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A Review of the Mammalian Unfolded Protein Response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping Endoplasmic Reticulum Quality Control through the Unfolded Protein Response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 Interaction Regulates ER Stress-Induced Autophagy in Auditory Cells. Sci. Rep. 2017, 7, 4442. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Karagöz, G.E.; Fröhlich, F.; Li, H.; Walther, T.C.; Walter, P. The Unfolded Protein Response and Endoplasmic Reticulum Protein Targeting Machineries Converge on the Stress Sensor IRE1. eLife 2018, 7, e43036. [Google Scholar] [CrossRef]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a Consensus Element Recognized and Cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER Stress Sensor and Cell Fate Executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat Shock Protein 90 Modulates the Unfolded Protein Response by Stabilizing IRE1α. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 Protects Cells from ER Stress-Induced Apoptosis via Enhancement of IRE1α-XBP1 Signaling through a Physical Interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian Endoplasmic Reticulum Stress Sensor IRE1 Signals by Dynamic Clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical Role of Endogenous Akt/IAPs and MEK1/ERK Pathways in Counteracting Endoplasmic Reticulum Stress-Induced Cell Death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 Serves ER Stress-Triggered Apoptosis via Selective Activation of the IRE1–JNK Pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER Stress Activates NF-κB by Integrating Functions of Basal IKK Activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef]
- Yang, Q.; Kim, Y.; Lin, Y.; Lewis, J.; Neckers, L.; Liu, Z. Tumour Necrosis Factor Receptor 1 Mediates Endoplasmic Reticulum Stress-induced Activation of the MAP Kinase JNK. EMBO Rep. 2006, 7, 622–627. [Google Scholar] [CrossRef]
- Lei, Y.; Yu, H.; Ding, S.; Liu, H.; Liu, C.; Fu, R. Molecular Mechanism of ATF6 in Unfolded Protein Response and Its Role in Disease. Heliyon 2024, 10, e25937. [Google Scholar] [CrossRef]
- Schindler, A.J.; Schekman, R. In Vitro Reconstitution of ER-Stress Induced ATF6 Transport in COPII Vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef]
- Hillary, R.F.; FitzGerald, U. A Lifetime of Stress: ATF6 in Development and Homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef]
- Merksamer, P.I.; Papa, F.R. The UPR and Cell Fate at a Glance. J. Cell Sci. 2010, 123, 1003–1006. [Google Scholar] [CrossRef]
- Cui, A.; Ding, D.; Li, Y. Regulation of Hepatic Metabolism and Cell Growth by the ATF/CREB Family of Transcription Factors. Diabetes 2021, 70, 653–664. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic Reticulum-Tethered Transcription Factor cAMP Responsive Element-Binding Protein, Hepatocyte Specific, Regulates Hepatic Lipogenesis, Fatty Acid Oxidation, and Lipolysis upon Metabolic Stress in Mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef]
- Demaurex, N.; Frieden, M.; Arnaudeau, S. ER Calcium and ER Chaperones: New Players in Apoptosis? In Calreticulin; Eggleton, P., Michalak, M., Eds.; Molecular Biology Intelligence Unit; Springer: Boston, MA, USA, 2003; pp. 133–141. ISBN 978-1-4613-4862-7. [Google Scholar]
- Allen, D.; Seo, J. ER Stress Activates the TOR Pathway through Atf6. J. Mol. Signal. 2018, 13, 1. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the Isoform-Specific Characteristics of ATF6α and ATF6β on Endoplasmic Reticulum Stress Response Gene Expression and Cell Viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Morrison, L.E.; Hoover, H.; Glembotski, C.C. Coordination of ATF6-Mediated Transcription and ATF6 Degradation by a Domain That Is Shared with the Viral Transcription Factor, VP16. J. Biol. Chem. 2002, 277, 20734–20739. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Morrison, L.; Glembotski, C.C. Opposing Roles for ATF6α and ATF6β in Endoplasmic Reticulum Stress Response Gene Induction. J. Biol. Chem. 2004, 279, 21078–21084. [Google Scholar] [CrossRef]
