
The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis


Simple Summary
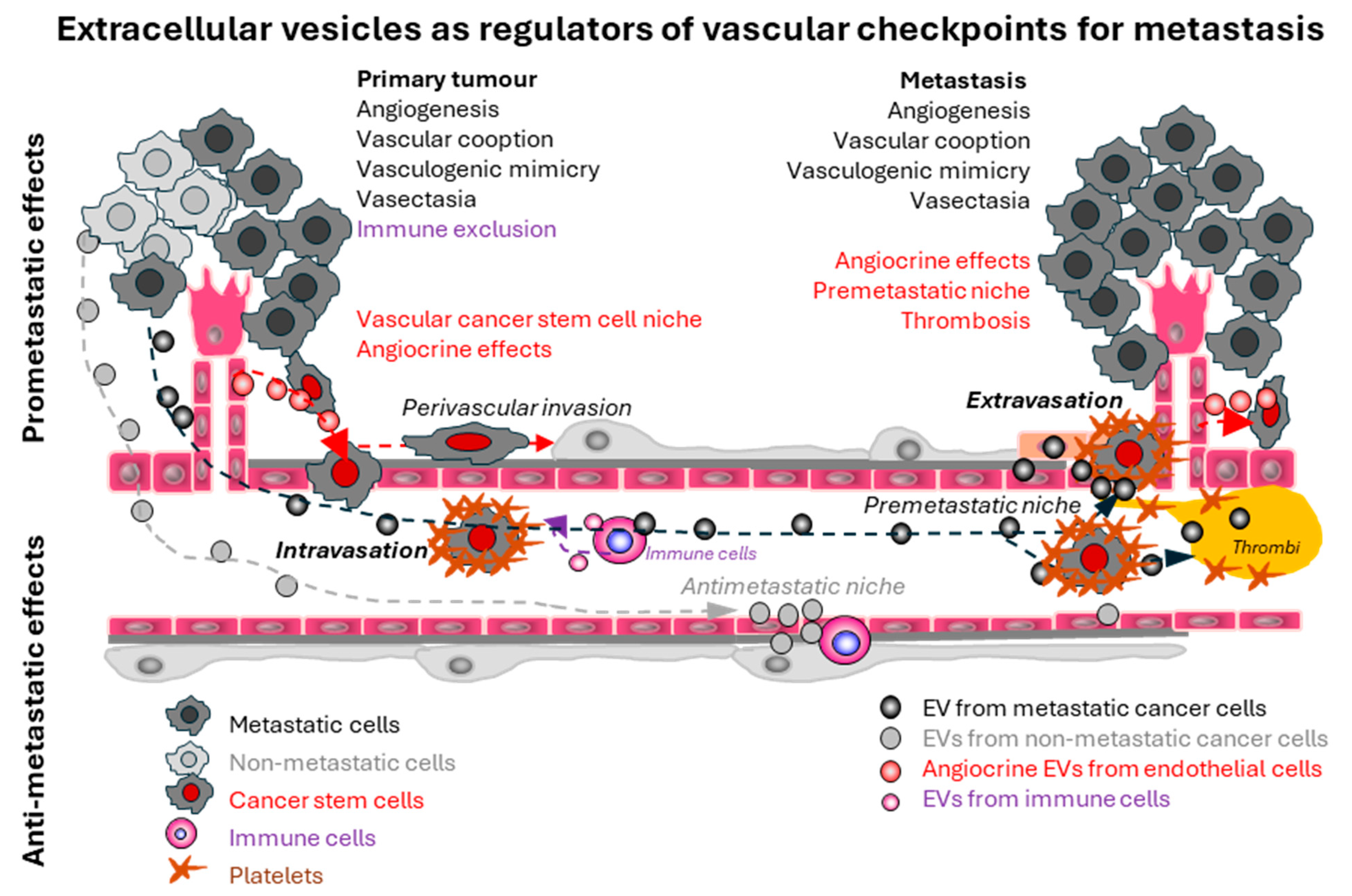
Abstract
1. Introduction—The Pivotal Role of the Vasculature in Systemic Manifestations of Cancer
2. Multifaceted Involvement of the Vasculature in Systemic Cancer Progression
3. Cancer-Associated Vascular Pathologies—Angiogenesis and Beyond
4. Alternative Neovascularization Pathways in Cancer
5. Vascular Checkpoints During Metastatic Dissemination
6. The Rise of the Particulate Secretome

7. The Emerging Biological Functions of Extracellular Vesicles and Particles
8. Extracellular Vesicle Communication as Regulatory Target of Oncogenic Pathways in Cancer
9. Impact of Oncogenic Extracellular Vesicles on the Tumour Vasculature
10. Particulate Mediators of Tumour–Vascular Communication in Cancer

{kind=link}
{kind=link}
{kind=link}
| Categories | EV Molecules | Cellular Origin | Effect on Vasculature | References |
|---|---|---|---|---|
| Angiogenesis (tumour EVs) | VEGF | Human umbilical vein endothelial cells (HUVECs) and human ovarian carcinoma A2780 cells | Promotes pro-angiogenic effect | Taraboletti, G. et al. (2006) [130] |
| VEGF | Glioblastoma cells | Promotes brain microvascular endothelial cell proliferation and migration in vitro | Skog, J. et al. (2008) [110] | |
| EGFRvIII | U373 (human astrocytoma) cells and U373-expressing EGFRvIII | Promotion of elevated VEGF secretion | Al-Nedawi, K. et al. (2008) [109] | |
| EGFR | Epidermoid carcinoma, lung adenocarcinoma and colorectal adenocarcinoma | Activation of MAPK and AKT signalling in endothelial cells and promotion VEGF autocrine signalling | Al-Nedawi, K. et al. (2009) [118] | |
| VEGF-A | Glioblastoma stem-like cells | Induction of brain EC permeability and angiogenesis | Treps, L. et al. (2017) [120] | |
| EPHB2 | Head and neck squamous cell carcinoma (OSC19, Detroit 562, SCC61, MOC1 and MOC2) | Induction of tumour angiogenesis by ephrin-B reverse signalling such as STAT3 phosphorylation (but without affecting phosphorylated VEGFR2) | Sato, S. et al. (2019) [170] | |
| Delta-like 4 | HUVECs and U87GM cells | Promotion of tip cell phenotype and endothelial tube formation | Sheldon, H. et al. (2010) [132] | |
| GGGU motif-containing miRNAs | HEK293T and CAL27 cells; oral squamous cell carcinoma (OSCC) patient tissues | EGFR boosts PCBP2 expression via transcriptional regulation, which then promotes the loading of specific miRNAs into sEVs by binding to the “GGGU” motif, thereby driving tumour angiogenesis | Xia, H. F. et al. (2025) [171] | |
| miR-210 | Breast cancer cells | Promotion of endothelial cell activation and cancer metastasis | Kosaka, N. et al. (2013) [172] | |
| miR-182-5p | Breast cancer cells (MDA-MB-231) | Promotes the proliferation, migration, and angiogenesis of HUVECs in vitro and in vivo; promotes tumourigenesis and metastasis of breast cancer cells by regulating the CMTM7/EGFR/AKT signalling axis . | Lu, C et al. (2021) [173] | |
| miR-205 | Ovarian cancer cells | Promotes angiogenesis via the PTEN-AKT pathway and induces tumour metastasis | He, L. et al. (2019) [174] | |
| FLT1 | Endothelial and nasopharyngeal carcinoma (NPC) cells | Induces a positive feedback loop between NPC cells and endothelial cells to promote tumour angiogenesis and tumour metastasis via PI3K/AKT pathway | Li, F. et al. (2025) [175] | |
| TGFb | Head and neck squamous cell carcinoma cells | TGFb-positive cancer EVs reprogram macrophages to proangiogenic phenotype | Ludwig, N. et al. (2022) [176] | |
| miR-155-5p | Melanoma cells (B16F10) | Melanoma EVs trigger proangiogenic switch in CAFs | Zhou et al. (2018) [177] | |
| Angiogenesis (stromal EVs) | VEGF, HGF, ANG-1 | Bone marrow-derived fibroblasts from multiple myeloma patients | Fibroblast-derived EVs were enriched in angiogenesis-regulating factors and impacted endothelial cells in uptake-dependent and independent manner | Lamanuzzi et al. (2022) [178] |
| miR-21-5p | Macrophages from head and neck squamous cell carcinoma (SCC25) | EVs from tumour-associated macrophages carry miR-21-5p to endothelial cells regulating YAP1/HIF1a pathway | Yan et al. (2024) [179] | |
| HOXD11 | Cancer-associated fibroblasts (CAFs) from ovarian carcinoma | CAF-EV-associated HOXD11 regulated FN1 and angiogenesis in ovarian cancer xenografts | Chen et al. (2025) [180] | |
| Non-angiogenic vascular growth | EGFR/EGFRvIII | Glioma stem cells (mesenchymal subtype) | Promotion of vasectasia (a novel neovascularization characterized by increased circumferential blood vessel growth) | Spinelli, C. et al. (2024) [60] |
| Cell migration-inducing and hyaluronan-binding protein (CEMIP) | Breast cancer cells and brain metastatic slices | Induces vascular remodelling, inflammation and vascular co-option, thereby promoting brain metastasis | Rodrigues, G. et al. (2019) [181] | |
| Endothelial barrier modification | miR-181c | Brain metastatic breast cancer cells and breast cancer patient sera | Promotion of blood–brain barrier destruction by altering tight junction proteins ZO-1, occludin and claudin-5 | Tominaga, N. et al. (2015) [35] |
