
TSGA10 as a Model of a Thermal Metabolic Regulator: Implications for Cancer Biology




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
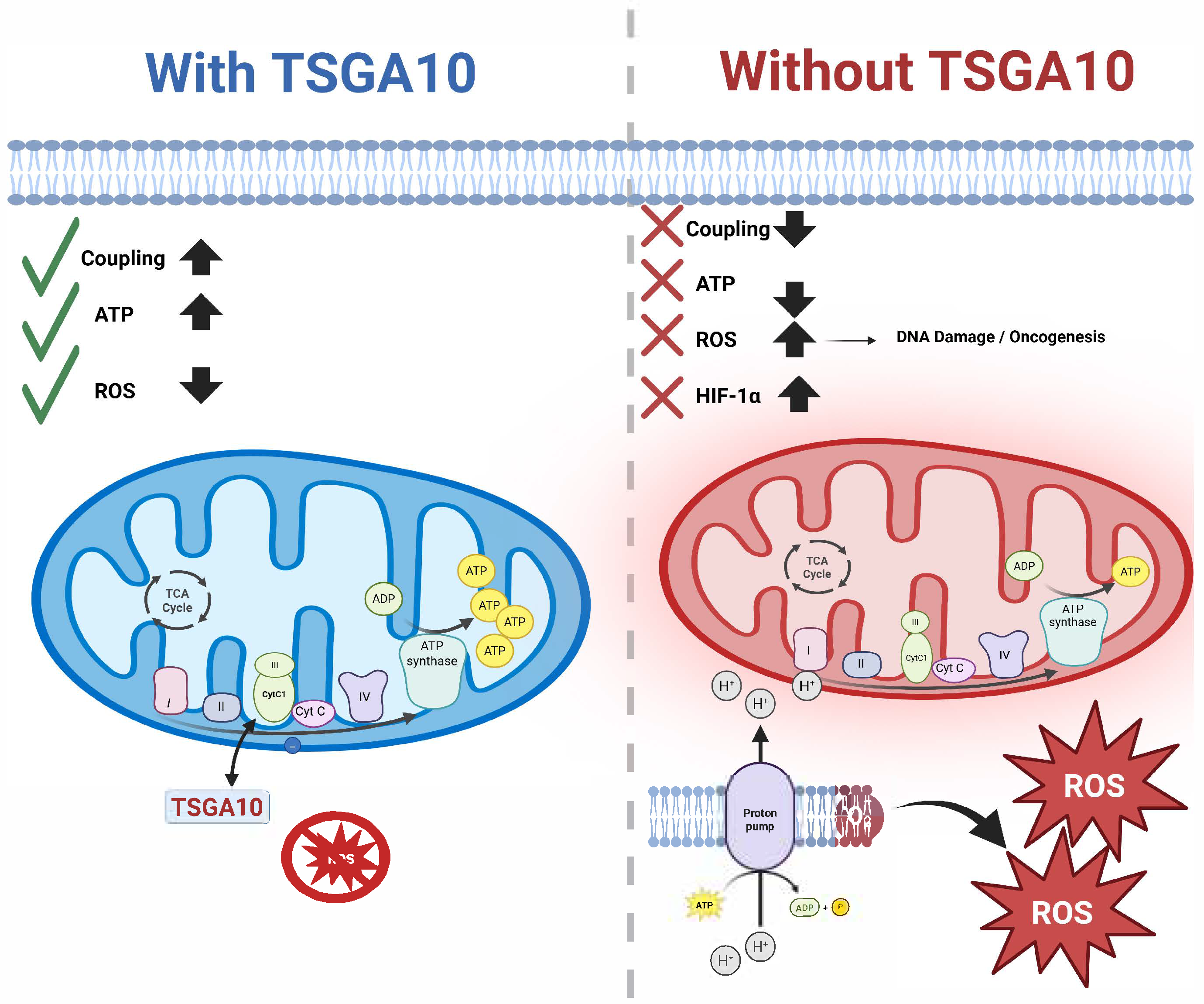
Abstract
1. Introduction
2. Materials and Methods
3. Mitochondrial Role in Heat Production and Uncoupling
3.1. Mitochondrial (De-)Coupling
3.2. Mitochondrial Coupling Across Healthy Tissues
3.3. Mitochondrial Decoupling in Brown Adipose Tissue
3.4. Mitochondrial Uncoupling and Cancer
4. TSGA10 and Metabolic Activity
4.1. Transcriptional and Post-Transcriptional Regulation of TSGA10
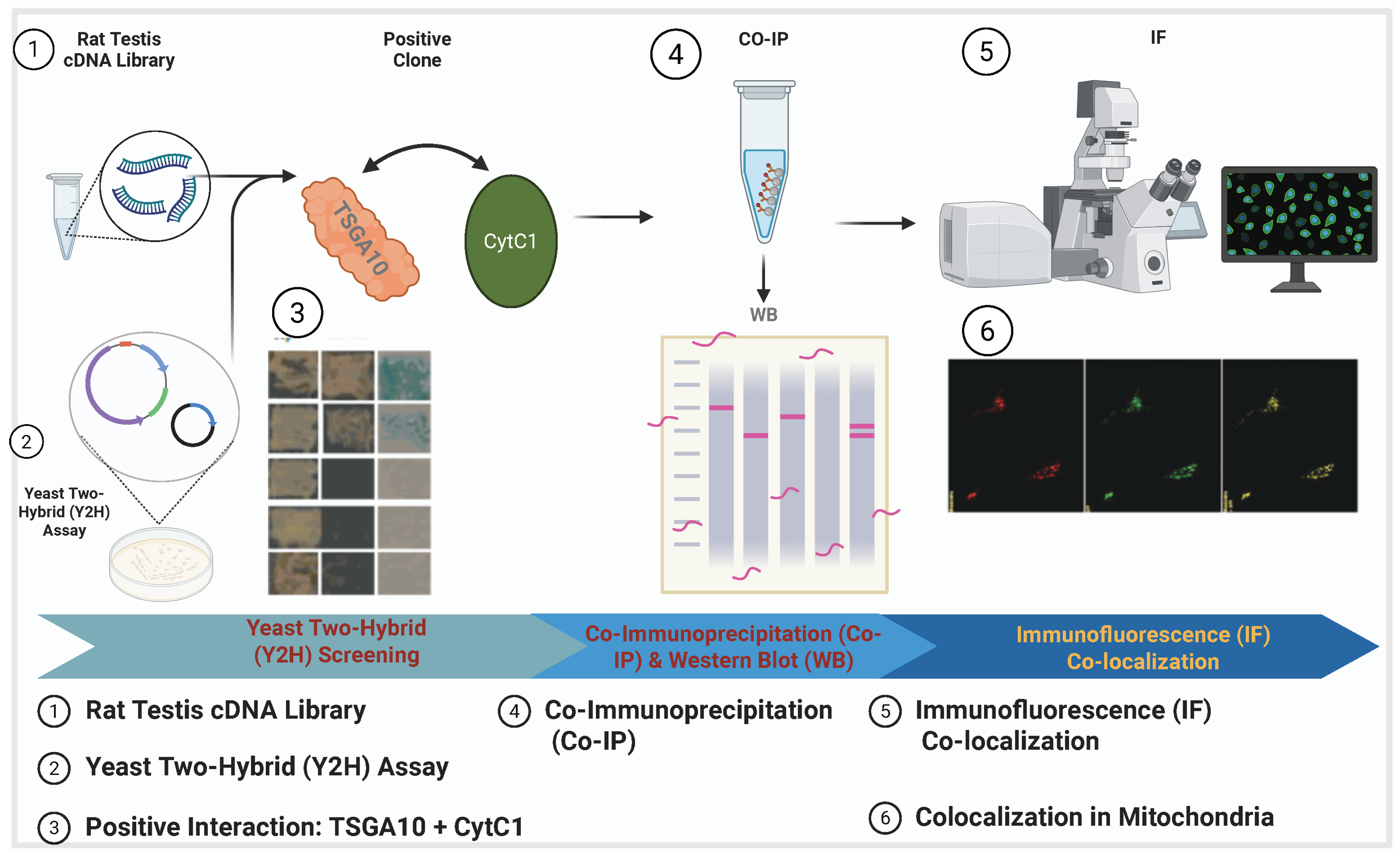
4.2. TSGA10 and Mitochondria
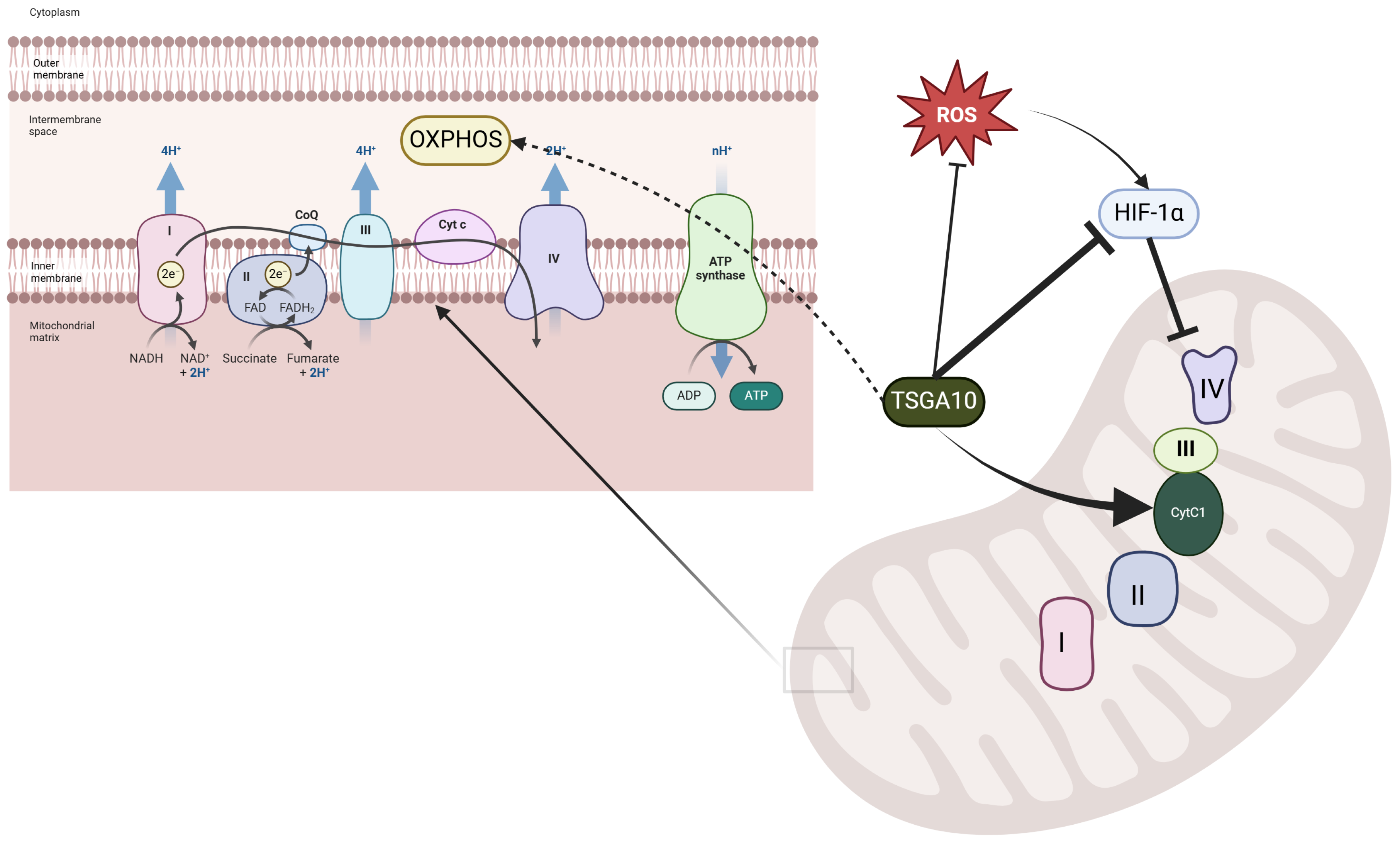
4.3. TSGA10 and Its Potential Role in Mitochondrial Coupling
4.3.1. Mitochondrial Complex III and the Electron Transport Chain
4.3.2. A Potential Thermal Role for TSGA10
4.3.3. A Potential Complex III Assembly Role for TSGA10
4.3.4. Disruption of Mitochondrial Coupling in Health and Disease