- Pontisso, I.; Ornelas-Guevara, R.; Combettes, L.; Dupont, G. A Journey in UPR Modelling. Biol. Cell 2023, 115, 2200111. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by Translational Block. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel Mechanism of Enhancing IRE1α-XBP1 Signalling via the PERK-ATF4 Pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 Kinase PERK and the Integrated Stress Response Facilitate Activation of ATF6 during Endoplasmic Reticulum Stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Düssmann, H.; Prehn, J.H.M. ER Stress Signaling Has an Activating Transcription Factor 6α (ATF6)-Dependent “off-Switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef]
- Wang, W.; Hawkridge, A.M.; Ma, Y.; Zhang, B.; Mangrum, J.B.; Hassan, Z.H.; He, T.; Blat, S.; Guo, C.; Zhou, H.; et al. Ubiquitin-like Protein 5 Is a Novel Player in the UPR–PERK Arm and ER Stress–Induced Cell Death. J. Biol. Chem. 2023, 299, 104915. [Google Scholar] [CrossRef]
- Hassink, G.C.; Zhao, B.; Sompallae, R.; Altun, M.; Gastaldello, S.; Zinin, N.V.; Masucci, M.G.; Lindsten, K. The ER-resident Ubiquitin-specific Protease 19 Participates in the UPR and Rescues ERAD Substrates. EMBO Rep. 2009, 10, 755–761. [Google Scholar] [CrossRef]
- Yang, L.; Xue, R.; Yang, C.; Lv, Y.; Li, S.; Xiang, W.; Guo, X.; Zhou, J. Endoplasmic Reticulum Stress on Glioblastoma: Tumor Growth Promotion and Immunosuppression. Int. Immunopharmacol. 2025, 157, 114806. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Y.; Oyang, L.; Cui, S.; Li, S.; Li, J.; Liu, L.; Li, Y.; Peng, M.; Tan, S.; et al. Endoplasmic Reticulum Stress—A Key Guardian in Cancer. Cell Death Discov. 2024, 10, 343. [Google Scholar] [CrossRef]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.A.; Lencioni, A.M.; Russell, R.L.; Madsen, H.; Nega, M.; et al. Induction of the Unfolded Protein Response Drives Enhanced Metabolism and Chemoresistance in Glioma Cells. PLoS ONE 2013, 8, e73267. [Google Scholar] [CrossRef]
- Jang, Y. Bioactive Compounds Targeting Dihydroceramide and Their Therapeutic Potential in Cancer Treatment. Cancers 2025, 17, 909. [Google Scholar] [CrossRef]
- Lan, B.; Zhuang, Z.; Zhang, J.; He, Y.; Wang, N.; Deng, Z.; Mei, L.; Li, Y.; Gao, Y. Triggering of Endoplasmic Reticulum Stress via ATF4-SPHK1 Signaling Promotes Glioblastoma Invasion and Chemoresistance. Cell Death Dis. 2024, 15, 552. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Tsymbal, D.O.; Minchenko, D.O.; Moenner, M.; Kovalevska, O.V.; Lypova, N.M. Inhibition of Kinase and Endoribonuclease Activity of ERN1/IRE1α Affects Expression of Proliferationrelated Genes in U87 Glioma Cells. Endoplasmic Reticulum Stress Dis. 2015, 2, 18–29. [Google Scholar] [CrossRef]
- White, B.E.; Liu, Y.; Hakonarson, H.; Buono, R.J. RNA Sequencing in Hypoxia-Adapted T98G Glioblastoma Cells Provides Supportive Evidence for IRE1 as a Potential Therapeutic Target. Genes 2023, 14, 841. [Google Scholar] [CrossRef] [PubMed]
- Ketkar, M.; Desai, S.; Rana, P.; Thorat, R.; Epari, S.; Dutt, A.; Dutt, S. Inhibition of PERK-Mediated Unfolded Protein Response Acts as a Switch for Reversal of Residual Senescence and as Senolytic Therapy in Glioblastoma. Neuro-Oncol. 2024, 26, 2027–2043. [Google Scholar] [CrossRef]
- Hou, X.; Liu, Y.; Liu, H.; Chen, X.; Liu, M.; Che, H.; Guo, F.; Wang, C.; Zhang, D.; Wu, J.; et al. PERK Silence Inhibits Glioma Cell Growth under Low Glucose Stress by Blockage of P-AKT and Subsequent HK2’s Mitochondria Translocation. Sci. Rep. 2015, 5, 9065. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Tsymbal, D.O.; Minchenko, D.O.; Riabovol, O.O.; Halkin, O.V.; Ratushna, O.O. IRE-1α Regulates Expression of Ubiquitin Specific Peptidases during Hypoxic Response in U87 Glioma Cells. Endoplasmic Reticulum Stress Dis. 2016, 3, 50–62. [Google Scholar] [CrossRef]
- Scholz, N.; Kurian, K.M.; Siebzehnrubl, F.A.; Licchesi, J.D.F. Targeting the Ubiquitin System in Glioblastoma. Front. Oncol. 2020, 10, 574011. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, X.; Liu, M.; Yang, Z.; Bi, Y.; Zou, H.; Wu, J.; Che, H.; Li, C.; Wang, X.; et al. XBP1 Silencing Decreases Glioma Cell Viability and Glycolysis Possibly by Inhibiting HK2 Expression. J. Neurooncol. 2016, 126, 455–462. [Google Scholar] [CrossRef]
- Obacz, J.; Archambeau, J.; Lafont, E.; Nivet, M.; Martin, S.; Aubry, M.; Voutetakis, K.; Pineau, R.; Boniface, R.; Sicari, D.; et al. IRE1 Endoribonuclease Signaling Promotes Myeloid Cell Infiltration in Glioblastoma. Neuro-Oncol. 2024, 26, 858–871. [Google Scholar] [CrossRef]
