
Cardiovascular Safety Profile of BRAF and MEK Inhibitors in Melanoma: FAERS Data Through a Retrospective Disproportionality Analysis (2014–2023)

 ,
,  ,
,  , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Data Processing and Duplicate Report Management
2.3. Case Selection
2.4. Data Analyses
3. Results
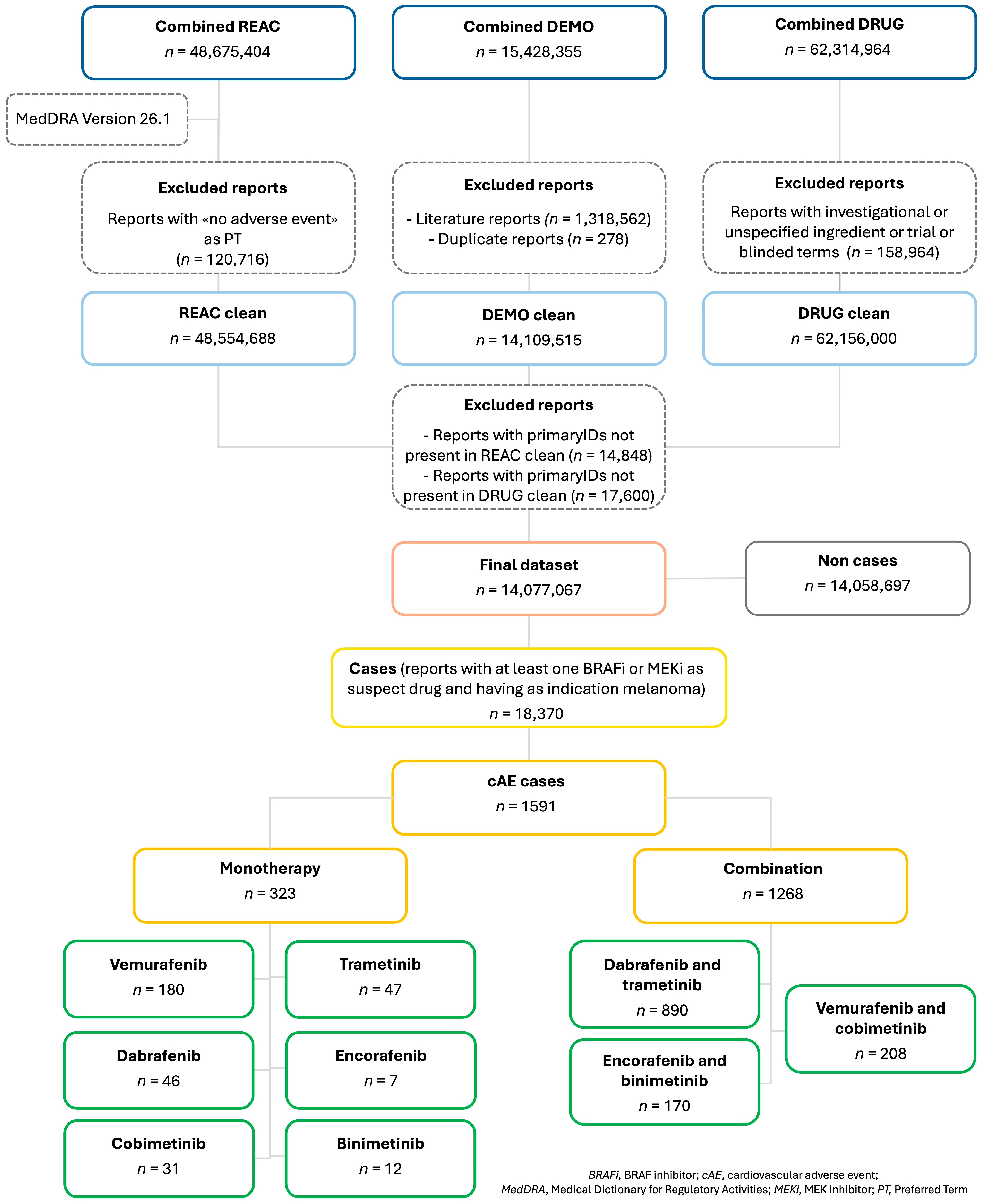
3.1. Cases Selection
3.2. Descriptive Analysis
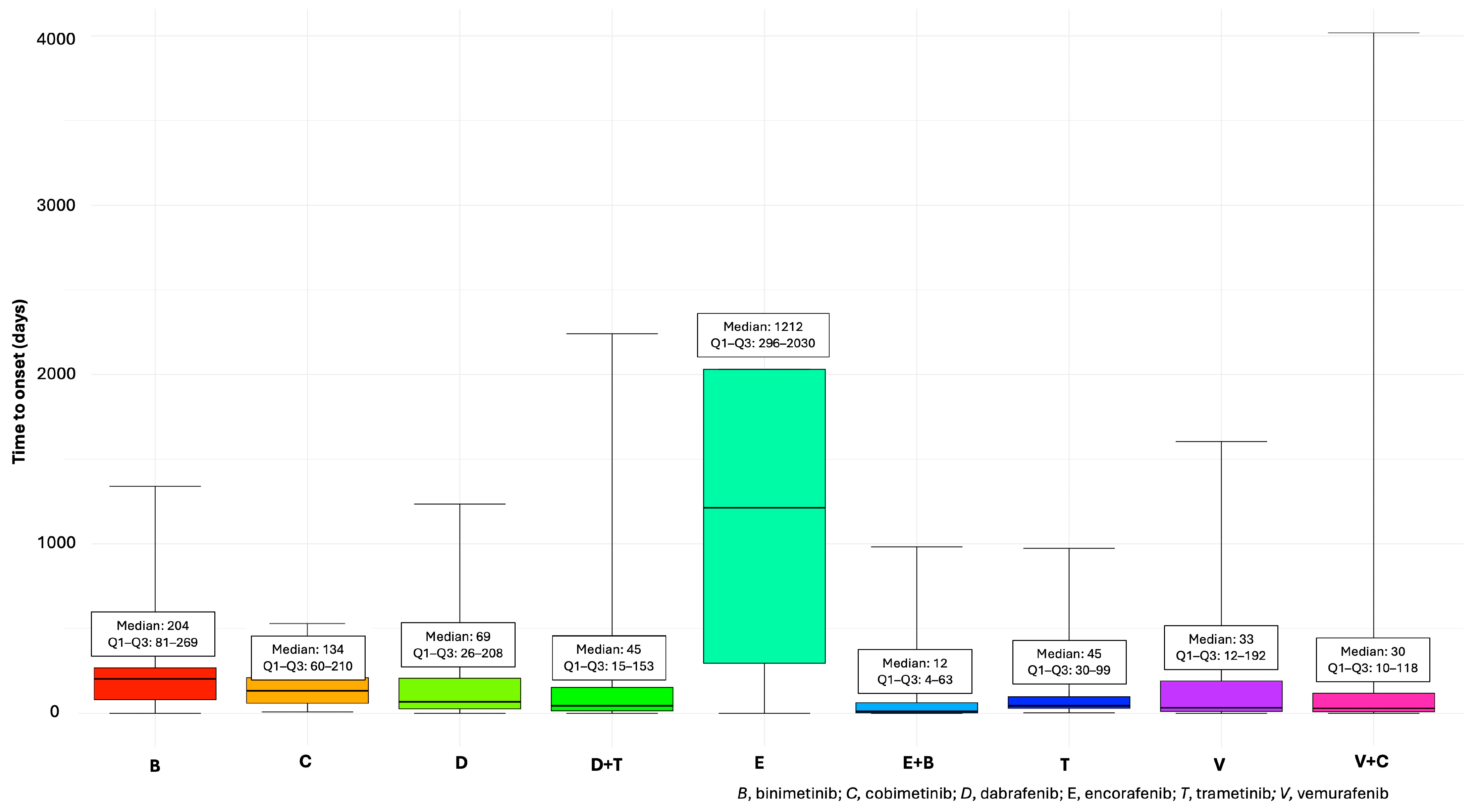
3.3. Cardiovascular Adverse Events
3.4. Disproportionality Analysis
4. Discussion
4.1. Descriptive Analysis
4.2. Disproportionality Analysis
4.2.1. Cardiomyopathy
4.2.2. Cardiac Failure
4.2.3. Arrhythmias and Ischaemic Heart Disease
4.2.4. Myocarditis and Pericarditis
4.2.5. Embolic and Thrombotic Events
4.3. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Agarwala, S.S.; Messersmith, H.; Alluri, K.C.; Ascierto, P.A.; Atkins, M.B.; Bollin, K.; Chacon, M.; Davis, N.; Faries, M.B.; et al. Systemic Therapy for Melanoma: ASCO Guideline Update. J. Clin. Oncol. 2023, 41, 4794–4820. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients with Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF Inhibition in Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib Combined with Vemurafenib in Advanced BRAF(V600)-Mutant Melanoma (CoBRIM): Updated Efficacy Results from a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF-Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; De Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Kirkwood, J.M.; Atkinson, V.; Mandala, M.; Merelli, B.; Sileni, V.C.; Nyakas, M.; Haydon, A.; et