4.4. TSGA10 and Oxygen Sensing
4.5. HIF-1 in Thermoregulation
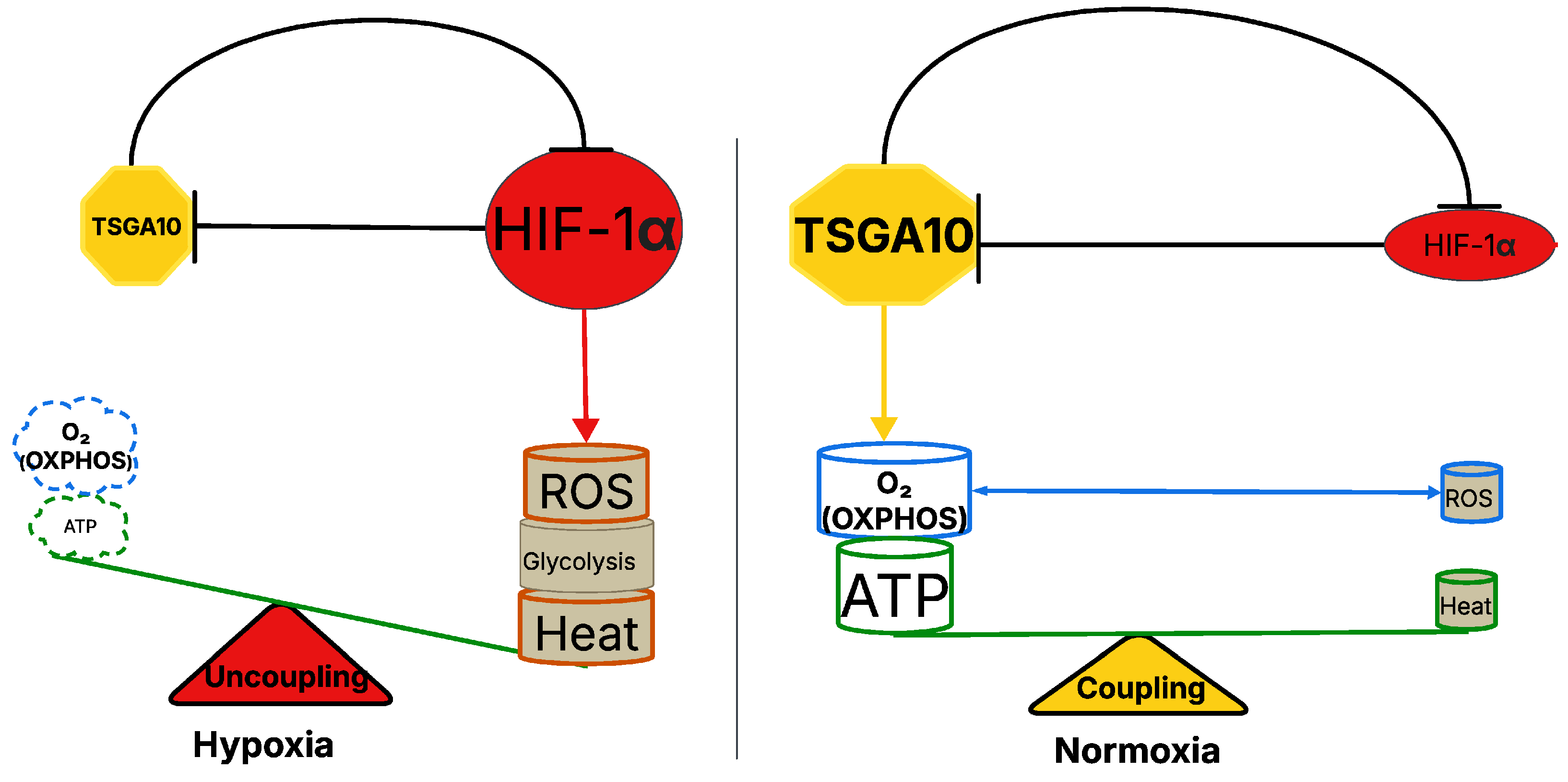
5. TSGA10 and HIF-1 Mutual Counter Repression in Thermoregulation
5.1. HIF-1-Dependent Regulation of ROS Generation and Thermogenic Mechanisms in Hypoxic Conditions
5.2. Role of Mitochondria Numbers and Blood Circulation in Thermal and Metabolic Stability
5.3. TSGA10 as a Potential Mitochondrial Regulator in Cancer
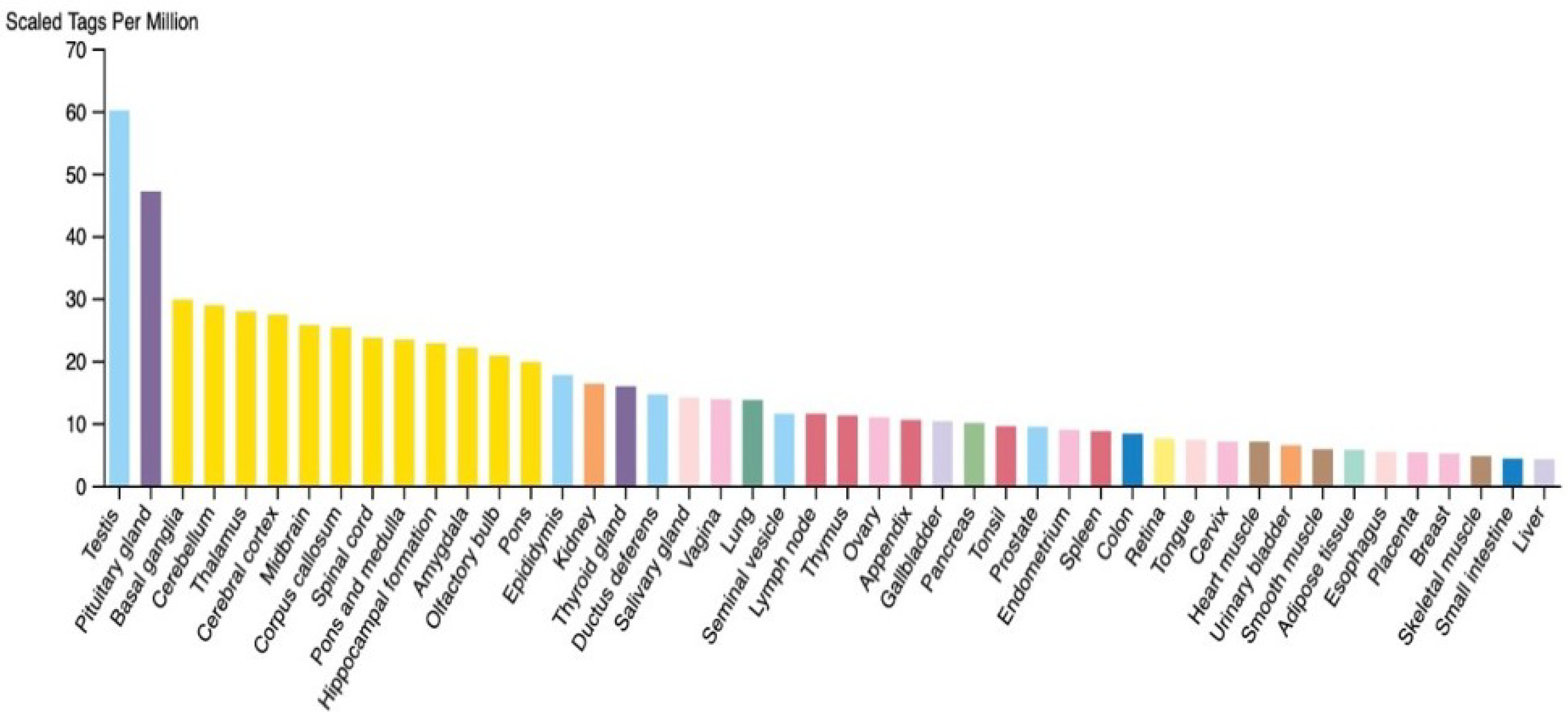
5.4. TSGA10 Is Expressed in Postmitotic Energy-Demanding Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TSGA10 | Testis-Specific Gene 10 |
| HIF-1 | Hypoxia-Inducible Factor 1-Alpha |
| VEGFA | Vascular Endothelial Growth Factor A |
| MMP2 | Matrix Metalloproteinase-2 |
| MMP9 | Matrix Metalloproteinase-9 |
| OXPHOS | Oxidative Phosphorylation |
| ROS | Reactive Oxygen Species |
| ETC | Electron Transport Chain |
| CytC1 | Cytochrome c1 |
| UCP1 | Uncoupling Protein 1 |