- Pelizzari-Raymundo, D.; Doultsinos, D.; Pineau, R.; Sauzay, C.; Koutsandreas, T.; Langlais, T.; Carlesso, A.; Gkotsi, E.; Negroni, L.; Avril, T.; et al. A Novel IRE1 Kinase Inhibitor for Adjuvant Glioblastoma Treatment. iScience 2023, 26, 106687. [Google Scholar] [CrossRef]
- Lorenz, N.I.; Sittig, A.C.M.; Urban, H.; Luger, A.-L.; Engel, A.L.; Münch, C.; Steinbach, J.P.; Ronellenfitsch, M.W. Activating Transcription Factor 4 Mediates Adaptation of Human Glioblastoma Cells to Hypoxia and Temozolomide. Sci. Rep. 2021, 11, 14161. [Google Scholar] [CrossRef]
- Dowdell, A.; Marsland, M.; Faulkner, S.; Gedye, C.; Lynam, J.; Griffin, C.P.; Marsland, J.; Jiang, C.C.; Hondermarck, H. Targeting XBP1 mRNA Splicing Sensitizes Glioblastoma to Chemotherapy. FASEB BioAdv. 2023, 5, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-C.; Liu, J.-W.; Li, K.; Zheng, J.; Xiong, Z.-Q. KPNB1 Inhibition Disrupts Proteostasis and Triggers Unfolded Protein Response-Mediated Apoptosis in Glioblastoma Cells. Oncogene 2018, 37, 2936–2952. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The Unfolded Protein Response Regulator GRP78/BiP as a Novel Target for Increasing Chemosensitivity in Malignant Gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, R.; Tian, C.; Wang, W.; Zhou, L.; Guo, T.; Yu, J.; Wu, C.; Shen, Y.; Liu, X.; et al. GRP78 Blockade Overcomes Intrinsic Resistance to UBA1 Inhibitor TAK-243 in Glioblastoma. Cell Death Discov. 2022, 8, 133. [Google Scholar] [CrossRef]
- Williams, C.H.; Neitzel, L.R.; Cornell, J.; Rea, S.; Mills, I.; Silver, M.S.; Ahmad, J.D.; Birukov, K.G.; Birukova, A.; Brem, H.; et al. GPR68-ATF4 Signaling Is a Novel Prosurvival Pathway in Glioblastoma Activated by Acidic Extracellular Microenvironment. Exp. Hematol. Oncol. 2024, 13, 13. [Google Scholar] [CrossRef]
- Wang, H.; Mi, K. Emerging Roles of Endoplasmic Reticulum Stress in the Cellular Plasticity of Cancer Cells. Front. Oncol. 2023, 13, 1110881. [Google Scholar] [CrossRef]
- Shah, S.S.; Rodriguez, G.A.; Musick, A.; Walters, W.M.; De Cordoba, N.; Barbarite, E.; Marlow, M.M.; Marples, B.; Prince, J.S.; Komotar, R.J.; et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers 2019, 11, 159. [Google Scholar] [CrossRef]
- Le Reste, P.J.; Pineau, R.; Voutetakis, K.; Samal, J.; Jégou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local Intracerebral Inhibition of IRE1 by MKC8866 Sensitizes Glioblastoma to Irradiation/Chemotherapy in Vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef]
- Oliveira, F.D.; De Campos, R.P.; Pereira, L.C.; Meira, L.B.; Lenz, G. Expression of Key Unfolded Protein Response Genes Predicts Patient Survival and an Immunosuppressive Microenvironment in Glioblastoma. Transl. Med. Commun. 2024, 9, 5. [Google Scholar] [CrossRef]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; Den Dunnen, W.F.A.; Kruyt, F.A.E. ER Stress and UPR Activation in Glioblastoma: Identification of a Noncanonical PERK Mechanism Regulating GBM Stem Cells through SOX2 Modulation. Cell Death Dis. 2019, 10, 690. [Google Scholar] [CrossRef]
- He, Y.; Meng, H.; Xu, H.; Fan, L.; Zhou, Z.; Xu, B.; Sun, L.; Gao, Y. Regulation of Integrated Stress Response Sensitizes U87MG Glioblastoma Cells to Temozolomide Through the Mitochondrial Apoptosis Pathway. Anat. Rec. 2018, 301, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Pan, P.-H.; Wu, C.-C.; Liao, S.-L.; Chen, W.-Y.; Kuan, Y.-H.; Wang, W.-Y.; Chen, C.-J. Endoplasmic Reticulum Stress Contributes to Gefitinib-Induced Apoptosis in Glioma. Int. J. Mol. Sci. 2021, 22, 3934. [Google Scholar] [CrossRef]
- Cao, L.; Lei, H.; Chang, M.-Z.; Liu, Z.-Q.; Bie, X.-H. Down-Regulation of 14-3-3β Exerts Anti-Cancer Effects through Inducing ER Stress in Human Glioma U87 Cells: Involvement of CHOP–Wnt Pathway. Biochem. Biophys. Res. Commun. 2015, 462, 389–395. [Google Scholar] [CrossRef]
- Kusaczuk, M.; Tyszka, N.; Krętowski, R.; Cechowska-Pasko, M. The Proteasome Inhibitor Marizomib Evokes Endoplasmic Reticulum Stress and Promotes Apoptosis in Human Glioblastoma Cells. Pharmaceuticals 2024, 17, 1089. [Google Scholar] [CrossRef]