al. Final Results for Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N. Engl. J. Med. 2024, 391, 1709–1720. [Google Scholar] [CrossRef]
- Garutti, M.; Bergnach, M.; Polesel, J.; Palmero, L.; Pizzichetta, M.A.; Puglisi, F. BRAF and MEK Inhibitors and Their Toxicities: A Meta-Analysis. Cancers 2023, 15, 141. [Google Scholar] [CrossRef]
- Gogas, H.J.; Flaherty, K.T.; Dummer, R.; Ascierto, P.A.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Adverse Events Associated with Encorafenib plus Binimetinib in the COLUMBUS Study: Incidence, Course and Management. Eur. J. Cancer 2019, 119, 97–106. [Google Scholar] [CrossRef]
- Bronte, E.; Bronte, G.; Novo, G.; Rinaldi, G.; Bronte, F.; Passiglia, F.; Russo, A. Cardiotoxicity Mechanisms of the Combination of BRAF-Inhibitors and MEK-Inhibitors. Pharmacol. Ther. 2018, 192, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Arangalage, D.; Degrauwe, N.; Michielin, O.; Monney, P.; Özdemir, B.C. Pathophysiology, Diagnosis and Management of Cardiac Toxicity Induced by Immune Checkpoint Inhibitors and BRAF and MEK Inhibitors. Cancer Treat. Rev. 2021, 100, 102282. [Google Scholar] [CrossRef]
- Glen, C.; Tan, Y.Y.; Waterston, A.; Evans, T.R.J.; Jones, R.J.; Petrie, M.C.; Lang, N.N. Mechanistic and Clinical Overview Cardiovascular Toxicity of BRAF and MEK Inhibitors: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol 2022, 4, 1–18. [Google Scholar] [CrossRef]
