
Breaking Cancer’s Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy

Simple Summary
Abstract
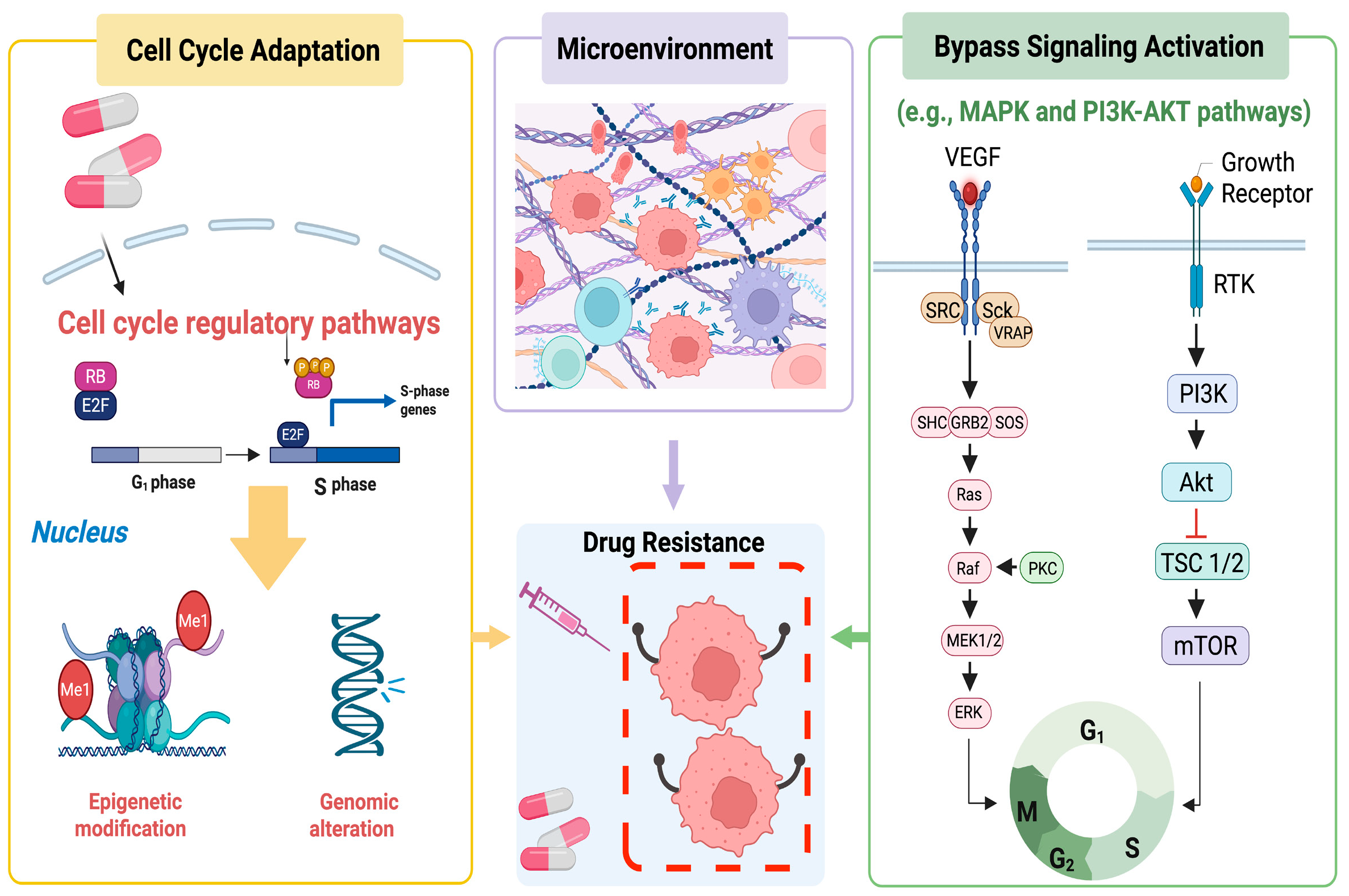
1. Introduction
2. Inhibition of CDK4/6 in Cancers
3. Development of Novel CDK4/6 Inhibitors
4. CDK4/6 Inhibition and Chemotherapy
5. CDK4/6 Inhibition and Other Targeted Therapies
5.1. CDK4/6 Inhibition and Estrogen Receptor Antagonist Therapy
5.2. CDK4/6 Inhibition and RAS-MAPK Inhibition
5.3. CDK4/6 Inhibition and PI3K-AKT-mTOR Inhibition
5.4. CDK4/6 Inhibition and Receptor Tyrosine Kinase Inhibition
5.5. CDK4/6 Inhibition and Autophagy Inhibition
6. CDK4/6 Inhibition and Immune Checkpoint Blockade
7. Cell Cycle Arrest and Senescence Induced by CDK4/6 Inhibition
8. Apoptosis Triggered by CDK4/6 Inhibition
9. Metabolism Alterations Induced by CDK4/6 Inhibition
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ER | Estrogen receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| CDK | Cyclin-dependent kinase |
| CAK | CDK-activating kinase |
| PFS | Progression-free survival |
| TNBC | Triple-negative breast cancer |
| FDA | Food and Drug Administration |
| RAS | Rat sarcoma |
| MAPK | Mitogen-activated protein kinase |
| RAF | Rapidly accelerated fibrosarcoma |
| MEK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| PDX | Patient-derived xenograft |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PDK | Phosphoinositide-dependent kinase |
| MTORC2 | Mechanistic target of rapamycin complex 2 |
| PTEN | Phosphatase and tensin homolog |
| RTK | Receptor tyrosine kinase |
| JAK-STAT | Janus kinase-signal transducer and activator of transcription |
| FGFR | Fibroblast growth factor receptor |
| EGFR | Epidermal growth factor receptor |
| ROS | Reactive oxygen species |
| HCQ | Hydroxychloroquine |
| ICB | Immune checkpoint blockade |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death-ligand 1 |
| SPOP | Speckle-type POZ protein |
| CXCL | C-X-C motif chemokine ligand |
| NFAT | Nuclear factor of activated T cell |
| HLA | Human leukocyte antigen |
| MHC | Major histocompatibility complex |
| NK cell | Natural killer cell |
| SASP | Senescence-associated secretory phenotype |
| MDM2 | Mouse double minute 2 |
| WD/DDLS | Well-differentiated and dedifferentiated liposarcoma |
| PDLIM7 | PDZ and LIM domain 7 |
| CDH18 | Cadherin 18 |
| FOXM1 | Forkhead box M1 |
| BH3 | Bcl-2 homology 3 |
| BCL-2 | B-cell lymphoma 2 |
| CG | Cardiac glycoside |
| FOXO4 | Forkhead box o4 |
| HSP90 | Heat shock protein 90 |
| AMPKα2 | AMP-activated protein kinase alpha 2 |
| GPX4 | Glutathione peroxidase 4 |
| Rb | Retinoblastoma protein |
References
- Pines, J. Cyclins: Wheels within wheels. Cell Growth Differ. 1991, 2, 305–310. [Google Scholar] [PubMed]
- Pellarin, I.; Dall’Acqua, A.; Favero, A.; Segatto, I.; Rossi, V.; Crestan, N.; Karimbayli, J.; Belletti, B.; Baldassarre, G. Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct. Target. Ther. 2025, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Mughal, M.J.; Bhadresha, K.; Kwok, H.F. CDK inhibitors from past to present: A new wave of cancer therapy. Semin. Cancer Biol. 2023, 88, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [Google Scholar] [CrossRef]