| BAT | Brown Adipose Tissue |
| MICOS | Mitochondrial Contact Site and Cristae Organizing System |
| ATP | Adenosine Triphosphate |
| HSPs | Heat Shock Proteins |
| RISP | Rieske Iron–Sulfur Protein |
| COX4 | Cytochrome c Oxidase Subunit 4 |
References
- Modarressi, M.H.; Cameron, J.; Taylor, K.E.; Wolfe, J. Identification and characterisation of a novel gene, TSGA10, expressed in testis. Gene 2001, 262, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh-Hesary, F.; Ghadyani, M.; Kashanchi, F.; Behnam, B. Exploring TSGA10 Function: A Crosstalk or Controlling Mechanism in the Signaling Pathway of Carcinogenesis? Cancers 2024, 16, 3044. [Google Scholar] [CrossRef] [PubMed]
- Behnam, B.; Modarressi, M.H.; Conti, V.; Taylor, K.E.; Puliti, A.; Wolfe, J. Expression of Tsga10 sperm tail protein in embryogenesis and neural development: From cilium to cell division. Biochem. Biophys. Res. Commun. 2006, 344, 1102–1110. [Google Scholar] [CrossRef]
- Salehipour, P.; Nematzadeh, M.; Mobasheri, M.B.; Afsharpad, M.; Mansouri, K.; Modarressi, M.H. Identification of new TSGA10 transcript variants in human testis with conserved regulatory RNA elements in 5’untranslated region and distinct expression in breast cancer. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 973–982. [Google Scholar] [CrossRef]
- Sha, Y.W.; Sha, Y.K.; Ji, Z.Y.; Mei, L.B.; Ding, L. TSGA10 is a novel candidate gene associated with acephalic spermatozoa. Clin. Genet. 2018, 93, 776–783. [Google Scholar] [CrossRef]
- Hägele, S.; Behnam, B.; Borter, E.; Wolfe, J.; Paasch, U.; Lukashev, D.; Sitkovsky, M.; Wenger, R.H.; Katschinski, D.M. TSGA10 prevents nuclear localization of the hypoxia-inducible factor (HIF)-1α. FEBS Lett. 2006, 580, 3731–3738. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Mostafie, A.; Rezazadeh, D.; Shahlaei, M.; Modarressi, M.H. New function of TSGA10 gene in angiogenesis and tumor metastasis: A response to a challengeable paradox. Hum. Mol. Genet. 2016, 25, 233–244. [Google Scholar] [CrossRef]