- Xipell, E.; Aragón, T.; Martínez-Velez, N.; Vera, B.; Idoate, M.A.; Martínez-Irujo, J.J.; Garzón, A.G.; Gonzalez-Huarriz, M.; Acanda, A.M.; Jones, C.; et al. Endoplasmic Reticulum Stress-Inducing Drugs Sensitize Glioma Cells to Temozolomide through Downregulation of MGMT, MPG, and Rad51. Neuro-Oncol. 2016, 18, 1109–1119. [Google Scholar] [CrossRef]
- Weatherbee, J.L.; Kraus, J.-L.; Ross, A.H. ER Stress in Temozolomide-Treated Glioblastomas Interferes with DNA Repair and Induces Apoptosis. Oncotarget 2016, 7, 43820–43834. [Google Scholar] [CrossRef]
- Hu, Y.; Chu, L.; Liu, J.; Yu, L.; Song, S.; Yang, H.; Han, F. Knockdown of CREB3 Activates Endoplasmic Reticulum Stress and Induces Apoptosis in Glioblastoma. Aging 2019, 11, 8156–8168. [Google Scholar] [CrossRef]
- Zielke, S.; Kardo, S.; Zein, L.; Mari, M.; Covarrubias-Pinto, A.; Kinzler, M.N.; Meyer, N.; Stolz, A.; Fulda, S.; Reggiori, F.; et al. ATF4 Links ER Stress with Reticulophagy in Glioblastoma Cells. Autophagy 2021, 17, 2432–2448. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, B.; Cheng, S.; Fan, H.; Liu, S.; Zhou, B.; Liu, W.; Liang, R.; Tang, Y.; Zhang, Y. KDELR2 Knockdown Synergizes with Temozolomide to Induce Glioma Cell Apoptosis through the CHOP and JNK/P38 Pathways. Transl. Cancer Res. 2021, 10, 3491–3506. [Google Scholar] [CrossRef]
- Ma, Y.; Di, Z.; Cao, Q.; Xu, W.; Bi, S.; Yu, J.; Shen, Y.; Yu, Y.; Shen, Y.; Feng, L. Xanthatin Induces Glioma Cell Apoptosis and Inhibits Tumor Growth via Activating Endoplasmic Reticulum Stress-Dependent CHOP Pathway. Acta Pharmacol. Sin. 2020, 41, 404–414. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Li, S.; Xu, J.; Ning, W.; Zhao, C.; Wang, J.; Qu, Y.; Zhang, M.; Zhou, W.; et al. Remdesivir Inhibits the Progression of Glioblastoma by Enhancing Endoplasmic Reticulum Stress. Biomed. Pharmacother. 2023, 157, 114037. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, J. Impact of Proteostasis Workload on Sensitivity to Proteasome Inhibitors in Multiple Myeloma. Clin. Exp. Med. 2025, 25, 176. [Google Scholar] [CrossRef] [PubMed]
- Hamedi, K.R.; Harmon, K.A.; Goodwin, R.L.; Arce, S. Autophagy and the Bone Marrow Microenvironment: A Review of Protective Factors in the Development and Maintenance of Multiple Myeloma. Front. Immunol. 2022, 13, 889954. [Google Scholar] [CrossRef]
- Schwestermann, J.; Besse, A.; Driessen, C.; Besse, L. Contribution of the Tumor Microenvironment to Metabolic Changes Triggering Resistance of Multiple Myeloma to Proteasome Inhibitors. Front. Oncol. 2022, 12, 899272. [Google Scholar] [CrossRef]
- Bagratuni, T.; Patseas, D.; Mavrianou-Koutsoukou, N.; Liacos, C.I.; Sklirou, A.D.; Rousakis, P.; Gavriatopoulou, M.; Terpos, E.; Tsitsilonis, O.E.; Trougakos, I.P.; et al. Characterization of a PERK Kinase Inhibitor with Anti-Myeloma Activity. Cancers 2020, 12, 2864. [Google Scholar] [CrossRef]
- Quwaider, D.; Corchete, L.A.; Martín-Izquierdo, M.; Hernández-Sánchez, J.M.; Rojas, E.A.; Cardona-Benavides, I.J.; García-Sanz, R.; Herrero, A.B.; Gutiérrez, N.C. RNA Sequencing Identifies Novel Regulated IRE1-Dependent Decay Targets That Affect Multiple Myeloma Survival and Proliferation. Exp. Hematol. Oncol. 2022, 11, 18. [Google Scholar] [CrossRef]
- Michallet, A.-S.; Mondiere, P.; Taillardet, M.; Leverrier, Y.; Genestier, L.; Defrance, T. Compromising the Unfolded Protein Response Induces Autophagy-Mediated Cell Death in Multiple Myeloma Cells. PLoS ONE 2011, 6, e25820. [Google Scholar] [CrossRef]
- Ninkovic, S.; Harrison, S.J.; Quach, H. Glucose-Regulated Protein 78 (GRP78) as a Potential Novel Biomarker and Therapeutic Target in Multiple Myeloma. Expert Rev. Hematol. 2020, 13, 1201–1210. [Google Scholar] [CrossRef]
- Steiner, N.; Borjan, B.; Hajek, R.; Jöhrer, K.; Göbel, G.; Willenbacher, W.; Kern, J.; Gunsilius, E.; Untergasser, G. Expression and Release of Glucose-Regulated Protein-78 (GRP78) in Multiple Myeloma. Oncotarget 2017, 8, 56243–56254. [Google Scholar] [CrossRef]
- Chhabra, S.; Jain, S.; Wallace, C.; Hong, F.; Liu, B. High Expression of Endoplasmic Reticulum Chaperone Grp94 Is a Novel Molecular Hallmark of Malignant Plasma Cells in Multiple Myeloma. J. Hematol. Oncol. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Arena, A.; Romeo, M.A.; Benedetti, R.; Gilardini Montani, M.S.; Cirone, M. Targeting C-Myc Unbalances UPR towards Cell Death and Impairs DDR in Lymphoma and Multiple Myeloma Cells. Biomedicines 2022, 10, 731. [Google Scholar] [CrossRef]