- Beykou, M.; Arias-Garcia, M.; Roumeliotis, T.I.; Choudhary, J.S.; Moser, N.; Georgiou, P.; Bakal, C. Proteomic characterisation of triple negative breast cancer cells following CDK4/6 inhibition. Sci. Data 2022, 9, 395. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Q.; Sun, W.; Zhou, J.; Zhou, J. Immunomodulatory effects of CDK4/6 inhibitors. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188912. [Google Scholar] [CrossRef]
- Peters, G. The D-type cyclins and their role in tumorigenesis. J. Cell Sci. Suppl. 1994, 18, 89–96. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Harris, A.W.; Bodrug, S.E.; Warner, B.J.; Bath, M.L.; Lindeman, G.J.; Adams, J.M. Cyclin D1 as the putative bcl-1 oncogene. Curr. Top. Microbiol. Immunol. 1995, 194, 347–353. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Rastogi, P.; Toi, M.; Martín, M.; O’Shaughnessy, J.; Headley, D.; Wei, J.; Cox, J.; Harbeck, N.; Johnston, S.R.D. Abstract OT2-02-02: MONARCH E: A phase 3 study of standard adjuvant endocrine therapy with or without abemaciclib in patients with high risk, node positive, hormone-receptor positive, human epidermal growth factor receptor 2-negative early-stage breast cancer. Cancer Res. 2020, 80, OT2-02-02. [Google Scholar]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Abramson, V.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Bailey, J.; Burstein, H.J.; et al. Breast Cancer, Version 3.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2024, 22, 331–357. [Google Scholar] [CrossRef]
- Parylo, S.; Vennepureddy, A.; Dhar, V.; Patibandla, P.; Sokoloff, A. Role of cyclin-dependent kinase 4/6 inhibitors in the current and future eras of cancer treatment. J. Oncol. Pharm. Pract. 2019, 25, 110–129. [Google Scholar] [CrossRef]
- Altenburg, J.D.; Farag, S.S. The potential role of PD0332991 (Palbociclib) in the treatment of multiple myeloma. Expert. Opin. Investig. Drugs 2015, 24, 261–271. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Desnoyers, A.; Nadler, M.B.; Kumar, V.; Saleh, R.; Amir, E. Comparison of treatment-related adverse events of different Cyclin-dependent kinase 4/6 inhibitors in metastatic breast cancer: A network meta-analysis. Cancer Treat. Rev. 2020, 90, 102086. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Differences of cyclin-dependent kinase 4/6 inhibitor, palbociclib and abemaciclib, in breast cancer. Jpn. J. Clin. Oncol. 2019, 49, 993–998. [Google Scholar] [CrossRef]
- Kappel, C.; Elliott, M.J.; Kumar, V.; Nadler, M.B.; Desnoyers, A.; Amir, E. Comparative overall survival of CDK4/6 inhibitors in combination with endocrine therapy in advanced breast cancer. Sci. Rep. 2024, 14, 3129. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Parashar, K.; Fajas, L. Beyond cell cycle regulation: The pleiotropic function of CDK4 in cancer. Semin. Cancer Biol. 2024, 98, 51–63. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Luo, Y.; Tong, Z.; Sun, T.; Shan, C.; Liu, X.; Yao, Y.; Zhao, B.; Wang, S.; et al. Lerociclib plus fulvestrant in patients with HR+/HER2− locally advanced or metastatic breast cancer who have progressed on prior endocrine therapy: LEONARDA-1 a phase III randomized trial. Nat. Commun. 2025, 16, 716. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Q.; Tong, Z.; Sun, T.; Li, W.; Ouyang, Q.; Hu, X.; Cheng, Y.; Yan, M.; Pan, Y.; et al. Dalpiciclib plus letrozole or anastrozole versus placebo plus letrozole or anastrozole as first-line treatment in patients with hormone receptor-positive, HER2-negative advanced breast cancer (DAWNA-2): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2023, 24, 646–657. [Google Scholar] [CrossRef]