| A disintegrin and metalloproteina-se 17 (ADAM17) | Colorectal cancer cells (HCT116) and patient plasma | Disrupts endothelial cell barrier and enhances vascular permeability by influencing vascular endothelial cadherin (VE-cadherin) cell membrane localization, thereby promoting lung and liver metastases | Li, K. et al. (2024) [182] | |
| Clathrin light chain A (CLTA) | Hepatocellular carcinoma cells and patient sera | Disrupts vascular endothelial barrier integrity by stabilizing basigin (BSG/CD147) to remodel microvascular niche and enhance pulmonary vessel leakage, thereby promoting liver and lung metastases | Xu, Y. et al. (2023) [183] | |
| Thrombosis/coagulation | Podoplanin, tissue factor | Glioblastoma cells | Promotion of thrombosis | Tawil, N. et al. (2021) [30] |
| Tissue factor | Colorectal cancer cells | Mediator of cancer-associated coagulation, promotion of angiogenesis | Yu, J. L. et al. (2005) [127] | |
| Others | Matrix metalloproteinases | HUVECs | Angiocrine reprogramming of glioma stem cells, contributing to tumour aggressiveness and therapy resistance | Adnani, L. et al. (2022) [19] |
| Fibronectin | HT1080 fibrosarcoma cell line | Promotion of cell adhesion and directional cell movement | Sung, B. H. et al. (2015) [129] | |
| NGFR | Melanoma cells and patient tissue samples | Promotion of lymphangiogenesis and lymph node metastasis | García-Silva, S. et al. (2024) [184] |
11. Extracellular Vesicles as Mediators of Pre-Metastatic Niche Formation
12. Therapeutic and Diagnostic Implications of EVP-Dependent Vascular Checkpoints in Metastatic Cancer—A Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Burke, K.; Cox, T.R.; Guise, T.; Jamal-Hanjani, M.; Janowitz, T.; Kaplan, R.; Lee, R.; Swanton, C.; Vander Heiden, M.G.; et al. Why do patients with cancer die? Nat. Rev. Cancer 2024, 24, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Rabas, N.; Ferreira, R.M.M.; Di Blasio, S.; Malanchi, I. Cancer-induced systemic pre-conditioning of distant organs: Building a niche for metastatic cells. Nat. Rev. Cancer 2024, 24, 829–849. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Wortzel, I.; Seo, Y.; Akano, I.; Shaashua, L.; Tobias, G.C.; Hebert, J.; Kim, K.A.; Kim, D.; Dror, S.; Liu, Y.; et al. Unique structural configuration of EV-DNA primes Kupffer cell-mediated antitumor immunity to prevent metastatic progression. Nat. Cancer 2024, 5, 1815–1833. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Cao, M.; Isaac, R.; Yan, W.; Ruan, X.; Jiang, L.; Wan, Y.; Wang, J.; Wang, E.; Caron, C.; Neben, S.; et al. Cancer-cell-secreted extracellular vesicles suppress insulin secretion through miR-122 to impair systemic glucose homeostasis and contribute to tumour growth. Nat. Cell Biol. 2022, 24, 954–967. [Google Scholar] [CrossRef]
- Wang, G.; Li, J.; Bojmar, L.; Chen, H.; Li, Z.; Tobias, G.C.; Hu, M.; Homan, E.A.; Lucotti, S.; Zhao, F.; et al. Tumour extracellular vesicles and particles induce liver metabolic dysfunction. Nature 2023, 618, 374–382. [Google Scholar] [CrossRef]
- Sardiña González, C.; Martínez Vivero, C.; López Castro, J. Paraneoplastic syndromes review: The great forgotten ones. Crit. Rev. Oncol. Hematol. 2022, 174, 103676. [Google Scholar] [CrossRef]
- Ma, G.M.; Chow, J.S.; Taylor, G.A. Review of paraneoplastic syndromes in children. Pediatr. Radiol. 2019, 49, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- Ager, A.; Watson, H.A.; Wehenkel, S.C.; Mohammed, R.N. Homing to solid cancers: A vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem. Soc. Trans. 2016, 44, 377–385. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef]
- Adnani, L.; Spinelli, C.; Tawil, N.; Rak, J. Role of extracellular vesicles in cancer-specific interactions between tumour cells and the vasculature. Semin. Cancer Biol. 2022, 87, 196–213. [Google Scholar] [CrossRef]
- Al-Zoughbi, W.; Al-Zhoughbi, W.; Huang, J.; Paramasivan, G.S.; Till, H.; Pichler, M.; Guertl-Lackner, B.; Hoefler, G. Tumor macroenvironment and metabolism. Semin. Oncol. 2014, 41, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br. J. Cancer 1968, 22, 258–273. [Google Scholar] [CrossRef]
- Rak, J.W.; Hegmann, E.J.; Lu, C.; Kerbel, R.S. Progressive loss of sensitivity to endothelium-derived growth inhibitors expressed by human melanoma cells during disease progression. J. Cell Physiol. 1994, 159, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Adnani, L.; Kassouf, J.; Meehan, B.; Spinelli, C.; Tawil, N.; Nakano, I.; Rak, J. Angiocrine extracellular vesicles impose mesenchymal reprogramming upon proneural glioma stem cells. Nat. Commun. 2022, 13, 5494. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial Cells Promote the Colorectal Cancer Stem Cell Phenotype through a Soluble Form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Rak, J. Is cancer stem cell a cell, or a multicellular unit capable of inducing angiogenesis? Med. Hypotheses 2006, 66, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Saidel, M.G.; Kleinerman, J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976, 36, 889–894. [Google Scholar]
- Nasr, M.M.; Lynch, C.C. How circulating tumor cluster biology contributes to the metastatic cascade: From invasion to dissemination and dormancy. Cancer Metastasis Rev. 2023, 42, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef]