- Yamashita, Y.; Morita, S.; Hosoi, H.; Kobata, H.; Kishimoto, S.; Ishibashi, T.; Mishima, H.; Kinoshita, A.; Backes, B.J.; Yoshiura, K.-I.; et al. Targeting Adaptive IRE1α Signaling and PLK2 in Multiple Myeloma: Possible Anti-Tumor Mechanisms of KIRA8 and Nilotinib. Int. J. Mol. Sci. 2020, 21, 6314. [Google Scholar] [CrossRef]
- Wu, C.; Liu, M.; Liu, J.; Jia, M.; Zeng, X.; Fu, Z.; Geng, Y.; He, Z.; Zhang, X.; Yan, H. Integrative Analysis of an Endoplasmic Reticulum Stress-related Signature in Multiple Myeloma. J. Gene Med. 2024, 26, e3595. [Google Scholar] [CrossRef]
- Kosako, H.; Yamashita, Y.; Morita, S.; Iwabuchi, S.; Hashimoto, S.; Matsuoka, T.-A.; Sonoki, T.; Tamura, S. Allosteric Inhibition of C-Abl to Induce Unfolded Protein Response and Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 16162. [Google Scholar] [CrossRef]
- Waad Sadiq, Z.; Brioli, A.; Al-Abdulla, R.; Çetin, G.; Schütt, J.; Murua Escobar, H.; Krüger, E.; Ebstein, F. Immunogenic Cell Death Triggered by Impaired Deubiquitination in Multiple Myeloma Relies on Dysregulated Type I Interferon Signaling. Front. Immunol. 2023, 14, 982720. [Google Scholar] [CrossRef]
- Fu, Y.-F.; Liu, X.; Gao, M.; Zhang, Y.-N.; Liu, J. Endoplasmic Reticulum Stress Induces Autophagy and Apoptosis While Inhibiting Proliferation and Drug Resistance in Multiple Myeloma through the PI3K/Akt/mTOR Signaling Pathway. Oncotarget 2017, 8, 61093–61106. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome Inhibitors Induce a Terminal Unfolded Protein Response in Multiple Myeloma Cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Dong, H.; Chen, L.; Chen, X.; Gu, H.; Gao, G.; Gao, Y.; Dong, B. Dysregulation of Unfolded Protein Response Partially Underlies Proapoptotic Activity of Bortezomib in Multiple Myeloma Cells. Leuk. Lymphoma 2009, 50, 974–984. [Google Scholar] [CrossRef]
- Nikesitch, N.; Tao, C.; Lai, K.; Killingsworth, M.; Bae, S.; Wang, M.; Harrison, S.; Roberts, T.L.; Ling, S.C.W. Predicting the Response of Multiple Myeloma to the Proteasome Inhibitor Bortezomib by Evaluation of the Unfolded Protein Response. Blood Cancer J. 2016, 6, e432. [Google Scholar] [CrossRef]
- Ling, S.C.W.; Lau, E.K.K.; Al-Shabeeb, A.; Nikolic, A.; Catalano, A.; Iland, H.; Horvath, N.; Ho, P.J.; Harrison, S.; Fleming, S.; et al. Response of Myeloma to the Proteasome Inhibitor Bortezomib Is Correlated with the Unfolded Protein Response Regulator XBP-1. Haematologica 2012, 97, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zou, J.; Zhang, H.; Fu, W.; Zeng, T.; Huang, H.; Zhou, F.; Hou, J. Unfolded Protein Response Inducers Tunicamycin and Dithiothreitol Promote Myeloma Cell Differentiation Mediated by XBP-1. Clin. Exp. Med. 2015, 15, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Bustany, S.; Cahu, J.; Guardiola, P.; Sola, B. Cyclin D1 Sensitizes Myeloma Cells to Endoplasmic Reticulum Stress-Mediated Apoptosis by Activating the Unfolded Protein Response Pathway. BMC Cancer 2015, 15, 262. [Google Scholar] [CrossRef] [PubMed]
- Olaisen, C.; Røst, L.M.; Sharma, A.; Søgaard, C.K.; Khong, T.; Berg, S.; Jang, M.; Nedal, A.; Spencer, A.; Bruheim, P.; et al. Multiple Myeloma Cells with Increased Proteasomal and ER Stress Are Hypersensitive to ATX-101, an Experimental Peptide Drug Targeting PCNA. Cancers 2024, 16, 3963. [Google Scholar] [CrossRef]
- Mitra, A.K.; Kumar, H.; Ramakrishnan, V.; Chen, L.; Baughn, L.; Kumar, S.; Rajkumar, S.V.; Van Ness, B.G. In Vitro and Ex Vivo Gene Expression Profiling Reveals Differential Kinetic Response of HSPs and UPR Genes Is Associated with PI Resistance in Multiple Myeloma. Blood Cancer J. 2020, 10, 78. [Google Scholar] [CrossRef]
- Borjan, B.; Kern, J.; Steiner, N.; Gunsilius, E.; Wolf, D.; Untergasser, G. Spliced XBP1 Levels Determine Sensitivity of Multiple Myeloma Cells to Proteasome Inhibitor Bortezomib Independent of the Unfolded Protein Response Mediator GRP78. Front. Oncol. 2020, 9, 1530. [Google Scholar] [CrossRef]