- Shi, C.; Ju, H.; Zhou, R.; Xu, S.-m.; Wu, Y.; Gu, Z.; Wang, Y.; Chen, W.; Huang, X.; Han, Y.; et al. The efficacy and safety of dalpiciclib, a cyclin-dependent kinase 4/6 inhibitor, in patients with advanced head and neck mucosal melanoma harboring CDK4 amplification. BMC Med. 2024, 22, 215. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Li, H.; Tong, Z.; Ouyang, Q.; Li, H.; Teng, Y.; Wang, B.; Sun, T.; Wang, J.; et al. Bireociclib plus fulvestrant for advanced HR+/HER2- breast cancer progressing after endocrine therapy: Interim analysis of a phase 3 trial (BRIGHT-2). J. Clin. Oncol. 2024, 42, 1058. [Google Scholar] [CrossRef]
- Young, J.A.; Tan, A.R. Trilaciclib: A First-in-class Therapy to Reduce Chemotherapy-induced Myelosuppression. Oncol. Haematol. 2022, 18, 152–158. [Google Scholar] [CrossRef]
- Palmer, C.L.; Boras, B.; Pascual, B.; Li, N.; Li, D.; Garza, S.; Huser, N.; Yuan, J.T.; Cianfrogna, J.A.; Sung, T.; et al. CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety. Cancer Cell 2025, 43, 464–481.e14. [Google Scholar] [CrossRef] [PubMed]
- Finley, R.S.; Grove, W.R.; Fortner, C.L. An Overview of Cancer Chemotherapy. J. Pharm. Technol. 1985, 1, 11–18. [Google Scholar] [CrossRef]
- Bagnyukova, T.V.; Serebriiskii, I.G.; Zhou, Y.; Hopper-Borge, E.A.; Golemis, E.A.; Astsaturov, I. Chemotherapy and signaling. Cancer Biol. Ther. 2010, 10, 839–853. [Google Scholar] [CrossRef]
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging Therapeutic Strategies to Overcome Drug Resistance in Cancer Cells. Cancers 2024, 16, 2478. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Song, D.; Xu, M.; Liebmen, M. New strategies for targeting drug combinations to overcome mutation-driven drug resistance. Semin. Cancer Biol. 2017, 42, 44–51. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef]
- Dall’Acqua, A.; Sonego, M.; Pellizzari, I.; Pellarin, I.; Canzonieri, V.; D’Andrea, S.; Benevol, S.; Sorio, R.; Giorda, G.; Califano, D.; et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med. 2017, 9, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Barbero, B.; Alvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 38, 584. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-White, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; van Hoogdalem, E.J.; et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci. Transl. Med. 2017, 9, eaal3986. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, S.; Xin, Q.; Zhang, Y.; Wang, K.; Li, M. Recent progress of CDK4/6 inhibitors’ current practice in breast cancer. Cancer Gene Ther. 2024, 31, 1283–1291. [Google Scholar] [CrossRef]
- Zhao, C.; Lam, E.W.; Sunters, A.; Enmark, E.; De Bella, M.T.; Coombes, R.C.; Gustafsson, J.A.; Dahlman-Wright, K. Expression of estrogen receptor beta isoforms in normal breast epithelial cells and breast cancer: Regulation by methylation. Oncogene 2003, 22, 7600–7606. [Google Scholar] [CrossRef]
