
Combined Radiation and Endocrine Therapies Elicit Benefit in ER+ Breast Cancer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Irradiation
2.2. Growth Assays
2.3. Western Blot
2.4. Antioxidant Intracellular State Measurement In Vitro
2.5. In Vitro Apoptosis Assay
2.6. Mouse Studies
2.7. Immunohistochemistry
2.8. Immunofluorescence
2.9. Oxidative Stress Response Score
2.10. Statistics
3. Results
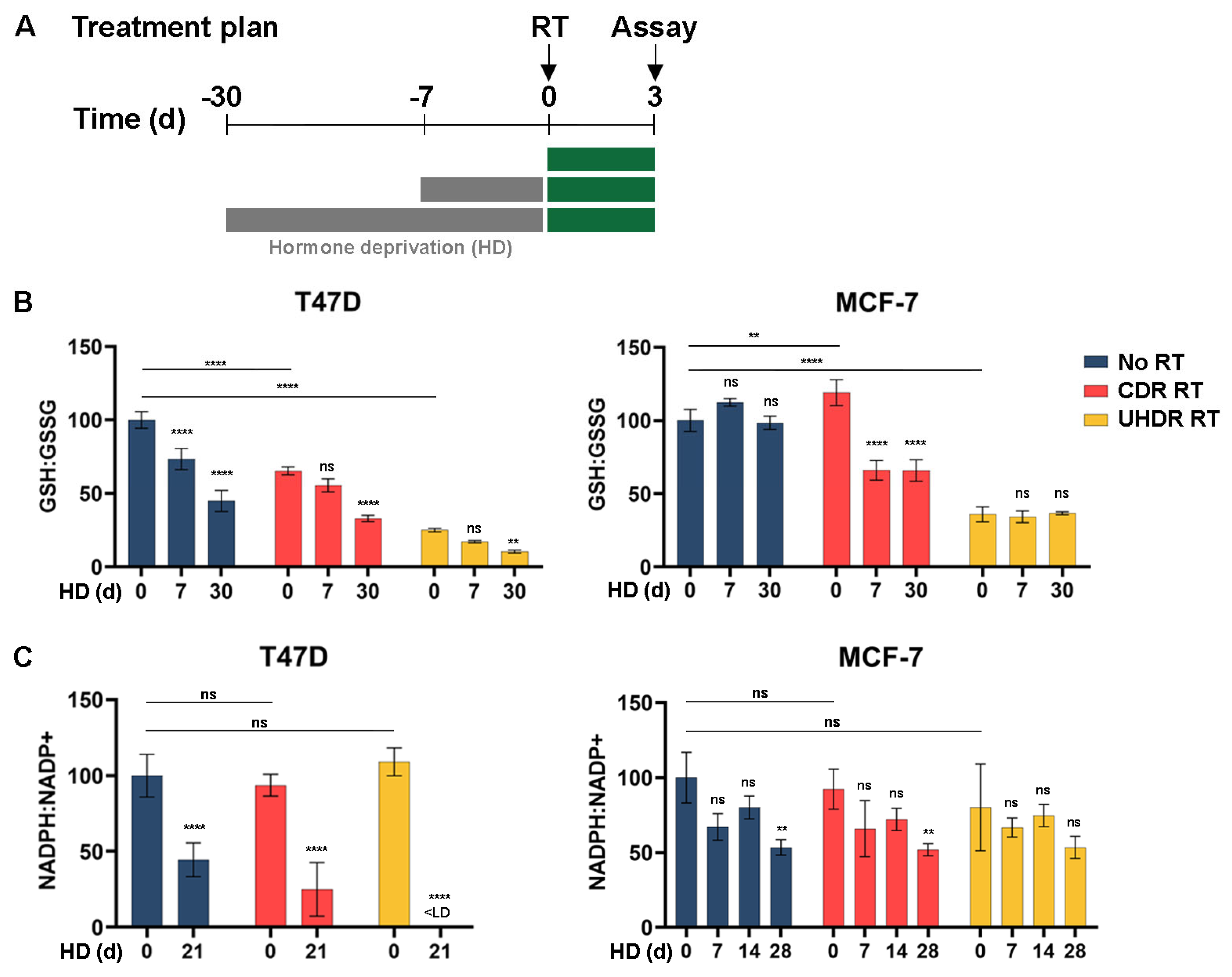
3.1. Estrogen Deprivation Slows Growth and Increases Oxidative Stress Response
3.2. Metabolic Analysis Indicates an Increased Oxidative State Due to Hormone Deprivation and Radiation
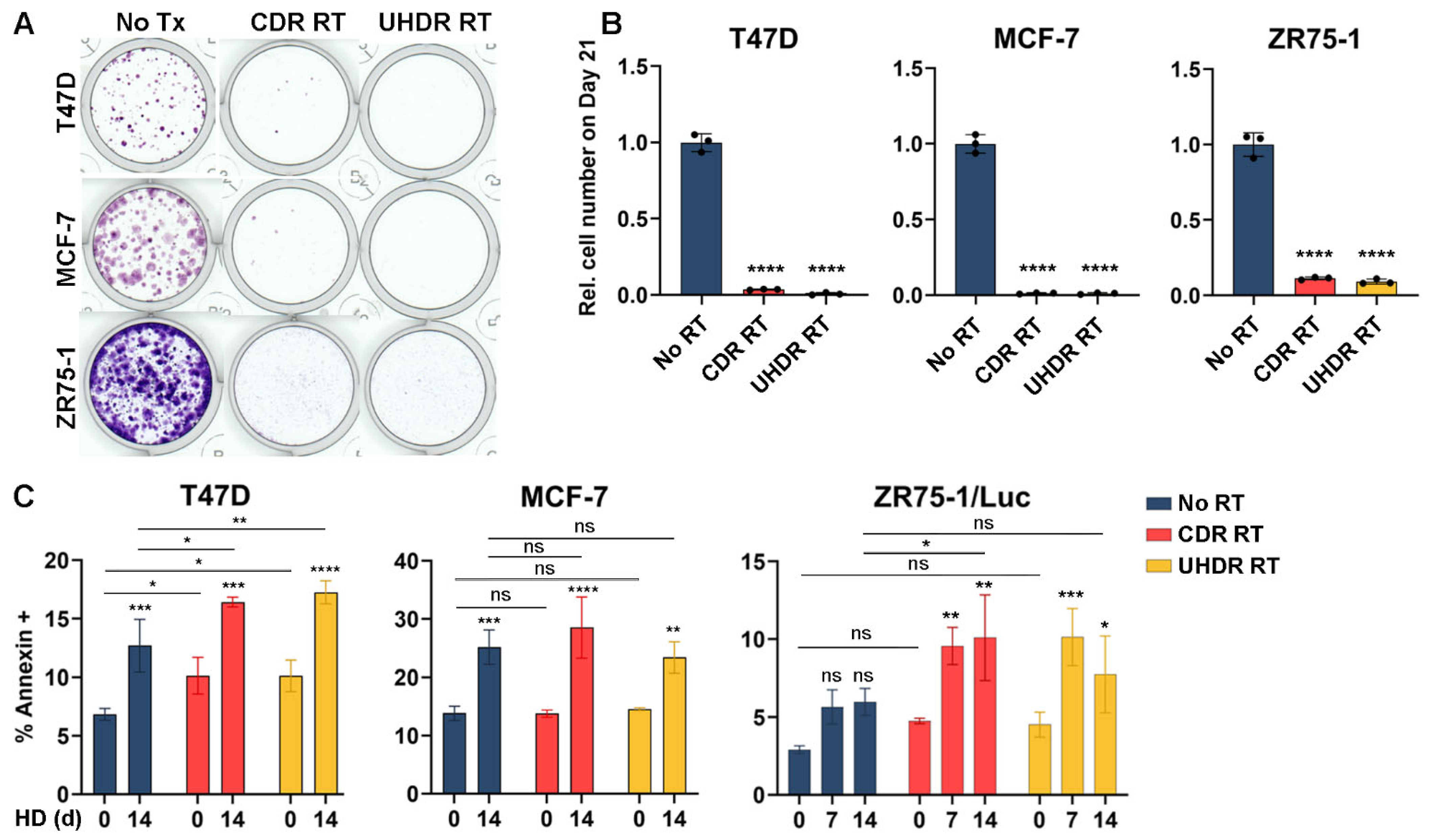
3.3. Radiation Inhibits Cell Growth and Hormone Deprivation Induces Apoptosis
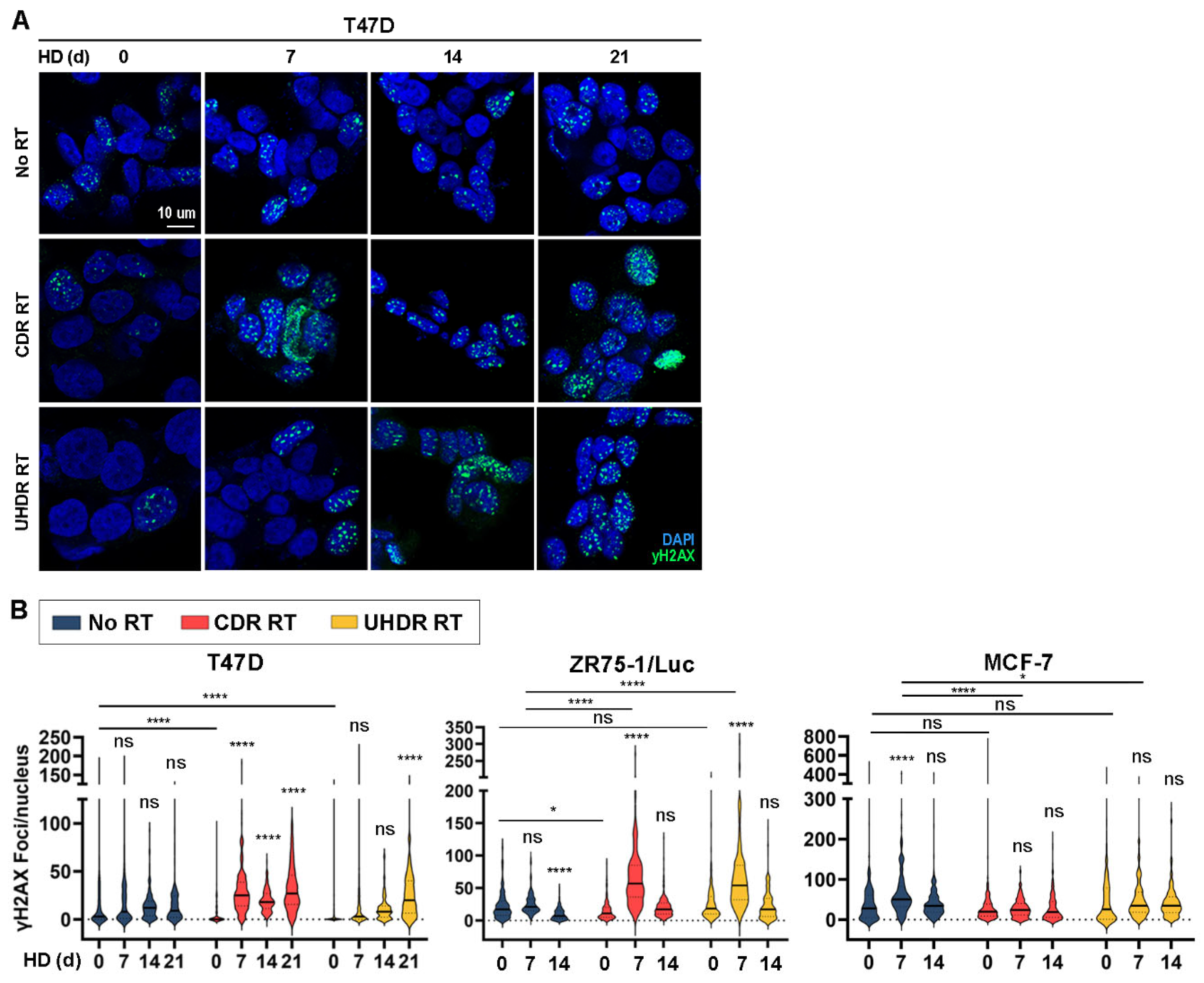
3.4. Combined Hormone Deprivation and Irradiation Induces DNA Damage
3.5. Co-Treatment with Hormone Deprivation and Radiation Is Sometimes Advantageous in Tumor Growth Studies Across Multiple Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.; McGale, P.; Correa, C.; Taylor, C.; Arriagada, R.; Clarke, M.; Cutter, D.; Davies, C.; Ewertz, M.; Godwin, J.; et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet 2011, 378, 1707–1716. [Google Scholar] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Burstein, H.J. Systemic Therapy for Estrogen Receptor-Positive, HER2-Negative Breast Cancer. N. Engl. J. Med. 2020, 383, 2557–2570. [Google Scholar] [CrossRef]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E.; Dees, E.C.; Goetz, M.P.; Olson, J.A.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Abe, O.; Abe, R.; Enomoto, K.; Kikuchi, K.; Koyama, H.; Masuda, H.; Nomura, Y.; Ohashi, Y.; Sakai, K.; Sugimachi, K.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar]