- Barbieri, M.A.; Sorbara, E.E.; Cicala, G.; Santoro, V.; Cutroneo, P.M.; Franchina, T.; Santarpia, M.; Silvestris, N.; Spina, E. Safety Profile of Tyrosine Kinase Inhibitors Used in Non-Small-Cell Lung Cancer: An Analysis from the Italian Pharmacovigilance Database. Front. Oncol. 2022, 12, 1005626. [Google Scholar] [CrossRef]
- Barbieri, M.A.; Russo, G.; Sorbara, E.E.; Cicala, G.; Franchina, T.; Santarpia, M.; Speranza, D.; Spina, E.; Silvestris, N. Neuropsychiatric Adverse Drug Reactions with Oral Tyrosine Kinase Inhibitors in Metastatic Colorectal Cancer: An Analysis from the FDA Adverse Event Reporting System. Front. Oncol. 2023, 13, 1268672. [Google Scholar] [CrossRef]
- Barbieri, M.A.; Sorbara, E.E.; Russo, G.; Cicala, G.; Franchina, T.; Santarpia, M.; Silvestris, N.; Spina, E. Neuropsychiatric Adverse Drug Reactions with Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors: An Analysis from the European Spontaneous Adverse Event Reporting System. Cancers 2023, 15, 1851. [Google Scholar] [CrossRef] [PubMed]
- Cicala, G.; Russo, G.; Santoro, V.; Franchina, T.; Silvestris, N.; Santarpia, M.; Spina, E.; Barbieri, M.A. Neuropsychiatric Adverse Events with Monoclonal Antibodies Approved for Multiple Myeloma: An Analysis from the FDA Adverse Event Reporting System. Pharmaceuticals 2024, 17, 1266. [Google Scholar] [CrossRef]
- READUS Statement—READUS-PV. Available online: https://readus-statement.org/readus-statement/ (accessed on 5 December 2024).