- Zhao, C.; Matthews, J.; Tujague, M.; Wan, J.; Ström, A.; Toresson, G.; Lam, E.W.; Cheng, G.; Gustafsson, J.A.; Dahlman-Wright, K. Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007, 67, 3955–3962. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.G.; Dickler, M.N. The Role of CDK4/6 Inhibition in Breast Cancer. Oncologist 2015, 20, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Lee, E.K.; Xiong, N.; Krasner, C.; Campos, S.; Kolin, D.L.; Liu, J.F.; Horowitz, N.; Wright, A.A.; Bouberhan, S.; et al. A Phase II, Two-Stage Study of Letrozole and Abemaciclib in Estrogen Receptor-Positive Recurrent Endometrial Cancer. J. Clin. Oncol. 2023, 41, 599–608. [Google Scholar] [CrossRef]
- Slomovitz, B.M.; Deng, W.; Killion, J.; Weroha, S.J.; Backes, F.; Miller, D.S.; Tenney, M.E.; Vogel, T.J.; Chen, L.-m.; Markham, M.J.; et al. GOG 3026 A phase II trial of letrozole + ribociclib in women with recurrent low-grade serous carcinoma of the ovary, fallopian tube or peritoneum: A GOG foundation study (001). Gynecol. Oncol. 2023, 176, S2. [Google Scholar] [CrossRef]
- Brighi, N.; Conteduca, V.; Lolli, C.; Gurioli, G.; Schepisi, G.; Palleschi, M.; Mariotti, M.; Casadei, C.; De Giorgi, U. The cyclin-dependent kinases pathway as a target for prostate cancer treatment: Rationale and future perspectives. Crit. Rev. Oncol. Hematol. 2021, 157, 103199. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Migliarese, C.; Sadeh, Y.; Torrini, C.; Turna Demir, F.; Nayyar, N.; Yamazawa, E.; Ishikawa, Y.; Ijad, N.; Summers, E.J.; Elliott, A.; et al. Combination therapy of adagrasib and abemaciclib in non-small cell lung cancer brain metastasis models genomically characterized by KRAS-G12C and homozygous loss of CDKN2A. Acta Neuropathol. Commun. 2025, 13, 88. [Google Scholar] [CrossRef]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef]
- Paulsohn, M.-B.; Frahnert, K.H.; Fang, X.; Schneider, C.; Tapia Contreras, C.; Schneider, G.; Hessmann, E.; Dobbelstein, M. Simultaneous Targeting of KRAS and CDK4 Synergistically Suppresses Pancreatic Cancer Cells. bioRxiv 2025. [Google Scholar] [CrossRef]
- Luo, C.W.; Hou, M.F.; Huang, C.W.; Wu, C.C.; Ou-Yang, F.; Li, Q.L.; Wu, C.C.; Pan, M.R. The CDK6-c-Jun-Sp1-MMP-2 axis as a biomarker and therapeutic target for triple-negative breast cancer. Am. J. Cancer Res. 2020, 10, 4325–4341. [Google Scholar] [PubMed]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar] [CrossRef]
- Shi, Y.; Zheng, H.; Wang, T.; Zhou, S.; Zhao, S.; Li, M.; Cao, B. Targeting KRAS: From metabolic regulation to cancer treatment. Mol. Cancer 2025, 24, 9. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Vadlakonda, L.; Pasupuleti, M.; Pallu, R. Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Front. Oncol. 2013, 3, 85. [Google Scholar] [CrossRef]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Toyosawa, D.; Nakamura, M.; Tsuboi, K.; Tokuda, E.; Niwa, T.; Ishida, T.; Hayashi, S.I. Decreased ER dependency after acquired resistance to CDK4/6 inhibitors. Breast Cancer 2020, 27, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef]
- Muranen, T.; Meric-Bernstam, F.; Mills, G.B. Promising rationally derived combination therapy with PI3K and CDK4/6 inhibitors. Cancer Cell 2014, 26, 7–9. [Google Scholar] [CrossRef]
- Zhang, N.; Li, Y. Receptor tyrosine kinases: Biological functions and anticancer targeted therapy. MedComm (2020) 2023, 4, e446. [Google Scholar] [CrossRef]
- Zhang, X.; Chang, A. Somatic mutations of the epidermal growth factor receptor and non-small-cell lung cancer. J. Med. Genet. 2007, 44, 166–172. [Google Scholar] [CrossRef]