- Sparano, J.; O’Neill, A.; Alpaugh, K.; Wolff, A.C.; Northfelt, D.W.; Dang, C.T.; Sledge, G.W.; Miller, K.D. Association of Circulating Tumor Cells with Late Recurrence of Estrogen Receptor-Positive Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1700–1706. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- O’Neill, P.; Wardman, P. Radiation chemistry comes before radiation biology. Int. J. Radiat. Biol. 2009, 85, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Favaudon, V.; Caplier, L.; Monceau, V.; Pouzoulet, F.; Sayarath, M.; Fouillade, C.; Poupon, M.F.; Brito, I.; Hupé, P.; Bourhis, J.; et al. Ultrahigh dose-rate FLASH irradiation increases the differential response between normal and tumor tissue in mice. Sci. Transl. Med. 2014, 6, 245ra93. [Google Scholar] [CrossRef] [PubMed]
- Schüler, E.; Acharya, M.; Montay-Gruel, P.; Loo, B.W.; Vozenin, M.C.; Maxim, P.G. Ultra-high dose rate electron beams and the FLASH effect: From preclinical evidence to a new radiotherapy paradigm. Med. Phys. 2022, 49, 2082–2095. [Google Scholar] [CrossRef] [PubMed]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER + Breast Cancer Cells. Clin. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Gligorich, K.M.; Wang, G.; Georgelas, A.; Bowman, P.; Courdy, S.J.; Welm, A.L.; Welm, B.E. Patient-derived Models of Human Breast Cancer: Protocols for In vitro and In vivo Applications in Tumor Biology and Translational Medicine. Curr. Protoc. Pharmacol. 2013, 60, 14–23. [Google Scholar] [CrossRef]
- Sloop, A.; Ashraf, M.R.; Rahman, M.; Sunnerberg, J.; Dexter, C.A.; Thompson, L.; Gladstone, D.J.; Pogue, B.W.; Bruza, P.; Zhang, R.; et al. Rapid Switching of a C-Series Linear Accelerator Between Conventional and Ultrahigh-Dose-Rate Research Mode with Beamline Modifications and Output Stabilization. Int. J. Radiat. Oncol. Biol. Phys. 2024, 119, 1317–1325. [Google Scholar] [CrossRef]
- Shousha, S. Oestrogen receptor status of breast carcinoma: Allred/H score conversion table. Histopathology 2008, 53, 346–347. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Leung, S.C.Y.; Rimm, D.L.; Dodson, A.; Acs, B.; Badve, S.; Denkert, C.; Ellis, M.J.; Fineberg, S.; Flowers, M.; et al. Assessment of Ki67 in Breast Cancer: Updated Recommendations from the International Ki67 in Breast Cancer Working Group. J. Natl. Cancer Inst. 2021, 113, 808–819. [Google Scholar] [CrossRef]