- Flaherty, L.; Hamid, O.; Linette, G.; Schuchter, L.; Hallmeyer, S.; Gonzalez, R.; Cowey, C.L.; Pavlick, A.; Kudrik, F.; Curti, B.; et al. A Single-Arm, Open-Label, Expanded Access Study of Vemurafenib in Patients with Metastatic Melanoma in the United States. Cancer J. 2014, 20, 18–24. [Google Scholar] [CrossRef]
- Larkin, J.; Del Vecchio, M.; Ascierto, P.A.; Krajsova, I.; Schachter, J.; Neyns, B.; Espinosa, E.; Garbe, C.; Sileni, V.C.; Gogas, H.; et al. Vemurafenib in Patients with BRAF(V600) Mutated Metastatic Melanoma: An Open-Label, Multicentre, Safety Study. Lancet Oncol. 2014, 15, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Dudnichenko, O.; Penkov, K.; McKean, M.; Mandalà, M.; Kukushkina, M.; Panella, T.; Csőszi, T.; Gerletti, P.; Thakur, M.; Polli, A.; et al. First-Line Encorafenib plus Binimetinib and Pembrolizumab for Advanced BRAF V600-Mutant Melanoma: Safety Lead-in Results from the Randomized Phase III STARBOARD Study. Eur. J. Cancer 2024, 213, 115070. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, E.; Zimmer, L.; Vaubel, J.; Schadendorf, D. BRAF, MEK and KIT Inhibitors for Melanoma: Adverse Events and Their Management. Chin. Clin. Oncol. 2014, 3, 29. [Google Scholar] [CrossRef]
- Banks, M.; Crowell, K.; Proctor, A.; Jensen, B.C. Cardiovascular Effects of the MEK Inhibitor, Trametinib: A Case Report, Literature Review, and Consideration of Mechanism. Cardiovasc. Toxicol. 2017, 17, 487–493. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernánde, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klei, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Cone, E.B.; Haeuser, L.; Reese, S.W.; Marchese, M.; Nguyen, D.D.; Nabi, J.; Chou, W.H.; Noldus, J.; McKay, R.R.; Kilbridge, K.L.; et al. Immune Checkpoint Inhibitor Monotherapy Is Associated with Less Cardiac Toxicity than Combination Therapy. PLoS ONE 2022, 17, e0272022. [Google Scholar] [CrossRef]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic Inhibition of Cardiac ERK1/2 Promotes Stress-Induced Apoptosis and Heart Failure but Has No Effect on Hypertrophy In Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Roberts, D.D.; Frazier, W.A. CD47 A New Target in Cardiovascular Therapy Thrombospondin-1 Regulation of Integrins Requires CD47. Arter. Thromb. Vasc. Biol. 2008, 28, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, K.A.K.; Madeddu, P.; Avolio, E. MEK Inhibitors: A Promising Targeted Therapy for Cardiovascular Disease. Front. Cardiovasc. Med. 2024, 11, 1404253. [Google Scholar] [CrossRef] [PubMed]
- Glen, C.; Adam, S.; McDowell, K.; Waterston, A.; Tan, Y.Y.; Petrie, M.C.; Coats, C.J.; Lang, N.N. Cardiotoxicity of BRAF/MEK Inhibitors: A Longitudinal Study Incorporating Contemporary Definitions and Risk Scores. JACC CardioOncol 2023, 5, 628–637. [Google Scholar] [CrossRef]
- Pedersen, S.; Larsen, K.O.; Christensen, A.H.; Svane, I.M.; Zerahn, B.; Ellebaek, E. Cardiotoxicity in Metastatic Melanoma Patients Treated with BRAF and MEK Inhibitors in a Real-World Setting. Acta Oncol. 2022, 61, 45–51. [Google Scholar] [CrossRef]
- Mincu, R.I.; Mahabadi, A.A.; Michel, L.; Mrotzek, S.M.; Schadendorf, D.; Rassaf, T.; Totzeck, M. Cardiovascular Adverse Events Associated With BRAF and MEK Inhibitors: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2019, 2, e198890. [Google Scholar] [CrossRef]