- Salimi, A.; Schroeder, K.M.; Schemionek-Reinders, M.; Vieri, M.; Maletzke, S.; Gezer, D.; Masouleh, B.K.; Appelmann, I. Targeting Autophagy Increases the Efficacy of Proteasome Inhibitor Treatment in Multiple Myeloma by Induction of Apoptosis and Activation of JNK. BMC Cancer 2022, 22, 735. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, Y.; Hu, K.; Lu, J. GRP78 Inhibitor HA15 Increases the Effect of Bortezomib on Eradicating Multiple Myeloma Cells through Triggering Endoplasmic Reticulum Stress. Heliyon 2023, 9, e19806. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 Synergistically Enhances Bortezomib-Induced Cytotoxicity in Multiple Myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.L.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Scandura, G.; Giallongo, C.; Puglisi, F.; Romano, A.; Parrinello, N.L.; Zuppelli, T.; Longhitano, L.; Giallongo, S.; Di Rosa, M.; Musumeci, G.; et al. TLR4 Signaling and Heme Oxygenase-1/Carbon Monoxide Pathway Crosstalk Induces Resiliency of Myeloma Plasma Cells to Bortezomib Treatment. Antioxidants 2022, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Sklirou, A.D.; Kastritis, E.; Liacos, C.I.; Spilioti, C.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Gavriatopoulou, M.; Terpos, E.; Trougakos, I.P.; et al. Toll-Like Receptor 4 Activation Promotes Multiple Myeloma Cell Growth and Survival Via Suppression of The Endoplasmic Reticulum Stress Factor Chop. Sci. Rep. 2019, 9, 3245. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, M.N.; Hewett, D.R.; Mrozik, K.M.; Panagopoulos, V.; Wallington-Gates, C.T.; Spencer, A.; Dold, S.M.; Engelhardt, M.; Vandyke, K.; Zannettino, A.C.W. Expression of the Chemokine Receptor CCR1 Decreases Sensitivity to Bortezomib in Multiple Myeloma Cell Lines. Leuk. Res. 2024, 139, 107469. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Abdul Aziz, A.; Bowles, K.; Rushworth, S. High NRF2 Expression Controls Endoplasmic Reticulum Stress Induced Apoptosis in Multiple Myeloma. Cancer Lett. 2018, 412, 37–45. [Google Scholar] [CrossRef]
- Vu, T.; Wang, Y.; Fowler, A.; Simieou, A.; McCarty, N. TRIM44, a Novel Prognostic Marker, Supports the Survival of Proteasome-Resistant Multiple Myeloma Cells. Cells 2024, 13, 1431. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The Extracellular Matrix as a Multitasking Player in Disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A Guide to the Composition and Functions of the Extracellular Matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Boot-Handford, R.P.; Briggs, M.D. The Unfolded Protein Response and Its Relevance to Connective Tissue Diseases. Cell Tissue Res. 2010, 339, 197–211. [Google Scholar] [CrossRef]
- Takasugi, M.; Ohtani, N.; Takemura, K.; Emmrich, S.; Zakusilo, F.T.; Yoshida, Y.; Kutsukake, N.; Mariani, J.N.; Windrem, M.S.; Chandler-Militello, D.; et al. CD44 Correlates with Longevity and Enhances Basal ATF6 Activity and ER Stress Resistance. Cell Rep. 2023, 42, 113130. [Google Scholar] [CrossRef]
- Wu, M.; Wang, C.; Gong, Y.; Huang, Y.; Jiang, L.; Zhang, M.; Gao, R.; Dang, B. Potential Mechanism of TMEM2/CD44 in Endoplasmic Reticulum Stress-induced Neuronal Apoptosis in a Rat Model of Traumatic Brain Injury. Int. J. Mol. Med. 2023, 52, 119. [Google Scholar] [CrossRef]
- Koopman, M.; Hetz, C.; Nollen, E.A.A. Saved by the Matrix: UPR Independent Survival under ER Stress. Cell 2019, 179, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, R.T.; Higuchi-Sanabria, R.; Shalem, O.; Moehle, E.A.; Webster, B.M.; Joe, L.; Bar-Ziv, R.; Frankino, P.A.; Durieux, J.; Pender, C.; et al. The Hyaluronidase, TMEM2, Promotes ER Homeostasis and Longevity Independent of the UPRER. Cell 2019, 179, 1306–1318.e18. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-Specific PERK Signaling Drives Invasion and Metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, X.; Chen, B.; Chen, B.; Song, W.; Sun, D.; Zhao, Y. Activating Transcription Factor 4 Promotes Esophageal Squamous Cell Carcinoma Invasion and Metastasis in Mice and Is Associated with Poor Prognosis in Human Patients. PLoS ONE 2014, 9, e103882. [Google Scholar] [CrossRef]
- Xia, T.; Tong, S.; Fan, K.; Zhai, W.; Fang, B.; Wang, S.-H.; Wang, J.-J. XBP1 Induces MMP-9 Expression to Promote Proliferation and Invasion in Human Esophageal Squamous Cell Carcinoma. Am. J. Cancer Res. 2016, 6, 2031–2040. [Google Scholar]
- Sun, Y.; Jiang, F.; Pan, Y.; Chen, X.; Chen, J.; Wang, Y.; Zheng, X.; Zhang, J. XBP1 Promotes Tumor Invasion and Is Associated with Poor Prognosis in Oral Squamous Cell Carcinoma. Oncol. Rep. 2018, 40, 988–998. [Google Scholar] [CrossRef]