- Huinen, Z.R.; Huijbers, E.J.M.; van Beijnum, J.R.; Nowak-Sliwinska, P.; Griffioen, A.W. Anti-angiogenic agents-overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat. Rev. Clin. Oncol. 2021, 18, 527–540. [Google Scholar] [CrossRef]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massagué, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef]
- van de Walle, T.; Vaccaro, A.; Ramachandran, M.; Pietilä, I.; Essand, M.; Dimberg, A. Tertiary Lymphoid Structures in the Central Nervous System: Implications for Glioblastoma. Front. Immunol. 2021, 12, 724739. [Google Scholar] [CrossRef]
- Tawil, N.; Bassawon, R.; Meehan, B.; Nehme, A.; Montermini, L.; Gayden, T.; De Jay, N.; Spinelli, C.; Chennakrishnaiah, S.; Choi, D.; et al. Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv. 2021, 5, 1682–1694. [Google Scholar] [CrossRef]
- Khorana, A.A.; Mackman, N.; Falanga, A.; Pabinger, I.; Noble, S.; Ageno, W.; Moik, F.; Lee, A.Y.Y. Cancer-associated venous thromboembolism. Nat. Rev. Dis. Primers 2022, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S.; Ogitani, Y.; Kenific, C.M.; Geri, J.; Kim, Y.H.; Gu, J.; Balaji, U.; Bojmar, L.; Shaashua, L.; Song, Y.; et al. Extracellular vesicles from the lung pro-thrombotic niche drive cancer-associated thrombosis and metastasis via integrin beta 2. Cell 2025, 188, 1642–1661.e24. [Google Scholar] [CrossRef] [PubMed]
- Ades, S.; Kumar, S.; Alam, M.; Goodwin, A.; Weckstein, D.; Dugan, M.; Ashikaga, T.; Evans, M.; Verschraegen, C.; Holmes, C.E. Tumor oncogene (KRAS) status and risk of venous thrombosis in patients with metastatic colorectal cancer. J. Thromb. Haemost. 2015, 13, 998–1003. [Google Scholar] [CrossRef]
- Meehan, B.; Dombrovsky, A.; Magnus, N.; Rak, J. Arteriogenic expansion of extratumoral macrovessels-impact of vascular ageing. Neoplasma 2015, 62, 372–383. [Google Scholar] [CrossRef]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lotvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Hu, Z.; Barney, K.A.; Degen, J.L. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 2007, 110, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Betsholtz, C. Cell-cell signaling in blood vessel development and function. EMBO Mol. Med. 2018, 10, e8610. [Google Scholar] [CrossRef]
- Rak, J.; Emmenegger, U. Angiogenesis. In The Basic Science of Oncology, 6th ed.; Harrington, L.A., Tannock, I.F., Hill, R.P., Cescon, D.W., Eds.; McGraw-Hill Education: New York, NY, USA, 2021. [Google Scholar]
- Cao, Y.; Langer, R.; Ferrara, N. Targeting angiogenesis in oncology, ophthalmology and beyond. Nat. Rev. Drug Discov. 2023, 22, 476–495. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Rossant, J. Cell fate decisions in early blood vessel formation. Trends Cardiovasc. Med. 2003, 13, 254–259. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, M.J.; Kumar, A.; Lee, H.W.; Yang, Y.; Kim, Y. Vascular endothelial growth factor signaling in health and disease: From molecular mechanisms to therapeutic perspectives. Signal Transduct. Target. Ther. 2025, 10, 170. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Unruh, D.; Schwarze, S.R.; Khoury, L.; Thomas, C.; Wu, M.; Chen, L.; Chen, R.; Liu, Y.; Schwartz, M.A.; Amidei, C.; et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. 2016, 132, 917–930. [Google Scholar] [CrossRef]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Gesierich, S.; Berezovskiy, I.; Ryschich, E.; Zoller, M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. 2006, 66, 7083–7094. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Dome, B.; Hendrix, M.J.; Paku, S.; Tovari, J.; Timar, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Patan, S.; Munn, L.L.; Jain, R.K. Intussusceptive microvascular growth in a human colon adenocarcinoma xenograft: A novel mechanism of tumor angiogenesis. Microvasc. Res. 1996, 51, 260–272. [Google Scholar] [CrossRef]
- Mathivet, T.; Bouleti, C.; Van Woensel, M.; Stanchi, F.; Verschuere, T.; Phng, L.K.; Dejaegher, J.; Balcer, M.; Matsumoto, K.; Georgieva, P.B.; et al. Dynamic stroma reorganization drives blood vessel dysmorphia during glioma growth. EMBO Mol. Med. 2017, 9, 1629–1645. [Google Scholar] [CrossRef]
- Spinelli, C.; Adnani, L.; Meehan, B.; Montermini, L.; Huang, S.; Kim, M.; Nishimura, T.; Croul, S.E.; Nakano, I.; Riazalhosseini, Y.; et al. Mesenchymal glioma stem cells trigger vasectasia-distinct neovascularization process stimulated by extracellular vesicles carrying EGFR. Nat. Commun. 2024, 15, 2865. [Google Scholar] [CrossRef]
- Karakousi, T.; Mudianto, T.; Lund, A.W. Lymphatic vessels in the age of cancer immunotherapy. Nat. Rev. Cancer 2024, 24, 363–381. [Google Scholar] [CrossRef]
- Karnezis, T.; Shayan, R.; Ceasar, C.; Roufail, S.; Harris, N.C.; Ardipradja, K.; Zhang, Y.F.; Williams, S.P.; Farnsworth, R.H.; Chai, M.G.; et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 2012, in press. [CrossRef] [PubMed]
- Rak, J. VEGF-D(ilated) Lymphatics as Gateways to Metastasis. Cancer Cell 2012, 21, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef]