- Simon, R.; Nocito, A.; Hübscher, T.; Bucher, C.; Torhorst, J.; Schraml, P.; Bubendorf, L.; Mihatsch, M.M.; Moch, H.; Wilber, K.; et al. Patterns of her-2/neu amplification and overexpression in primary and metastatic breast cancer. J. Natl. Cancer Inst. 2001, 93, 1141–1146. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.M.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER(+) Breast Cancer. Clin. Cancer Res. 2017, 23, 6138–6150. [Google Scholar] [CrossRef]
- Haines, E.; Chen, T.; Kommajosyula, N.; Chen, Z.; Herter-Sprie, G.S.; Cornell, L.; Wong, K.K.; Shapiro, G.I. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 2018, 9, 31572–31589. [Google Scholar] [CrossRef]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Horpratraporn, K.; Adchariyasakulchai, P.; Sainamthip, P.; Ketchart, W. Combining lapatinib and palbociclib inhibits cell proliferation and invasion via AKT signaling pathway in endocrine-resistant breast cancer cells. Med. Oncol. 2024, 41, 58. [Google Scholar] [CrossRef]
- de Jager, V.D.; Stigt, J.A.; Niemantsverdriet, M.; Ter Elst, A.; van der Wekken, A.J. Osimertinib and palbociclib in an EGFR-mutated NSCLC with primary CDK4 amplification after progression under osimertinib. npj Precis. Oncol. 2024, 8, 113. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Raghavendra, A.S.; Kettner, N.M.; Kwiatkowski, D.; Damodaran, S.; Wang, Y.; Ramirez, D.; Gombos, D.S.; Hunt, K.K.; Shen, Y.; Keyomarsi, K.; et al. Phase I trial of hydroxychloroquine to enhance palbociclib and letrozole efficacy in ER+/HER2− breast cancer. npj Breast Cancer 2025, 11, 7. [Google Scholar] [CrossRef]
- Gong, C.; Lin, Q.; Qin, T.; Zeng, Y.; Xu, F.; Yang, Y.; Yin, D.; Duan, Z.; Chen, C.L.; Wing-Cheong Chow, L.; et al. Targeting autophagy plus high-dose CDK4/6 inhibitors in advanced HR+HER2- breast cancer: A phase 1b/2 trial. Med 2025, 6, 100559. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Li, J.; Jiang, K.; Wang, R.; Ge, J.L.; Yang, H.; Liu, S.J.; Jia, L.T.; Wang, L.; Chen, B.L. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics 2020, 10, 10619–10633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Charles, A.; Bourne, C.M.; Korontsvit, T.; Aretz, Z.E.H.; Mun, S.S.; Dao, T.; Klatt, M.G.; Scheinberg, D.A. Low-dose CDK4/6 inhibitors induce presentation of pathway specific MHC ligands as potential targets for cancer immunotherapy. Oncoimmunology 2021, 10, 1916243. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Ali, L.R.; Garrido-Castro, A.C.; Lenehan, P.J.; Bollenrucher, N.; Stump, C.T.; Dougan, M.; Goel, S.; Shapiro, G.I.; Tolaney, S.M.; Dougan, S.K. PD-1 blockade and CDK4/6 inhibition augment nonoverlapping features of T cell activation in cancer. J. Exp. Med. 2023, 220, e20220729. [Google Scholar] [CrossRef]
- Iannello, A.; Raulet, D.H. Immunosurveillance of senescent cancer cells by natural killer cells. Oncoimmunology 2014, 3, e27616. [Google Scholar] [CrossRef]
- Crozier, L.; Foy, R.; Mouery, B.L.; Whitaker, R.H.; Corno, A.; Spanos, C.; Ly, T.; Gowen Cook, J.; Saurin, A.T. CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J. 2022, 41, e108599. [Google Scholar] [CrossRef]