- Rzymski, T.; Petry, A.; Kračun, D.; Rieß, F.; Pike, L.; Harris, A.L.; Görlach, A. The Unfolded Protein Response Controls Induction and Activation of ADAM17/TACE by Severe Hypoxia and ER Stress. Oncogene 2012, 31, 3621–3634. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Cmoch, A.; Wolczyk, M.; Bugajski, L.; Tkaczyk, M.; Dadlez, M.; Nieborowska-Skorska, M.; Koromilas, A.E.; Skorski, T.; Piwocka, K. Increased Phosphorylation of eIF2α in Chronic Myeloid Leukemia Cells Stimulates Secretion of Matrix Modifying Enzymes. Oncotarget 2016, 7, 79706–79721. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-Requiring Enzyme 1α Is a Key Regulator of Angiogenesis and Invasion in Malignant Glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Pluquet, O.; Dejeans, N.; Bouchecareilh, M.; Lhomond, S.; Pineau, R.; Higa, A.; Delugin, M.; Combe, C.; Loriot, S.; Cubel, G.; et al. Posttranscriptional Regulation of PER1 Underlies the Oncogenic Function of IREα. Cancer Res. 2013, 73, 4732–4743. [Google Scholar] [CrossRef]
- Limia, C.; Sauzay, C.; Urra, H.; Hetz, C.; Chevet, E.; Avril, T. Emerging Roles of the Endoplasmic Reticulum Associated Unfolded Protein Response in Cancer Cell Migration and Invasion. Cancers 2019, 11, 631. [Google Scholar] [CrossRef] [PubMed]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK Integrates Autophagy and Oxidative Stress Responses To Promote Survival during Extracellular Matrix Detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Silvers, C.R.; Messing, E.M.; Lee, Y.-F. Bladder Cancer Extracellular Vesicles Drive Tumorigenesis by Inducing the Unfolded Protein Response in Endoplasmic Reticulum of Nonmalignant Cells. J. Biol. Chem. 2019, 294, 3207–3218. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Fontana, S.; Amodio, N.; Costa, V.; Carina, V.; Bellavia, D.; Raimondo, S.; Siragusa, S.; Monteleone, F.; et al. Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis. Cancers 2020, 12, 2167. [Google Scholar] [CrossRef]
- Maltseva, D.; Raygorodskaya, M.; Knyazev, E.; Zgoda, V.; Tikhonova, O.; Zaidi, S.; Nikulin, S.; Baranova, A.; Turchinovich, A.; Rodin, S.; et al. Knockdown of the A5 Laminin Chain Affects Differentiation of Colorectal Cancer Cells and Their Sensitivity to Chemotherapy. Biochimie 2020, 174, 107–116. [Google Scholar] [CrossRef]
- Pereira, E.R.; Frudd, K.; Awad, W.; Hendershot, L.M. Endoplasmic Reticulum (ER) Stress and Hypoxia Response Pathways Interact to Potentiate Hypoxia-Inducible Factor 1 (HIF-1) Transcriptional Activity on Targets Like Vascular Endothelial Growth Factor (VEGF). J. Biol. Chem. 2014, 289, 3352–3364. [Google Scholar] [CrossRef]
- Vellanki, R.N.; Zhang, L.; Volchuk, A. OASIS/CREB3L1 Is Induced by Endoplasmic Reticulum Stress in Human Glioma Cell Lines and Contributes to the Unfolded Protein Response, Extracellular Matrix Production and Cell Migration. PLoS ONE 2013, 8, e54060. [Google Scholar] [CrossRef]
- Dejeans, N.; Pluquet, O.; Lhomond, S.; Grise, F.; Bouchecareilh, M.; Juin, A.; Meynard-Cadars, M.; Bidaud-Meynard, A.; Gentil, C.; Moreau, V.; et al. Autocrine Control of Glioma Cells Adhesion/Migration through Inositol Requiring Enzyme 1α (IRE1α)-Mediated Cleavage of Secreted Protein Acidic Rich in Cysteine (SPARC) mRNA. J. Cell Sci. 2012, 125, 4278–4287. [Google Scholar] [CrossRef]
- Khoonkari, M.; Liang, D.; Lima, M.T.; Van Der Land, T.; Liang, Y.; Sun, J.; Dolga, A.; Kamperman, M.; Van Rijn, P.; Kruyt, F.A.E. The Unfolded Protein Response Sensor PERK Mediates Stiffness-Dependent Adaptation in Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 6520. [Google Scholar] [CrossRef]
- Khoonkari, M.; Liang, D.; Kamperman, M.; Van Rijn, P.; Kruyt, F.A.E. The Unfolded Protein Response Sensor PERK Mediates Mechanical Stress-induced Maturation of Focal Adhesion Complexes in Glioblastoma Cells. FEBS Lett. 2024, 598, 3021–3035. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the Endoplasmic Reticulum: Atypical GRP78 in Cell Viability, Signalling and Therapeutic Targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto Binding to Cell Surface GRP78 Inhibits Oncogenic Cripto Signaling via MAPK/PI3K and Smad2/3 Pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Wang, W.; Zhu, Y.; Chen, Y.; Tian, B. Gemcitabine Treatment Induces Endoplasmic Reticular (ER) Stress and Subsequently Upregulates Urokinase Plasminogen Activator (uPA) to Block Mitochondrial-Dependent Apoptosis in Panc-1 Cancer Stem-like Cells (CSCs). PLoS ONE 2017, 12, e0184110. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Zhao, Y.; Li, H.; Xiao, H.; Fu, R.; Zhao, C.; Wu, H.; Li, Z. Cell-Surface GRP78 Facilitates Colorectal Cancer Cell Migration and Invasion. Int. J. Biochem. Cell Biol. 2013, 45, 987–994. [Google Scholar] [CrossRef]