- Chuang, Y.-Y.E.; Chen, Y.; Gadisetti; Chandramouli, V.R.; Cook, J.A.; Coffin, D.; Tsai, M.; DeGraff, W.; Yan, H.; Zhao, S.; et al. Gene Expression after Treatment with Hydrogen Peroxide, Menadione, or t-Butyl Hydroperoxide in Breast Cancer Cells. Cancer Res. 2002, 62, 6246–6254. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Selli, C.; Turnbull, A.K.; Pearce, D.A.; Li, A.; Fernando, A.; Wills, J.; Renshaw, L.; Thomas, J.S.; Dixon, J.M.; Sims, A.H. Molecular changes during extended neoadjuvant letrozole treatment of breast cancer: Distinguishing acquired resistance from dormant tumours. Breast Cancer Res. 2019, 21, 2. [Google Scholar] [CrossRef]
- Patani, N.; Dunbier, A.K.; Anderson, H.; Ghazoui, Z.; Ribas, R.; Anderson, E.; Gao, Q.; A’hern, R.; Mackay, A.; Lindemann, J.; et al. Differences in the transcriptional response to fulvestrant and estrogen deprivation in ER-positive breast cancer. Clin. Cancer Res. 2014, 20, 3962–3973. [Google Scholar] [CrossRef]
- Miller, W.R.; Larionov, A. Changes in expression of oestrogen regulated and proliferation genes with neoadjuvant treatment highlight heterogeneity of clinical resistance to the aromatase inhibitor, letrozole. Breast Cancer Res. 2010, 12, R52. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides—Small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H. A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1826, 423–433. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.L.; Rangaswamy, S.; Wicker, C.A.; Izumi, T. Repair of oxidative DNA damage and cancer: Recent progress in DNA base excision repair. Antioxid. Redox Signal 2014, 20, 708–726. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Traphagen, N.A.; Schwartz, G.N.; Tau, S.; Roberts, A.M.; Jiang, A.; Hosford, S.R.; Marotti, J.D.; Goen, A.E.; Romo, B.A.; Johnson, A.L.; et al. Estrogen Therapy Induces Receptor-Dependent DNA Damage Enhanced by PARP Inhibition in ER+ Breast Cancer. Clin. Cancer Res. 2023, 29, 3717–3728. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tau, S.; Chamberlin, M.D.; Yang, H.; Marotti, J.D.; Muskus, P.C.; Roberts, A.M.; Carmichael, M.M.; Cressey, L.; Dragnev, C.P.C.; Demidenko, E.; et al. Oxidative Phosphorylation is a Metabolic Vulnerability of Endocrine Therapy-Tolerant Persister Cells in ER+ Breast Cancer. Cancer Res. 2025, 85, 1145–1161. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Yan, F.; Zhao, Q.; Li, Y.; Zheng, Z.; Kong, X.; Shu, C.; Liu, Y.; Shi, Y. The role of oxidative stress in ovarian aging: A review. J. Ovarian Res. 2022, 15, 100. [Google Scholar] [CrossRef]
- Price, D.K. Efficacy of androgen deprivation therapy and the role of oxidative stress. Ann. Oncol. 2017, 28, 451–453. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Kashiwagi, E.; Masubuchi, D.; Eto, M.; Uchiumi, T.; Naito, S. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 2010, 29, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Hurt, E.M.; Thomas, S.B.; Farrar, W.L. Effects of manganese superoxide dismutase silencing on androgen receptor function and gene regulation: Implications for castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6073–6080. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Song, Y.; Zhan, Y.; Zhou, S.; Ke, J.; Ao, W.; Zhang, Y.; Liang, Q.; He, M.; Li, S.; et al. Fangchinoline inhibits non-small cell lung cancer metastasis by reversing epithelial-mesenchymal transition and suppressing the cytosolic ROS-related Akt-mTOR signaling pathway. Cancer Lett. 2022, 543, 215783. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ren, S.; Chen, Y.; Zhang, A.; Zhu, Y.; Zhang, Z.; Li, Z.; Piao, D. Fangchinoline exerts antitumour activity by suppressing the EGFR-PI3K/AKT signalling pathway in colon adenocarcinoma. Oncol. Rep. 2020, 45, 139–150. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal 2014, 21, 251–259. [Google Scholar] [CrossRef]