- Wälchli, T.; Ghobrial, M.; Schwab, M.; Takada, S.; Zhong, H.; Suntharalingham, S.; Vetiska, S.; Gonzalez, D.R.; Wu, R.; Rehrauer, H.; et al. Single-cell atlas of the human brain vasculature across development, adulthood and disease. Nature 2024, 632, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e720. [Google Scholar] [CrossRef]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.C.; Pircher, A.; Geldhof, V.; de Rooij, L.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, C.Y.; Lawson, D.A.; Kwek, S.; Velozo, H.G.; Owyong, M.; Lai, M.D.; Fong, L.; Wilson, M.; Su, H.; et al. Single-cell RNA sequencing reveals gene expression signatures of breast cancer-associated endothelial cells. Oncotarget 2018, 9, 10945–10961. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Fidler, I.J. Biological heterogeneity of cancer: Implication to therapy. Hum. Vaccin. Immunother. 2012, 8, 1141–1142. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef]
- Dankner, M.; Maritan, S.M.; Priego, N.; Kruck, G.; Nkili-Meyong, A.; Nadaf, J.; Zhuang, R.; Annis, M.G.; Zuo, D.; Nowakowski, A.; et al. Invasive growth of brain metastases is linked to CHI3L1 release from pSTAT3-positive astrocytes. Neuro Oncol. 2024, 26, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic, M.M.; Molina, H.; Kohsaka, S.; Di, G.A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Steeg, P.S. The blood-tumour barrier in cancer biology and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Tsamchoe, M.; Lazaris, A.; Kim, D.; Krzywon, L.; Bloom, J.; Mayer, T.; Petrillo, S.K.; Dejgaard, K.; Gao, Z.-H.; Rak, J.; et al. Circulating extracellular vesicles containing S100A9 reflect histopathology, immunophenotype and therapeutic responses of liver metastasis in colorectal cancer patients. BJC Rep. 2023, 1, 8. [Google Scholar] [CrossRef]
- Garzia, L.; Kijima, N.; Morrissy, A.S.; De Antonellis, P.; Guerreiro-Stucklin, A.; Holgado, B.L.; Wu, X.; Wang, X.; Parsons, M.; Zayne, K.; et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 2018, 173, 1549. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Skobe, M.; Rockwell, P.; Goldstein, N.; Vosseler, S.; Fusenig, N.E. Halting angiogenesis suppresses carcinoma cell invasion. Nat. Med. 1997, 3, 1222–1227. [Google Scholar] [CrossRef]
- Jung, E.; Osswald, M.; Ratliff, M.; Dogan, H.; Xie, R.; Weil, S.; Hoffmann, D.C.; Kurz, F.T.; Kessler, T.; Heiland, S.; et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat. Commun. 2021, 12, 1014. [Google Scholar] [CrossRef]
- Barnhill, R.L.; Fandrey, K.; Levy, M.A.; Mihm, M.C., Jr.; Hyman, B. Angiogenesis and tumor progression of melanoma. Quantification of vascularity in melanocytic nevi and cutaneous malignant melanoma. Lab. Investig. 1992, 67, 331–337. [Google Scholar] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef]
- Miller, F.R. Tumor subpopulation interactions in metastasis. Inv. Metast 1983, 3, 234–242. [Google Scholar]
- Gorelik, E.; Beere, W.W.; Herberman, R.B. Role of the NK cells in the anti-metastatic effect of anti-coagulant drug. Int. J. Cancer 1984, 33, 87. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 833–891. [Google Scholar] [CrossRef]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Thetchinamoorthy, K.; Wierzbicka, D.; Konopko, A.; Ratajczak, J.; Kucia, M. Extracellular microvesicles/exosomes-magic bullets in horizontal transfer between cells of mitochondria and molecules regulating mitochondria activity. Stem Cells 2025, 43, sxae086. [Google Scholar] [CrossRef]
- Hausmann, D.; Hoffmann, D.C.; Venkataramani, V.; Jung, E.; Horschitz, S.; Tetzlaff, S.K.; Jabali, A.; Hai, L.; Kessler, T.; Azoŕin, D.D.; et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 2023, 613, 179–186. [Google Scholar] [CrossRef]
- Winkler, F.; Venkatesh, H.S.; Amit, M.; Batchelor, T.; Demir, I.E.; Deneen, B.; Gutmann, D.H.; Hervey-Jumper, S.; Kuner, T.; Mabbott, D.; et al. Cancer neuroscience: State of the field, emerging directions. Cell 2023, 186, 1689–1707. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Dixson, A.C.; Dawson, T.R.; Di Vizio, D.; Weaver, A.M. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell Biol. 2023, 24, 454–476. [Google Scholar] [CrossRef]
- D’Asti, E.; Kool, M.; Korshunov, A.; Huang, A.; Pfister, S.; Rak, J. Molecular subtypes define the composition of coagulome in medulloblastoma. 2013; Manuscript in preparation. [Google Scholar]
- Jeppesen, D.K.; Sanchez, Z.C.; Kelley, N.M.; Hayes, J.B.; Ambroise, J.; Koory, E.N.; Krystofiak, E.; Taneja, N.; Zhang, Q.; Dungan, M.M.; et al. Blebbisomes are large, organelle-rich extracellular vesicles with cell-like properties. Nat. Cell Biol. 2025, 27, 438–448. [Google Scholar] [CrossRef]
- Rak, J.; Strzadala, L. Heterogeneity of Extracellular Vesicles and Particles: Molecular Voxels in the Blood Borne “Hologram” of Organ Function, Disfunction and Cancer. Arch. Immunol. Ther. Exp. 2023, 71, 5. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Montermini, L.; Jeong, H.; Sharma, S.; Meehan, B.; Rak, J. Mapping Subpopulations of Cancer Cell-Derived Extracellular Vesicles and Particles by Nano-Flow Cytometry. ACS Nano 2019, 13, 10499–10511. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Van Der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles 2020, 9, 1713526. [Google Scholar] [CrossRef]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Jalali, M.; Del Real Mata, C.; Montermini, L.; Jeanne, O.; Hosseini, I.I.; Gu, Z.; Spinelli, C.; Lu, Y.; Tawil, N.; Guiot, M.C.; et al. MoS(2)-Plasmonic Nanocavities for Raman Spectra of Single Extracellular Vesicles Reveal Molecular Progression in Glioblastoma. ACS Nano 2023, 17, 12052–12071. [Google Scholar] [CrossRef] [PubMed]