- Roghanian, A.; Jones, D.C.; Pattisapu, J.V.; Wolfe, J.; Young, N.T.; Behnam, B. Filament-associated TSGA10 protein is expressed in professional antigen presenting cells and interacts with vimentin. Cell. Immunol. 2010, 265, 120–126. [Google Scholar] [CrossRef]
- Tanaka, R.; Ono, T.; Sato, S.; Nakada, T.; Koizumi, F.; Hasegawa, K.; Nakagawa, K.; Okumura, H.; Yamashita, T.; Ohtsuka, M.; et al. Over-expression of the testis-specific gene TSGA10 in cancers and its immunogenicity. Microbiol. Immunol. 2004, 48, 339–345. [Google Scholar] [CrossRef]
- Mobasheri, M.B.; Modarressi, M.H.; Shabani, M.; Asgarian, H.; Sharifian, R.A.; Vossough, P.; Shokri, F. Expression of the testis-specific gene, TSGA10, in Iranian patients with acute lymphoblastic leukemia (ALL). Leuk. Res. 2006, 30, 883–889. [Google Scholar] [CrossRef]
- Luo, G.; Hou, M.; Wang, B.; Liu, Z.; Liu, W.; Han, T.; Zhang, D.; Zhou, X.; Jia, W.; Tan, Y.; et al. Tsga10 is essential for arrangement of mitochondrial sheath and male fertility in mice. Andrology 2021, 9, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.M.; Wei, L.S.; Ye, J.F. Advancements in mitochondrial-targeted nanotherapeutics: Overcoming biological obstacles and optimizing drug delivery. Front. Immunol. 2024, 15, 1451989. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Yeagle, P.L. The Membranes of Cells, 2nd ed.; Academic Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Walker, B.R.; Moraes, C.T. Nuclear-Mitochondrial Interactions. Biomolecules 2022, 12, 427. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M. Principles of Neural Science, 4th ed.; McGraw-Hill: New York, NY, USA, 2000. [Google Scholar]