- Jayakumar, S.; Patwardhan, R.S.; Pal, D.; Sharma, D.; Sandur, S.K. Dimethoxycurcumin, a metabolically stable analogue of curcumin enhances the radiosensitivity of cancer cells: Possible involvement of ROS and thioredoxin reductase. Biochem. Biophys. Res. Commun. 2016, 478, 446–454. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Lerner, L.M.; Pesch, A.M.; Ward, C.; Schwartz, R.; Wilder-Romans, K.; Liu, M.; Nino, C.; Jungles, K.; Azaria, R.; et al. Estrogen receptor inhibition mediates radiosensitization of ER-positive breast cancer models. NPJ Breast Cancer 2022, 8, 31. [Google Scholar] [CrossRef]
- Villalobos, M.; Aranda, M.; Nunez, M.I.; Becerra, D.; Olea, N.; de Almodovar, M.R.; Pedraza, V. Interaction between ionizing radiation, estrogens and antiestrogens in the modification of tumor microenvironment in estrogen dependent multicellular spheroids. Acta Oncol. 1995, 34, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.H.U.; Strickert, T.; Marthinsen, A.B.L.; Lundgren, S. Changes in radiation sensitivity and steroid receptor content induced by hormonal agents and ionizing radiation in breast cancer cells in vitro. Acta Oncol. 1996, 35, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Böhninga, K.; Buschb, M.; Rave-Fränk, M.; Dühmkeb, E. Effect of Tamoxifen and Estrogen Treatment on the Radiosensitivity of MCF-7 Breast Cancer Cells. Onkologie 1996, 19, 430–434. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Miller, E.M.; Parker, C.J.; Jordan, V.C.; Mulcahy, R.T. 4-Hydroxytamoxifen, an active metabolite of tamoxifen, does not alter the radiation sensitivity of MCF-7 breast carcinoma cells irradiated in vitro. Breast Cancer Res. Treat. 1994, 30, 159–165. [Google Scholar] [CrossRef]
- Ahn, P.H.; Vu, H.T.; Lannin, D.; Obedian, E.; DiGiovanna, M.P.; Burtness, B.; Haffty, B.G. Sequence of radiotherapy with tamoxifen in conservatively managed breast cancer does not affect local relapse rates. J. Clin. Oncol. 2005, 23, 17–23. [Google Scholar] [CrossRef]