- Barman, B.; Sung, B.H.; Krystofiak, E.; Ping, J.; Ramirez, M.; Millis, B.; Allen, R.; Prasad, N.; Chetyrkin, S.; Calcutt, M.W.; et al. VAP-A and its binding partner CERT drive biogenesis of RNA-containing extracellular vesicles at ER membrane contact sites. Dev. Cell 2022, 57, 974–994.e978. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Han, T.; Song, P.; Wu, Z.; Liu, Y.; Ying, W.; Shen, C. Inflammation Modifies miR-21 Expression Within Neuronal Extracellular Vesicles to Regulate Remyelination Following Spinal Cord Injury. Stem Cell Rev. Rep. 2023, 19, 2024–2037. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Graves-Deal, R.; Trinh, V.Q.; Ramirez, M.A.; Sohn, Y.; Neininger, A.C.; Taneja, N.; McKinley, E.T.; et al. Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nat. Cell Biol. 2021, 23, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.A.; Petrova, S.; Pound, J.D.; Voss, J.J.; Melville, L.; Paterson, M.; Farnworth, S.L.; Gallimore, A.M.; Cuff, S.; Wheadon, H.; et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr. Biol. 2015, 25, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.D.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef]
- Zhang, C.; Li, T.; Yin, S.; Gao, M.; He, H.; Li, Y.; Jiang, D.; Shi, M.; Wang, J.; Yu, L. Monocytes deposit migrasomes to promote embryonic angiogenesis. Nat. Cell Biol. 2022, 24, 1726–1738. [Google Scholar] [CrossRef]
- Kim, C.W.; Lee, H.M.; Lee, T.H.; Kang, C.; Kleinman, H.K.; Gho, Y.S. Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer Res. 2002, 62, 6312–6317. [Google Scholar] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef]
- Wang, J.G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J.; et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood 2012, 19, 5543–5552. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Rak, J. Extracellular vesicles-biomarkers and effectors of the cellular interactome in cancer. Front. Pharmacol. 2013, 4, 21. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A brief history of nearly EV-erything-The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Quay, S.C.; Orenstein, N.S.; Dvorak, A.M.; Hahn, P.; Bitzer, A.M.; Carvalho, A.C. Tumor shedding and coagulation. Science 1981, 212, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; May, L.; Lhotak, V.; Shahrzad, S.; Shirasawa, S.; Weitz, J.I.; Coomber, B.L.; Mackman, N.; Rak, J.W. Oncogenic events regulate tissue factor expression in colorectal cancer cells: Implications for tumor progression and angiogenesis. Blood 2005, 105, 1734–1741. [Google Scholar] [CrossRef]
- Hendrix, A.; Westbroek, W.; Bracke, M.; De, W.O. An ex(o)citing machinery for invasive tumor growth. Cancer Res. 2010, 70, 9533–9537. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef]
- Taraboletti, G.; D’Ascenzo, S.; Giusti, I.; Marchetti, D.; Borsotti, P.; Millimaggi, D.; Giavazzi, R.; Pavan, A.; Dolo, V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia 2006, 8, 96–103. [Google Scholar] [CrossRef]
- Yang, B.; Lin, Y.; Huang, Y.; Zhu, N.; Shen, Y.Q. Extracellular vesicles modulate key signalling pathways in refractory wound healing. Burns Trauma. 2023, 11, tkad039. [Google Scholar] [CrossRef]
- Sheldon, H.; Heikamp, E.; Turley, H.; Dragovic, R.; Thomas, P.; Oon, C.E.; Leek, R.; Edelmann, M.; Kessler, B.; Sainson, R.C.; et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood 2010, 116, 2385–2394. [Google Scholar] [CrossRef]
- Higginbotham, J.N.; Demory, B.M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef]
- Ikeda, H.; Kawase, K.; Nishi, T.; Watanabe, T.; Takenaga, K.; Inozume, T.; Ishino, T.; Aki, S.; Lin, J.; Kawashima, S.; et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 2025, 638, 225–236. [Google Scholar] [CrossRef]
- Ridder, K.; Keller, S.; Dams, M.; Rupp, A.K.; Schlaudraff, J.; Turco, D.D.; Starmann, J.; Macas, J.; Karpova, D.; Devraj, K.; et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS. Biol. 2014, 12, e1001874. [Google Scholar] [CrossRef]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.; et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- de Jong, O.G.; Murphy, D.E.; Mäger, I.; Willms, E.; Garcia-Guerra, A.; Gitz-Francois, J.J.; Lefferts, J.; Gupta, D.; Steenbeek, S.C.; van Rheenen, J.; et al. A CRISPR-Cas9-based reporter system for single-cell detection of extracellular vesicle-mediated functional transfer of RNA. Nat. Commun. 2020, 11, 1113. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Poste, G.; Nicolson, G.L. Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc. Natl. Acad. Sci. USA 1980, 77, 399–403. [Google Scholar] [CrossRef]
- Bastón, E.; García-Agulló, J.; Peinado, H. The influence of extracellular vesicles on tumor evolution and resistance to therapy. Physiol. Rev. 2025, 105, 1173–1212. [Google Scholar] [CrossRef] [PubMed]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Lee, T.H.; Spinelli, C.; Chennakrishnaiah, S.; D’Asti, E.; Rak, J. Extracellular vesicle communication pathways as regulatory targets of oncogenic transformation. Semin. Cell Dev. Biol. 2017, 67, 11–22. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-regulated release of extracellular vesicles. Dev. Cell 2021, 56, 1989–2006.e1986. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.; Boutros, M. Extracellular vesicles and oncogenic signaling. Mol. Oncol. 2021, 15, 3–26. [Google Scholar] [CrossRef]