- Wang, B.; Varela-Eirin, M.; Brandenburg, S.M.; Hernandez-Segura, A.; van Vliet, T.; Jongbloed, E.M.; Wilting, S.M.; Ohtani, N.; Jager, A.; Demaria, M. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 2022, 41, e108946. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Tanaka, Y.; Okada, R.; Takahashi, A. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr. Gerontol. Int. 2020, 20, 539–546. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Hao, X.; Wang, C.; Zhang, R. Chromatin basis of the senescence-associated secretory phenotype. Trends Cell Biol. 2022, 32, 513–526. [Google Scholar] [CrossRef]
- Zhang, J.W.; Zhang, D.; Yu, B.P. Senescent cells in cancer therapy: Why and how to remove them. Cancer Lett. 2021, 520, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.; Prats, N.; López-Domínguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov. 2023, 13, 410–431. [Google Scholar] [CrossRef]
- Hanna, A.; Balko, J.M. No rest for the wicked: Tumor cell senescence reshapes the immune microenvironment. Cancer Cell 2023, 41, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef]
- Klein, M.E.; Dickson, M.A.; Antonescu, C.; Qin, L.X.; Dooley, S.J.; Barlas, A.; Manova, K.; Schwartz, G.K.; Crago, A.M.; Singer, S.; et al. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene 2018, 37, 5066–5078. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.C. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol. 2006, 26, 7269–7282. [Google Scholar] [CrossRef] [PubMed]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; O’Connor, R.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Schwartz, G.K.; Singer, S.; et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 2015, 6, 8226–8243. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C.; et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Piskorz, W.M.; Cechowska-Pasko, M. Senescence of Tumor Cells in Anticancer Therapy-Beneficial and Detrimental Effects. Int. J. Mol. Sci. 2022, 23, 11082. [Google Scholar] [CrossRef]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 4120–4134. [Google Scholar] [CrossRef]
- Muttiah, C.; Whittle, J.R.; Oakman, C.; Lindeman, G.J. PALVEN: Phase Ib Trial of Palbociclib, Letrozole and Venetoclax in Estrogen Receptor- and BCL2-Positive Advanced Breast Cancer. Future Oncol. 2022, 18, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Galiana, I.; Lozano-Torres, B.; Sancho, M.; Alfonso, M.; Bernardos, A.; Bisbal, V.; Serrano, M.; Martínez-Máñez, R.; Orzáez, M. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanoSenolytic. J. Control. Release 2020, 323, 624–634. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Bousset, L.; Gil, J. Targeting senescence as an anticancer therapy. Mol. Oncol. 2022, 16, 3855–3880. [Google Scholar] [CrossRef]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. CDK4 inhibitors and apoptosis: A novel mechanism requiring nucleolar targeting of RelA. Cell Cycle 2007, 6, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, C.; Boopathi, E.; Liu, Y.; McNair, C.; Haber, A.; Perepelyuk, M.; Bhardwaj, A.; Addya, S.; Ertel, A.; Shoyele, S.; et al. Therapeutic Challenge with a CDK 4/6 Inhibitor Induces an RB-Dependent SMAC-Mediated Apoptotic Response in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 1402–1414. [Google Scholar] [CrossRef]