- Berger, M.; Amini-Adlé, M.; Maucort-Boulch, D.; Robinson, P.; Thomas, L.; Dalle, S.; Courand, P.Y. Left Ventricular Ejection Fraction Decrease Related to BRAF and/or MEK Inhibitors in Metastatic Melanoma Patients: A Retrospective Analysis. Cancer Med. 2020, 9, 2611–2620. [Google Scholar] [CrossRef]
- Guha, A.; Jain, P.; Fradley, M.G.; Lenihan, D.; Gutierrez, J.M.; Jain, C.; de Lima, M.; Barnholtz-Sloan, J.S.; Oliveira, G.H.; Dowlati, A.; et al. Cardiovascular Adverse Events Associated with BRAF versus BRAF/MEK Inhibitor: Cross-Sectional and Longitudinal Analysis Using Two Large National Registries. Cancer Med. 2021, 10, 3862–3872. [Google Scholar] [CrossRef]
- Nebot, N.; Arkenau, H.T.; Infante, J.R.; Chandler, J.C.; Weickhardt, A.; Lickliter, J.D.; Sarantopoulos, J.; Gordon, M.S.; Mak, G.; St-Pierre, A.; et al. Evaluation of the Effect of Dabrafenib and Metabolites on QTc Interval in Patients with BRAF V600-Mutant Tumours. Br. J. Clin. Pharmacol. 2018, 84, 764–775. [Google Scholar] [CrossRef]
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity Associated with Immune Checkpoint Inhibitor Therapy: A Meta-Analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Kim, A.S.; Khorana, A.A.; McCrae, K.R. Mechanisms and Biomarkers of Cancer-Associated Thrombosis. Transl. Res. 2020, 225, 33–53. [Google Scholar] [CrossRef]
- Mackin, A.G.; Pecen, P.E.; Dinsmore, A.L.; Patnaik, J.L.; Gonzalez, R.; Robinson, W.A.; Palestine, A.G. Inflammatory Side Effects of BRAF and MEK Inhibitors. Melanoma Res. 2019, 29, 522–526. [Google Scholar] [CrossRef]
- Beckmann, M.; Schlüter, J.; Erdmann, M.; Kramer, R.; Cunningham, S.; Hackstein, H.; Zimmermann, R.; Heinzerling, L. Interdependence of Coagulation with Immunotherapy and BRAF/MEK Inhibitor Therapy: Results from a Prospective Study. Cancer Immunol. Immunother. 2024, 74, 5. [Google Scholar] [CrossRef]
- Cann, C.G.; Tillman, B.F.; Davis, E.J.; Johnson, D.B. Empiric Therapy with BRAF and MEK Inhibitors in Metastatic Melanoma. Oncologist 2019, 24, 1495–1496. [Google Scholar] [CrossRef]
- Haggstrom, L.; Duong, T.A.; Thomas, B.; Brungs, D.; Aghmesheh, M.; Parmar, G. Disseminated Intravascular Coagulation and Melanoma: A Novel Case Occurring in Metastatic Melanoma with BRAF and NRAS Mutations and Systematic Review. Melanoma Res. 2019, 29, 533–538. [Google Scholar] [CrossRef]
- Chuang, J.; Uche, A.; Gupta, R.; Margolin, K.; Kim, P. Fulminant Disseminated Intravascular Coagulation as Initial Presentation of BRAF-Mutated Melanoma. Case Rep. Oncol. Med. 2019, 2019, 1–2. [Google Scholar] [CrossRef]
- Zito, C.; Manganaro, R.; Ciappina, G.; Spagnolo, C.C.; Racanelli, V.; Santarpia, M.; Silvestris, N.; Carerj, S. Cardiotoxicity Induced by Immune Checkpoint Inhibitors: What a Cardio-Oncology Team Should Know and Do. Cancers 2022, 14, 5403. [Google Scholar] [CrossRef]
- Senechal, I.; Andres, M.S.; Tong, J.; Ramalingam, S.; Nazir, M.S.; Rosen, S.D.; Young, K.; Idaikkadar, P.; Larkin, J.; Lyon, A.R. Risk Stratification, Screening and Treatment of BRAF/MEK Inhibitors-Associated Cardiotoxicity. Curr. Oncol. Rep. 2024, 26, 1431–1441. [Google Scholar] [CrossRef]
- Karakasis, P.; Patoulias, D.; Fragakis, N. Balancing Promise and Evidence: Critical Perspectives on SGLT2 Inhibitors for Cardiotoxicity Prevention. JACC CardioOncol 2025, 7, 314–315. [Google Scholar] [CrossRef]