- Choi, D.; Montermini, L.; Kim, D.K.; Meehan, B.; Roth, F.P.; Rak, J. The Impact of Oncogenic EGFRvIII on the Proteome of Extracellular Vesicles Released from Glioblastoma Cells. Mol. Cell Proteom. 2018, 17, 1948–1964. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Hoshino, D.; Hong, N.H.; Cha, D.J.; Franklin, J.L.; Coffey, R.J.; Patton, J.G.; Weaver, A.M. KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep. 2016, 15, 978–987. [Google Scholar] [CrossRef]
- Cha, D.J.; Franklin, J.L.; Dou, Y.; Liu, Q.; Higginbotham, J.N.; Demory Beckler, M.; Weaver, A.M.; Vickers, K.; Prasad, N.; Levy, S.; et al. KRAS-dependent sorting of miRNA to exosomes. Elife 2015, 4, e07197. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Meehan, B.; Rak, J.; Di Vizio, D. Oncosomes-large and small: What are they, where they came from? J. Extracell. Vesicles 2016, 5, 33109. [Google Scholar] [CrossRef]
- Montermini, L.; Meehan, B.; Garnier, D.; Lee, W.J.; Lee, T.H.; Guha, A.; Al-Nedawi, K.; Rak, J. Inhibition of oncogenic epidermal growth factor receptor kinase triggers release of exosome-like extracellular vesicles and impacts their phosphoprotein and DNA content. J. Biol. Chem. 2015, 290, 24534–24546. [Google Scholar] [CrossRef]
- Spinelli, C.; Montermini, L.; Meehan, B.; Brisson, A.R.; Tan, S.; Choi, D.; Nakano, I.; Rak, J. Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J. Extracell. Vesicles 2018, 7, 1490144. [Google Scholar] [CrossRef]
- Chennakrishnaiah, S.; Tsering, T.; Gregory, C.; Tawil, N.; Spinelli, C.; Montermini, L.; Karatzas, N.; Aprikian, S.; Choi, D.; Klewes, L.; et al. Extracellular vesicles from genetically unstable, oncogene-driven cancer cells trigger micronuclei formation in endothelial cells. Sci. Rep. 2020, 10, 8532. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Kobayashi, N.B.; Takatani-Nakase, T.; Yoshida, T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep. 2015, 5, 10300. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef]
- Choi, D.; Montermini, L.; Meehan, B.; Lazaris, A.; Metrakos, P.; Rak, J. Oncogenic RAS drives the CRAF-dependent extracellular vesicle uptake mechanism coupled with metastasis. J. Extracell. Vesicles 2021, 10, e12091. [Google Scholar] [CrossRef]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef]
- Viloria-Petit, A.; Miquerol, L.; Yu, J.L.; Gertsenstein, M.; Sheehan, C.; May, L.; Henkin, J.; Lobe, C.; Nagy, A.; Kerbel, R.S.; et al. Contrasting effects of VEGF gene disruption in embryonic stem cell-derived versus oncogene-induced tumors. EMBO J. 2003, 22, 4091–4102. [Google Scholar] [CrossRef] [PubMed]
- Magnus, N.; Garnier, D.; Rak, J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 2010, 116, 815–818. [Google Scholar] [CrossRef]
- Chinot, O.L.; Reardon, D.A. The future of antiangiogenic treatment in glioblastoma. Curr. Opin. Neurol. 2014, 27, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. NPJ Genom. Med. 2020, 5, 2. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef]
- Wei, X.; Meel, M.H.; Breur, M.; Bugiani, M.; Hulleman, E.; Phoenix, T.N. Defining tumor-associated vascular heterogeneity in pediatric high-grade and diffuse midline gliomas. Acta Neuropathol. Commun. 2021, 9, 142. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Santi, A.; Zanivan, S. Extracellular vesicles as central regulators of blood vessel function in cancer. Sci. Signal 2022, 15, eaaz4742. [Google Scholar] [CrossRef]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular Vesicles in Angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Delle Monache, S.; Di Francesco, M.; Sanità, P.; D’Ascenzo, S.; Gravina, G.L.; Festuccia, C.; Dolo, V. From glioblastoma to endothelial cells through extracellular vesicles: Messages for angiogenesis. Tumour Biol. 2016, 37, 12743–12753. [Google Scholar] [CrossRef]
- Sato, S.; Vasaikar, S.; Eskaros, A.; Kim, Y.; Lewis, J.S.; Zhang, B.; Zijlstra, A.; Weaver, A.M. EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. JCI Insight 2019, 4, e132447. [Google Scholar] [CrossRef]
- Xia, H.F.; Wang, X.L.; Zhang, H.J.; Wang, K.M.; Zhang, L.Z.; Yang, Y.; Shi, X.; Chen, G. PCBP2-dependent secretion of miRNAs via extracellular vesicles contributes to the EGFR-driven angiogenesis. Theranostics 2025, 15, 1255–1271. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- Lu, C.; Zhao, Y.; Wang, J.; Shi, W.; Dong, F.; Xin, Y.; Zhao, X.; Liu, C. Breast cancer cell-derived extracellular vesicles transfer miR-182-5p and promote breast carcinogenesis via the CMTM7/EGFR/AKT axis. Mol. Med. 2021, 27, 78. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhu, W.; Chen, Q.; Yuan, Y.; Wang, Y.; Wang, J.; Wu, X. Ovarian cancer cell-secreted exosomal miR-205 promotes metastasis by inducing angiogenesis. Theranostics 2019, 9, 8206–8220. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, L.; He, Y.; Chen, P.; Wang, J.; Zeng, M.; Li, C.; Chen, J.; Chen, H.; Guo, Q.; et al. FLT1-enriched extracellular vesicles induce a positive feedback loop between nasopharyngeal carcinoma cells and endothelial cells to promote angiogenesis and tumour metastasis. Oncogene 2025. ahead of print. [Google Scholar] [CrossRef]