- Ishio, T.; Kumar, S.; Shimono, J.; Daenthanasanmak, A.; Dubois, S.; Lin, Y.; Bryant, B.; Petrus, M.N.; Bachy, E.; Huang, D.W.; et al. Genome-wide CRISPR screen identifies CDK6 as a therapeutic target in adult T-cell leukemia/lymphoma. Blood 2022, 139, 1541–1556. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Guan, J.; Khalid, U.; Ning, J.; Costa, M.R.; Chan, J.; Li, Q.; Fortin, J.P.; Wong, W.R.; Perampalam, P.; et al. Inhibition of GPX4 enhances CDK4/6 inhibitor and endocrine therapy activity in breast cancer. Nat. Commun. 2024, 15, 9550. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336–349.e6. [Google Scholar] [CrossRef]
- Lee, Y.; Dominy, J.E.; Choi, Y.J.; Jurczak, M.; Tolliday, N.; Camporez, J.P.; Chim, H.; Lim, J.H.; Ruan, H.B.; Yang, X.; et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014, 510, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Terenziani, R.; Zoppi, S.; Fumarola, C.; La Monica, S.; Cretella, D.; Alfieri, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; et al. Dual Inhibition of CDK4/6 and PI3K/AKT/mTOR Signaling Impairs Energy Metabolism in MPM Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5165. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Oral Bioavailability | IC50 in Cell-Free Assay (nM) | Time to Peak Concentration (h) | Half-Life (h) | Metabolism | Excretion | Standard Dose | Schedule |
|---|---|---|---|---|---|---|---|---|---|
| Palbociclib (PD0332991) |  | approximately 46% | CDK4 (11) CDK6 (15) | 6–12 | 29 | CYP3A4; SULT2A1 | feces; urine | 125 mg once daily | 21 days on, 7 days off |
| Ribociclib (LEE011) |  | approximately 65.8% | CDK4 (10) CDK6 (39) | 1–4 | 29.7–54.7 | CYP3A4 | feces; urine | 600 mg once daily | 21 days on, 7 days off |
| Abemaciclib (LY2835219) |  | approximately 45% | CDK4 (2) CDK6 (9.9) | 4–6 | 18.3 | CYP3A4 | feces; urine | 150 mg twice daily | Continuous dosing |
| Identifier | CDK4/6 Inhibitor | RAS-MAPK Inhibitor | Conditions | Phase |
|---|---|---|---|---|
| NCT05358249 | LEE011 | MEK inhibitor: Trametinib; KRAS G12C inhibitor: JDQ443; EGFR inhibitor: Cetuximab | KRAS G12C mutant solid tumours | Ib/II |
| NCT05178888 | Palbociclib | KRAS G12C inhibitor: MRTX849 | Advanced solid tumour with KRAS G12C mutation | 1/1b |
| NCT03170206 | Palbociclib | MEK inhibitor: Binimetinib (MEK162) | Advanced KRAS mutant non-small-cell lung cancer | I |
| NCT02159066 | LEE011 | BRAF inhibitor: LGX818 and MEK inhibitor: MEK162 | Locally advanced or metastatic BRAF V600 melanoma | II |
| NCT01543698 | LEE011 | BRAF inhibitor: LGX818 and MEK inhibitor: MEK162 | BRAF V600-dependent advanced solid tumours | Ib/II |
| NCT03981614 | Palbociclib | MEK inhibitor: Binimetinib (MEK162) | KRAS and NRAS mutant metastatic colorectal cancers | II |
| NCT01781572 | LEE011 | MEK inhibitor: Binimetinib (MEK162) | NRAS mutant melanoma | Ib/II |
| NCT03454035 | Palbociclib | ERK1/2 inhibitor: Ulixertinib (BVD-523) | Pancreatic cancer and metastatic melanoma | I |
| NCT03132454 | Palbociclib | Multi-kinase inhibitor: Sorafenib | Relapsed and refractory leukemias | I |
| NCT02065063 | Palbociclib | MEK inhibitor: Trametinib | Solid tumours | I |
| NCT02022982 | Palbociclib | MEK inhibitor PD-0325901 | KRAS mutant non-small-cell lung cancer and other solid tumours | I |
| Identifier | CDK4/6 Inhibitor | PI3K-AKT-mTOR Inhibitor | Conditions | Phase |
|---|---|---|---|---|
| NCT05563220 | Palbociclib or Ribociclib or Abemaciclib | PI3K inhibitor: Alpelisib or mTOR inhibitor: Everolimus or AKT inhibitor: Capivasertib | Metastatic breast cancer | I/II |