- Giunchi, V.; Fusaroli, M.; Hauben, M.; Raschi, E.; Poluzzi, E. Challenges and Opportunities in Accessing and Analysing FAERS Data: A Call Towards a Collaborative Approach. Drug Saf. 2023, 46, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Alatawi, Y.M.; Hansen, R.A. Empirical Estimation of Under-Reporting in the U.S. Food and Drug Administration Adverse Event Reporting System (FAERS). Expert. Opin. Drug Saf. 2017, 16, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Montastruc, J.L.; Sommet, A.; Bagheri, H.; Lapeyre-Mestre, M. Benefits and Strengths of the Disproportionality Analysis for Identification of Adverse Drug Reactions in a Pharmacovigilance Database. Br. J. Clin. Pharmacol. 2011, 72, 905–908. [Google Scholar] [CrossRef]
- Cutroneo, P.M.; Sartori, D.; Tuccori, M.; Crisafulli, S.; Battini, V.; Carnovale, C.; Rafaniello, C.; Capuano, A.; Poluzzi, E.; Moretti, U.; et al. Conducting and Interpreting Disproportionality Analyses Derived from Spontaneous Reporting Systems. Front. Drug Saf. Regul. 2024, 3, 1323057. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Characteristic | Combination | Monotherapy | ||
|---|---|---|---|---|
| All AEs (n = 14,103) | cAEs (n = 1268) | All AEs (n = 4267) | cAEs (n = 323) | |
| Age (years), median (Q1–Q3) | 61 (51–70) | 66 (55–74) | 62 (51–71) | 65 (58–74) |
| Age group, n (%) | ||||
| Neonate | 1 (<0.1) | 3 (0.1) | ||
| Child | 20 (0.1) | 6 (0.5) | 14 (0.3) | |
| Adolescent | 7 (<0.1) | 1 (0.1) | 10 (0.2) | 5 (1.5) |
| Adult | 5493 (38.9) | 469 (37) | 1633 (38.3) | 121 (37.5) |
| Elderly | 3937 (27.9) | 549 (43.3) | 1233 (28.9) | 146 (45.2) |
| Not available | 4645 (32.9) | 243 (19.2) | 1374 (32.2) | 51 (15.8) |
| Sex, n (%) | ||||
| Female | 5624 (39.9) | 519 (40.9) | 1729 (40.5) | 134 (41.5) |
| Male | 6501 (46.1) | 662 (52.2) | 2266 (53.1) | 171 (52.9) |
| Not available | 1978 (14) | 87 (6.9) | 272 (6.4) | 18 (5.6) |
| Weight (kg), median (Q1–Q3) | 77 (65–90) | 79 (66–90) | 74 (62–87) | 72 (59–86) |
| Type of reporter, n (%) | ||||
| Consumer | 4370 (31) | 235 (18.5) | 1211 (28.4) | 35 (10.8) |
| Physician | 6144 (43.6) | 692 (54.6) | 1887 (44.2) | 225 (69.7) |
| Pharmacist | 689 (4.9) | 38 (3) | 360 (8.4) | 10 (3.1) |
| Health-professional | 1457 (10.3) | 147 (11.6) | 237 (5.6) | 16 (5) |
| Other health-professional | 1167 (8.3) | 129 (10.2) | 443 (10.4) | 34 (10.5) |
| Not available | 276 (2) | 27 (2.1) | 129 (3) | 3 (0.9) |
| Outcome, n (%) | ||||
| Death | 1717 (12.2) | 114 (9) | 615 (14.4) | 33 (10.2) |
| Disability | 152 (1.1) | 22 (1.7) | 57 (1.3) | 3 (0.9) |
| Life-threatening | 228 (1.6) | 55 (4.3) | 72 (1.7) | 12 (3.7) |
| Hospitalization—initial or prolonged | 4215 (29.9) | 460 (36.3) | 1078 (25.3) | 149 (46.1) |
| Other serious (IME) | 5251 (37.2) | 602 (47.5) | 1232 (28.9) | 117 (36.2) |
| Required intervention to prevent permanent impairment/damage | 2 (<0.1) | 1 (<0.1) | 1 (0.3) | |
| Not available | 2538 (18) | 15 (1.2) | 1212 (28.4) | 8 (2.5) |
| Reporter country, n (%) | ||||
| Africa | 28 (0.2) | 25 (0.6) | ||
| Asia | 1595 (11.3) | 159 (12.5) | 309 (7.2) | 42 (13) |
| Europe | 5596 (39.7) | 762 (60.1) | 1323 (31) | 175 (54.2) |
| North America | 5102 (36.2) | 206 (16.2) | 2337 (54.8) | 85 (26.3) |
| Central America | 17 (0.1) | 2 (0.2) | 17 (0.4) | 3 (0.9) |
| South America | 420 (3) | 39 (3.1) | 60 (1.4) | 6 (1.9) |
| Oceania | 200 (1.4) | 23 (1.8) | 76 (1.8) | 11 (3.4) |
| Not available | 1145 (8.1) | 77 (6.1) | 120 (2.8) | 1 (0.3) |
| Year of reporting, n (%) | ||||
| 2014 | 207 (1.5) | 21 (1.7) | 394 (9.2) | 35 (10.8) |
| 2015 | 1055 (7.5) | 72 (5.7) | 904 (21.2) | 78 (24.1) |
| 2016 | 1130 (8) | 96 (7.6) | 975 (22.8) | 55 (17) |
| 2017 | 1226 (8.7) | 126 (9.9) | 352 (8.2) | 34 (10.5) |