- Ludwig, N.; Yerneni, S.S.; Azambuja, J.H.; Pietrowska, M.; Widłak, P.; Hinck, C.S.; Głuszko, A.; Szczepański, M.J.; Kärmer, T.; Kallinger, I.; et al. TGFβ(+) small extracellular vesicles from head and neck squamous cell carcinoma cells reprogram macrophages towards a pro-angiogenic phenotype. J. Extracell. Vesicles 2022, 11, e12294. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef]
- Lamanuzzi, A.; Saltarella, I.; Reale, A.; Melaccio, A.; Solimando, A.G.; Altamura, C.; Tamma, G.; Storlazzi, C.T.; Tolomeo, D.; Desantis, V.; et al. Uptake-Dependent and -Independent Effects of Fibroblasts-Derived Extracellular Vesicles on Bone Marrow Endothelial Cells from Patients with Multiple Myeloma: Therapeutic and Clinical Implications. Biomedicines 2023, 11, 1400. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Liu, J.; Liu, Y.; Wen, Z.; Jin, D.; Wang, F.; Gao, L. Tumor-associated macrophage-derived exosomal miR21-5p promotes tumor angiogenesis by regulating YAP1/HIF-1α axis in head and neck squamous cell carcinoma. Cell Mol. Life Sci. 2024, 81, 179. [Google Scholar] [CrossRef]
- Chen, C.; Wang, F.; Cheng, C.; Li, H.; Fan, Y.; Jia, L. Cancer-associated Fibroblasts-derived Exosomes with HOXD11 Overexpression Promote Ovarian Cancer Cell Angiogenesis via FN1. Reprod. Sci. 2025, 32, 1530–1544. [Google Scholar] [CrossRef]
- Rodrigues, G.; Hoshino, A.; Kenific, C.M.; Matei, I.R.; Steiner, L.; Freitas, D.; Kim, H.S.; Oxley, P.R.; Scandariato, I.; Casanova-Salas, I.; et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat. Cell Biol. 2019, 21, 1403–1412. [Google Scholar] [CrossRef]
- Li, K.; Xue, W.; Lu, Z.; Wang, S.; Zheng, J.; Lu, K.; Li, M.; Zong, Y.; Xu, F.; Dai, J.; et al. Tumor-derived exosomal ADAM17 promotes pre-metastatic niche formation by enhancing vascular permeability in colorectal cancer. J. Exp. Clin. Cancer Res. 2024, 43, 59. [Google Scholar] [CrossRef]
- Xu, Y.; Yao, Y.; Yu, L.; Zhang, X.; Mao, X.; Tey, S.K.; Wong, S.W.K.; Yeung, C.L.S.; Ng, T.H.; Wong, M.Y.; et al. Clathrin light chain A-enriched small extracellular vesicles remodel microvascular niche to induce hepatocellular carcinoma metastasis. J. Extracell. Vesicles 2023, 12, e12359. [Google Scholar] [CrossRef]
- García-Silva, S.; Peinado, H. Mechanisms of lymph node metastasis: An extracellular vesicle perspective. Eur. J. Cell Biol. 2024, 103, 151447. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, C.; Adnani, L.; Choi, D.; Rak, J. Extracellular Vesicles as Conduits of Non-Coding RNA Emission and Intercellular Transfer in Brain Tumors. Noncoding RNA 2018, 5, 1. [Google Scholar] [CrossRef]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074.e2064. [Google Scholar] [CrossRef] [PubMed]
- Peña-Flores, J.A.; Muela-Campos, D.; Guzmán-Medrano, R.; Enríquez-Espinoza, D.; González-Alvarado, K. Functional Relevance of Extracellular Vesicle-Derived Long Non-Coding and Circular RNAs in Cancer Angiogenesis. Noncoding RNA 2024, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Ehnfors, J.; Kost-Alimova, M.; Persson, N.L.; Bergsmedh, A.; Castro, J.; Levchenko-Tegnebratt, T.; Yang, L.; Panaretakis, T.; Holmgren, L. Horizontal transfer of tumor DNA to endothelial cells in vivo. Cell Death. Differ. 2009, 16, 749–757. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Naito, Y.; Yoshioka, Y.; Ochiya, T. Intercellular crosstalk between cancer cells and cancer-associated fibroblasts via extracellular vesicles. Cancer Cell Int. 2022, 22, 367. [Google Scholar] [CrossRef]
- Kuriyama, N.; Yoshioka, Y.; Kikuchi, S.; Azuma, N.; Ochiya, T. Extracellular Vesicles Are Key Regulators of Tumor Neovasculature. Front. Cell Dev. Biol. 2020, 8, 611039. [Google Scholar] [CrossRef] [PubMed]
- Miaomiao, S.; Xiaoqian, W.; Yuwei, S.; Chao, C.; Chenbo, Y.; Yinghao, L.; Yichen, H.; Jiao, S.; Kuisheng, C. Cancer-associated fibroblast-derived exosome microRNA-21 promotes angiogenesis in multiple myeloma. Sci. Rep. 2023, 13, 9671. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhu, H.; Jiang, H.; Yue, H.; Yuan, F.; Wang, F. Cancer-associated fibroblasts-derived exosomes from chemoresistant patients regulate cisplatin resistance and angiogenesis by delivering VEGFA in colorectal cancer. Anticancer. Drugs 2023, 34, 422–430. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Wang, X.; Zou, B.; Mei, J.; Peng, X.; Wu, Z. Extracellular vesicles-encapsulated microRNA-10a-5p shed from cancer-associated fibroblast facilitates cervical squamous cell carcinoma cell angiogenesis and tumorigenicity via Hedgehog signaling pathway. Cancer Gene Ther. 2021, 28, 529–542. [Google Scholar] [CrossRef]
- Njock, M.S.; O’Grady, T.; Nivelles, O.; Lion, M.; Jacques, S.; Cambier, M.; Herkenne, S.; Muller, F.; Christian, A.; Remacle, C.; et al. Endothelial extracellular vesicles promote tumour growth by tumour-associated macrophage reprogramming. J. Extracell. Vesicles 2022, 11, e12228. [Google Scholar] [CrossRef]
- Wortzel, I. EV-DNA is uniquely chromatinized and primes anti-tumor immunity to prevent metastatic progression. In Proceedings of the ISEV2023 Annual Meeting, Seattle, WA, USA, 17–21 May 2023. [Google Scholar]