- Katanasaka, Y.; Ishii, T.; Asai, T.; Naitou, H.; Maeda, N.; Koizumi, F.; Miyagawa, S.; Ohashi, N.; Oku, N. Cancer Antineovascular Therapy with Liposome Drug Delivery Systems Targeted to BiP/GRP78. Int. J. Cancer 2010, 127, 2685–2698. [Google Scholar] [CrossRef]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. GRP-78 Secreted by Tumor Cells Blocks the Antiangiogenic Activity of Bortezomib. Blood 2009, 114, 3960–3967. [Google Scholar] [CrossRef]
- Peñaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A.E. The Endoplasmic Reticulum Stress/Unfolded Protein Response in Gliomagenesis, Tumor Progression and as a Therapeutic Target in Glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Li, X.; Zhang, K.; Li, Z. Unfolded Protein Response in Cancer: The Physician’s Perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent(s)—Combined Treatment | Tumor | Phase (ID) |
|---|---|---|
| Nelfinavir i. UPR activation ii. Sensitization to BTZ | Multiple myeloma | II (NCT02188537) |
| Nelfinavir and lenalidomide/ dexamethasone i. UPR activation ii. Sensitization to BTZ | Multiple myeloma | I/II (NCT01555281) |
| 1. Ixazomib (MLN9708) i. Proteasome inhibition 2. Fulvestrant i. Estrogen receptor-downregulating antiestrogen ii. Estrogen receptor block UPR induction | ER-positive breast cancer | Ib (NCT02384746) |
| 1. Pevonedistat i. ER stress/terminal UPR induction 2. Vincristine, dexamethasone, PEG-asparaginase, and doxorubicin (VXLD) i. Overall cytotoxicity | Acute lymphoblastic leukemia and Lymphoblastic non-Hodgkin lymphoma | I (NCT03349281) |
| Monoclonal immunoglobulin M antibody PAT-SM6 i. BiP recognition/inhibition | Multiple myeloma | I (NCT01727778) |
| Fenofibrate i. Terminal UPR activation | Multiple myeloma | II (NCT01965834) |
| 1. Minnelide i. ER stress induction 2. Abraxane and gemcitabine i. Standard of care drugs | Metastatic adenocarcinoma of the pancreas | Ib (NCT05557851) |
| 1. Botensilimab (AGEN1811) and Balstilimab (AGEN2034) i. Immune-based therapy for ER stress/UPR induction 2. Nab-paclitaxel, gemcitabine and cisplatin i. Triple chemotherapy 3. Chloroquine i. Autophagy inhibition 4. Celecoxib i. Inflammation inhibition | Metastatic pancreatic cancer | I (NCT06076837) |
| NMS-03597812 i. PERK inhibition | Multiple myeloma | I (NCT05027594) |
| BTZ i. UPS inhibition | Recurrent glioma | I (NCT00006773) |
| 1. Marizomib i. UPS inhibition (BBB permeable) 2. TMZ i. Alkylating agent 3. Radiotherapy | Brain cancer | I (NCT02903069) |
| 1. Marizomib i. Alkylating agent 2. Bevacizumab i. Angiogenesis inhibition | Glioblastoma grade IV | I/II (NCT02330562) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanasopoulos, E.N.; Natsiou, A.; Kyriazopoulou, M.; Manou, D.; Theocharis, A.D.; Labropoulou, V.T. Activation of Unfolded Protein Response Pathway in Malignancies: Interplay with Extracellular Matrix and Targeting Perspectives. Cancers 2025, 17, 1972. https://doi.org/10.3390/cancers17121972
Athanasopoulos EN, Natsiou A, Kyriazopoulou M, Manou D, Theocharis AD, Labropoulou VT. Activation of Unfolded Protein Response Pathway in Malignancies: Interplay with Extracellular Matrix and Targeting Perspectives. Cancers. 2025; 17(12):1972. https://doi.org/10.3390/cancers17121972
Chicago/Turabian StyleAthanasopoulos, Eleftherios N., Angeliki Natsiou, Maria Kyriazopoulou, Dimitra Manou, Achilleas D. Theocharis, and Vassiliki T. Labropoulou. 2025. "Activation of Unfolded Protein Response Pathway in Malignancies: Interplay with Extracellular Matrix and Targeting Perspectives" Cancers 17, no. 12: 1972. https://doi.org/10.3390/cancers17121972
APA StyleAthanasopoulos, E. N., Natsiou, A., Kyriazopoulou, M., Manou, D., Theocharis, A. D., & Labropoulou, V. T. (2025). Activation of Unfolded Protein Response Pathway in Malignancies: Interplay with Extracellular Matrix and Targeting Perspectives. Cancers, 17(12), 1972. https://doi.org/10.3390/cancers17121972