| 2018 | 1786 (12.7) | 159 (12.5) | 340 (8) | 35 (10.8) |
| 2019 | 2430 (17.2) | 209 (16.5) | 312 (7.3) | 35 (10.8) |
| 2020 | 1887 (13.4) | 190 (15) | 350 (8.2) | 19 (5.9) |
| 2021 | 1714 (12.2) | 132 (10.4) | 230 (5.4) | 14 (4.3) |
| 2022 | 1317 (9.3) | 150 (11.8) | 236 (5.5) | 9 (2.8) |
| 2023 | 1351 (9.6) | 113 (8.9) | 174 (4.1) | 9 (2.8) |
| D + T | V + C | E + B | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N | ROR (95% CI) | Expected | N | ROR (95% CI) | Expected | N | ROR (95% CI) | Expected | |
| Bradyarrhythmias | |||||||||
| Atrioventricular block complete | 6 | 3.24 (1.45–7.22) | Yes | ||||||
| Brugada syndrome | 4 | 25.12 (9.35–67.45) | No | ||||||
| Electrocardiogram QRS complex prolonged | 5 | 6.53 (2.71–15.73) | No | ||||||
| Electrocardiogram QT prolonged | 59 | 5.09 (3.94–6.58) | No | 26 | 9.5 (6.45–13.98) | Yes | 14 | 3.46 (2.04–5.84) | Yes |
| Electrocardiogram repolarisation abnormality | 8 | 32.89 (16.33–66.27) | No | ||||||
| Atrioventricular block * | 4 | - | |||||||
| Atrioventricular block first degree * | 5 | 3.98 (1.66–9.58) | Yes | ||||||
| Atrioventricular block second degree * | 3 | 3.52 (1.13–10.92) | Yes | ||||||
| Bundle branch block left * | 5 | 4.14 (1.72–9.97) | Yes | ||||||
| Bundle branch block right * | 5 | 3.97 (1.65–9.56) | Yes | 5 | 16.78 (6.97–40.4) | No | |||
| Defect conduction intraventricular * | 6 | 49.2 (21.83–110.91) | No | 3 | 69.62 (22.25–217.87) | No | |||
| Sinus arrhythmia * | 3 | 7.37 (2.37–22.92) | No | ||||||
| Sinus bradycardia * | 10 | 3.64 (1.95–6.76) | Yes | 6 | 6.27 (2.81–13.97) | No | |||
| Cardiac failure | |||||||||
| Cardiac failure | 67 | 2.05 (1.61–2.6) | Yes | 29 | 3.76 (2.6–5.42) | No | 18 | - | |
| Cardiac failure chronic | 3 | - | |||||||
| Cardiac failure congestive | 9 | - | |||||||
| Cardiogenic shock | 5 | 3.91 (1.63–9.4) | No | ||||||
| Left ventricular failure | 10 | 32.56 (17.47–60.7) | No | 4 | 8.82 (3.31–23.54) | Yes | |||
| Pulmonary oedema | 15 | - | |||||||
| Cardiomyopathy | |||||||||
| Cardiomyopathy | 16 | 3.7 (2.26–6.04) | Yes | 4 | 3.89 (1.46–10.37) | Yes | 3 | - | |
| Cardiotoxicity | 7 | 4.22 (2.01–8.87) | No | ||||||
| Dilated cardiomyopathy | 12 | 7.17 (4.07–12.65) | Yes | ||||||
| Ischaemic cardiomyopathy | 6 | 6.44 (2.89–14.37) | Yes | ||||||
| Tachycardia induced cardiomyopathy * | 7 | 135.12 (62.43–292.43) | Yes | ||||||
| Ejection fraction abnormal * | 4 | 5.95 (2.23–15.9) | Yes | ||||||
| Ejection fraction decreased * | 148 | 24.58 (20.87–28.95) | Yes | 36 | 24.93 (17.92–34.67) | Yes | 17 | 7.91 (4.91–12.75) | Yes |
| Stress cardiomyopathy * | 5 | 16.61 (6.9–39.98) | Yes | ||||||
| Tachyarrhythmias | |||||||||
| Atrial fibrillation | 99 | 2.37 (1.94–2.89) | No | 27 | 2.73 (1.86–3.99) | Yes | 8 | - | |
| Ventricular arrhythmia | 4 | 4.13 (1.55–11.01) | No | 5 | 21.8 (9.05–52.49) | Yes | |||
| Atrial flutter * | 5 | - | 3 | - | |||||
| Extrasystoles * | 3 | - | 4 | 6.39 (2.39–17.05) | No | ||||
| Sinus tachycardia * | 8 | 2.04 (1.02–4.08) | No | ||||||
| Supraventricular tachycardia * | 5 | ||||||||
| Tachyarrhythmia * | 8 | 9.07 (4.53–18.18) | No | ||||||
| Ventricular extrasystoles * | 3 | - | 4 | 5.44 (2.04–14.52) | No | ||||
| Ischaemic heart disease | |||||||||
| Acute myocardial infarction | 8 | - | 5 | - | 3 | - | |||
| Myocardial infarction | 30 | - | 5 | - | 8 | - | |||
| Angina pectoris | 14 | - | 7 | - | |||||
| Coronary artery stenosis | 10 | 7.28 (3.91–13.55) | No | ||||||