- Asao, T.; Tobias, G.C.; Lucotti, S.; Jones, D.R.; Matei, I.; Lyden, D. Extracellular vesicles and particles as mediators of long-range communication in cancer: Connecting biological function to clinical applications. Extracell. Vesicles Circ. Nucl. Acids 2023, 4, 461–485. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Urabe, F.; Patil, K.; Ramm, G.A.; Ochiya, T.; Soekmadji, C. Extracellular vesicles in the development of organ-specific metastasis. J. Extracell. Vesicles 2021, 10, e12125. [Google Scholar] [CrossRef]
- Fidler, I.J. Selection of successive cell lines for metastasis. Nature 1973, 242, 148–149. [Google Scholar]
- Hunter, K. Host genetics influence tumour metastasis. Nat. Rev. Cancer 2006, 6, 141–146. [Google Scholar] [CrossRef]
- Strzadala, L.; Rak, I.; Ziolo, E.; Paprocka, M.; Radzikowski, C.; Den, O.W. Non-cytotoxic asialo-GM1-positive cells exert antimetastatic activity. Cancer Immunol. Immunother. 1989, 30, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, G.; Auer, R. “Don’t let it to air”: A cautionary tale of the potential consequences of surgery of residual cancer. Brain Behav. Immun. 2023, 111, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Rak, J. Anti-metastatic extracellular vesicles carrying DNA. Nat. Cancer 2024, 5, 1793–1795. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Whiteside, T.L. Potential roles of tumor-derived exosomes in angiogenesis. Expert. Opin. Ther. Targets 2018, 22, 409–417. [Google Scholar] [CrossRef]
- Hyenne, V.; Ghoroghi, S.; Collot, M.; Bons, J.; Follain, G.; Harlepp, S.; Mary, B.; Bauer, J.; Mercier, L.; Busnelli, I.; et al. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Dev. Cell 2019, 48, 554–572.e557. [Google Scholar] [CrossRef]
- Chennakrishnaiah, S.; Meehan, B.; D’Asti, E.; Montermini, L.; Lee, T.H.; Karatzas, N.; Buchanan, M.; Tawil, N.; Choi, D.; Divangahi, M.; et al. Leukocytes as a reservoir of circulating oncogenic DNA and regulatory targets of tumor-derived extracellular vesicles. J. Thromb. Haemost. 2018, 16, 1800–1813. [Google Scholar] [CrossRef]
- Liu, K.; Dou, R.; Yang, C.; Di, Z.; Shi, D.; Zhang, C.; Song, J.; Fang, Y.; Huang, S.; Xiang, Z.; et al. Exosome-transmitted miR-29a induces colorectal cancer metastasis by destroying the vascular endothelial barrier. Carcinogenesis 2023, 44, 356–367. [Google Scholar] [CrossRef]
- Cen, J.; Feng, L.; Ke, H.; Bao, L.; Li, L.Z.; Tanaka, Y.; Weng, J.; Su, L. Exosomal Thrombospondin-1 Disrupts the Integrity of Endothelial Intercellular Junctions to Facilitate Breast Cancer Cell Metastasis. Cancers 2019, 11, 1946. [Google Scholar] [CrossRef]
- Dieudé, M.; Turgeon, J.; Karakeussian Rimbaud, A.; Beillevaire, D.; Qi, S.; Patey, N.; Gaboury, L.A.; Boilard, É.; Hébert, M.J. Extracellular vesicles derived from injured vascular tissue promote the formation of tertiary lymphoid structures in vascular allografts. Am. J. Transplant. 2020, 20, 726–738. [Google Scholar] [CrossRef]
- Neves, K.B.; Rios, F.J.; Jones, R.; Evans, T.R.J.; Montezano, A.C.; Touyz, R.M. Microparticles from vascular endothelial growth factor pathway inhibitor-treated cancer patients mediate endothelial cell injury. Cardiovasc. Res. 2019, 115, 978–988. [Google Scholar] [CrossRef]
- Adnani, L.; Meehan, B.; Kim, M.; Choi, D.; Rudd, C.E.; Riazalhosseini, Y.; Rak, J. Immune cell infiltration into brain tumor microenvironment is mediated by Rab27-regulated vascular wall integrity. Sci. Adv. 2025, 11, eadr6940. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Wiederschain, D.; Stetler-Stevenson, W.G.; Folkman, J.; Moses, M.A. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J. Biol. Chem. 1999, 274, 29568–29571. [Google Scholar] [CrossRef]
- Di Ianni, E.; Obuchi, W.; Breyne, K.; Breakefield, X.O. Extracellular vesicles for the delivery of gene therapy. Nat. Rev. Bioeng. 2025, 3, 360–373. [Google Scholar] [CrossRef]
- Lucotti, S.; Kenific, C.M.; Zhang, H.; Lyden, D. Extracellular vesicles and particles impact the systemic landscape of cancer. Embo J. 2022, 41, e109288. [Google Scholar] [CrossRef]
- Ghufran, S.M.; Brown, M.L.; Beierle, E.A. Role of exosomes in diagnosis, prognostication, and treatment of pediatric solid tumors. Mol. Ther. Oncol. 2025, 33, 200930. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Carney, R.P.; Mizenko, R.R.; Bozkurt, B.T.; Lowe, N.; Henson, T.; Arizzi, A.; Wang, A.; Tan, C.; George, S.C. Harnessing extracellular vesicle heterogeneity for diagnostic and therapeutic applications. Nat. Nanotechnol. 2024, 20, 14–25. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, F.C.; Rak, J. The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis. Cancers 2025, 17, 1966. https://doi.org/10.3390/cancers17121966
Wong FC, Rak J. The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis. Cancers. 2025; 17(12):1966. https://doi.org/10.3390/cancers17121966
Chicago/Turabian StyleWong, Fang Cheng, and Janusz Rak. 2025. "The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis" Cancers 17, no. 12: 1966. https://doi.org/10.3390/cancers17121966
APA StyleWong, F. C., & Rak, J. (2025). The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis. Cancers, 17(12), 1966. https://doi.org/10.3390/cancers17121966