| NCT03065062 | Palbociclib | Dual PI3K/mTOR inhibitor: Gedatolisib | Advanced squamous cell lung, pancreatic, head and neck, and other solid tumours | I |
| NCT02626507 | Palbociclib | Dual PI3K/mTOR inhibitor: Gedatolisib | ER+/HER2− breast cancer | I |
| NCT03006172 | Palbociclib | PI3Kα inhibitor: Inavolisib | Locally advanced or metastatic PIK3CA mutant breast cancer | I |
| NCT02985125 | LEE011 | mTOR inhibitor: Everolimus | Metastatic pancreatic adenocarcinoma | I/II |
| NCT03008408 | Ribociclib | mTOR inhibitor: Everolimus | Advanced or recurrent endometrial carcinoma | II |
| NCT03114527 | Ribociclib | mTOR inhibitor: Everolimus | Advanced dedifferentiated liposarcoma (DDL) and leiomyosarcoma (LMS) | II |
| NCT01872260 | LEE011 | PI3Kα inhibitor: Alpelisib (BYL719) | ER+/HER2− locally advanced or metastatic breast cancer | I/II |
| Identifier | CDK4/6 Inhibitor | RTK Inhibitor | Conditions | Phase |
|---|---|---|---|---|
| NCT03304080 | Palbociclib | HER2 inhibitor: trastuzumab and pertuzumab | HR-positive, HER2-positive metastatic breast cancer | I/II |
| NCT03132454 | Palbociclib | Multikinase RTK inhibitor: Sorafenib | Relapsed and refractory leukemias | I |
| Identifier | CDK4/6 Inhibitor | ICI Inhibitor | Conditions | Phase |
|---|---|---|---|---|
| NCT02791334 | Abemaciclib | LY3300054 | HR+/HER2− breast cancer | I |
| NCT04118036 | Abemaciclib | Pembrolizumab | Recurrent glioblastoma | II |
| NCT02778685 | Palbociclib | Pembrolizumab | Newly diagnosed metastatic stage IV estrogen receptor-positive breast cancer | II |
| NCT04075604 | Palbociclib | Nivolumab | ER+/HER2− breast cancer | II |
| NCT02779751 | Abemaciclib | Pembrolizumab | Non-small-cell lung cancer or breast cancer | I |
| NCT03147287 | Palbociclib | Avelumab | HR+/HER2− breast cancer | II |
| NCT03280563 | Abemaciclib | Atezolizumab | Hormone receptor-positive, HER2-negative breast cancer | Ib/II |
| NCT04272645 | Abemaciclib | Atezolizumab | Metastatic castration-resistant prostate cancer | II |
| NCT04220892 | Abemaciclib | Pembrolizumab | High-grade glioma | I |
| NCT03997448 | Abemaciclib | Pembrolizumab | Advanced gastric, gastroesophageal junction, esophageal adenocarcinoma | II |
| NCT04088032 | Abemaciclib | Durvalumab | Locally advanced hormone receptor-positive breast cancer | I |
| NCT03294694 | Ribociclib | PDR001 | Metastatic hormone receptor-positive breast cancer and metastatic ovarian cancer | Is |
| NCT04360941 | Palbociclib | Avelumab | Metastatic AR+ triple-negative breast cancer | Ib |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Park, S.; Li, Y. Breaking Cancer’s Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy. Cancers 2025, 17, 1941. https://doi.org/10.3390/cancers17121941
Liu Y, Park S, Li Y. Breaking Cancer’s Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy. Cancers. 2025; 17(12):1941. https://doi.org/10.3390/cancers17121941
Chicago/Turabian StyleLiu, Yanbiao, Seohyun Park, and Yan Li. 2025. "Breaking Cancer’s Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy" Cancers 17, no. 12: 1941. https://doi.org/10.3390/cancers17121941
APA StyleLiu, Y., Park, S., & Li, Y. (2025). Breaking Cancer’s Momentum: CDK4/6 Inhibitors and the Promise of Combination Therapy. Cancers, 17(12), 1941. https://doi.org/10.3390/cancers17121941