- Pierce, L.J.; Hutchins, L.F.; Green, S.R.; Lew, D.L.; Gralow, J.R.; Livingston, R.B.; Osborne, C.K.; Albain, K.S. Sequencing of tamoxifen and radiotherapy after breast-conserving surgery in early-stage breast cancer. J. Clin. Oncol. 2005, 23, 24–29. [Google Scholar] [CrossRef]
- Liu, W.L.; Gao, M.; Tzen, K.Y.; Tsai, C.L.; Hsu, F.M.; Cheng, A.L.; Cheng, J.C.H. Targeting Phosphatidylinositide3-Kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget 2014, 5, 3662–3672. [Google Scholar] [CrossRef]
- Prabakaran, P.J.; Javaid, A.M.; Swick, A.D.; Werner, L.R.; Nickel, K.P.; Sampene, E.; Hu, R.; Ong, I.M.; Bruce, J.Y.; Hartig, G.K.; et al. Radiosensitization of adenoid cystic carcinoma with MDM2 inhibition. Clin. Cancer Res. 2017, 23, 6044–6053. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, A.L.; Tau, S.; Sloop, A.M.; Dai, T.; Roberts, A.M.; Muskus, P.; Warren, A.; Kleist, S.A.; Hampsch, R.A.; Jorns, J.M.; et al. Combined Radiation and Endocrine Therapies Elicit Benefit in ER+ Breast Cancer. Cancers 2025, 17, 1921. https://doi.org/10.3390/cancers17121921
Johnson AL, Tau S, Sloop AM, Dai T, Roberts AM, Muskus P, Warren A, Kleist SA, Hampsch RA, Jorns JM, et al. Combined Radiation and Endocrine Therapies Elicit Benefit in ER+ Breast Cancer. Cancers. 2025; 17(12):1921. https://doi.org/10.3390/cancers17121921
Chicago/Turabian StyleJohnson, Anneka L., Steven Tau, Austin M. Sloop, Tianyuan Dai, Alyssa M. Roberts, Patricia Muskus, Alexa Warren, Sierra A. Kleist, Riley A. Hampsch, Julie M. Jorns, and et al. 2025. "Combined Radiation and Endocrine Therapies Elicit Benefit in ER+ Breast Cancer" Cancers 17, no. 12: 1921. https://doi.org/10.3390/cancers17121921
APA StyleJohnson, A. L., Tau, S., Sloop, A. M., Dai, T., Roberts, A. M., Muskus, P., Warren, A., Kleist, S. A., Hampsch, R. A., Jorns, J. M., Zhang, R., Jarvis, L. A., & Miller, T. W. (2025). Combined Radiation and Endocrine Therapies Elicit Benefit in ER+ Breast Cancer. Cancers, 17(12), 1921. https://doi.org/10.3390/cancers17121921