| Myocardial ischaemia | 3 | - | |||||||
| Coronary artery disease * | 6 | - | |||||||
| Blood creatine phosphokinase MB increased * | 7 | 25.95 (12.29–54.77) | No | ||||||
| Troponin T increased * | 11 | 24.36 (13.43–44.2) | No | ||||||
| Troponin increased * | 6 | - | |||||||
| Embolism and thrombotic events | |||||||||
| Disseminated intravascular coagulation | 38 | 10.22 (7.42–14.06) | No | 4 | 3.06 (1.15–8.17) | No | |||
| Splenic infarction | 9 | 15.78 (8.18–30.43) | No | ||||||
| Blindness transient | 5 | 4.78 (1.99–11.49) | No | ||||||
| Retinal vein occlusion | 14 | 14.93 (8.82–25.29) | Yes | 4 | 17.87 (6.69–47.72) | Yes | 3 | 9.12 (2.94–28.33) | Yes |
| Cerebral infarction | 11 | - | 3 | - | |||||
| Cerebral ischaemia | 12 | 7.24 (4.11–12.77) | No | 8 | 20.37 (10.16–40.83) | No | |||
| Cerebrovascular accident | 77 | - | 11 | - | 15 | - | |||
| Haemorrhagic stroke | 6 | - | |||||||
| Hemiparesis | 29 | 4.42 (3.07–6.37) | No | 7 | 3.06 (1.46–6.42) | No | |||
| Hemiplegia | 10 | 3.06 (1.64–5.68) | No | ||||||
| Ischaemic stroke | 6 | - | |||||||
| Monoplegia | 4 | - | |||||||
| Paraparesis | 7 | 11.1 (5.28–23.35) | No | ||||||
| Paraplegia | 9 | 8.47 (4.4–16.3) | No | ||||||
| Quadriplegia | 3 | 4.84 (1.56–15.04) | No | ||||||
| Transient ischaemic attack | 7 | - | 6 | - | |||||
| Pulmonary embolism | 111 | 3.35 (2.78–4.04) | Yes | 22 | 2.79 (1.83–4.24) | No | 16 | - | |
| Pulmonary infarction | 4 | 17.07 (6.39–45.6) | No | ||||||
| Pulmonary thrombosis | 6 | - | |||||||
| Deep vein thrombosis | 35 | 1.59 (1.14–2.21) | Yes | 10 | 1.91 (1.03–3.55) | No | 4 | - | |
| Embolism | 4 | - | 9 | 8.95 (4.65–17.23) | Yes | ||||
| Subclavian vein thrombosis | 3 | 7.88 (2.54–24.52) | Yes | ||||||
| Thrombosis | 52 | 1.47 (1.12–1.93) | Yes | 4 | - | 10 | - | ||
| Monoparesis * | 8 | 43.49 (21.67–87.28) | No | ||||||
| Paradoxical embolism * | 7 | 230.83 (104.42–510.27) | No | ||||||
| Superficial vein thrombosis * | 3 | - | |||||||
| Thrombophlebitis * | 7 | 5.47 (2.6–11.49) | Yes | ||||||
| Venous thrombosis * | 10 | 7.28 (3.91–13.55) | Yes | ||||||
| Venous thrombosis limb * | 4 | 4.18 (1.57–11.15) | Yes | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbieri, M.A.; Russo, G.; Cicala, G.; Zito, C.; Spina, E.; Silvestris, N.; Santarpia, M. Cardiovascular Safety Profile of BRAF and MEK Inhibitors in Melanoma: FAERS Data Through a Retrospective Disproportionality Analysis (2014–2023). Cancers 2025, 17, 1755. https://doi.org/10.3390/cancers17111755
Barbieri MA, Russo G, Cicala G, Zito C, Spina E, Silvestris N, Santarpia M. Cardiovascular Safety Profile of BRAF and MEK Inhibitors in Melanoma: FAERS Data Through a Retrospective Disproportionality Analysis (2014–2023). Cancers. 2025; 17(11):1755. https://doi.org/10.3390/cancers17111755
Chicago/Turabian StyleBarbieri, Maria Antonietta, Giulia Russo, Giuseppe Cicala, Concetta Zito, Edoardo Spina, Nicola Silvestris, and Mariacarmela Santarpia. 2025. "Cardiovascular Safety Profile of BRAF and MEK Inhibitors in Melanoma: FAERS Data Through a Retrospective Disproportionality Analysis (2014–2023)" Cancers 17, no. 11: 1755. https://doi.org/10.3390/cancers17111755
APA StyleBarbieri, M. A., Russo, G., Cicala, G., Zito, C., Spina, E., Silvestris, N., & Santarpia, M. (2025). Cardiovascular Safety Profile of BRAF and MEK Inhibitors in Melanoma: FAERS Data Through a Retrospective Disproportionality Analysis (2014–2023). Cancers, 17(11), 1755. https://doi.org/10.3390/cancers17111755