- Hertzler, J.I.; Bernard, A.R.; Rolls, M.M. Dendrite regeneration mediates functional recovery after complete dendrite removal. Dev. Biol. 2023, 497, 18–25. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Sokolova, I. Mitochondrial adaptations to variable environments and their role in animals’ stress tolerance. Integr. Comp. Biol. 2018, 58, 519–531. [Google Scholar] [CrossRef]
- Hamilton, T.R.; Mendes, C.M.; de Castro, L.S.; de Assis, P.M.; Siqueira, A.F.; de Carvalho Delgado, J.; Goissis, M.D.; Muiño-Blanco, T.; Cebrián-Pérez, J.Á.; Nichi, M.; et al. Evaluation of Lasting Effects of Heat Stress on Sperm Profile and Oxidative Status of Ram Semen and Epididymal Sperm. Oxidative Med. Cell. Longev. 2016, 2016, 1687657. [Google Scholar] [CrossRef]
- Park, A.; Kim, K.E.; Park, I.; Lee, S.H.; Park, K.Y.; Jung, M.; Li, X.; Sleiman, M.B.; Lee, S.J.; Kim, D.-S.; et al. Mitochondrial matrix protein LETMD1 maintains thermogenic capacity of brown adipose tissue in male mice. Nat. Commun. 2023, 14, 3746. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Mahani, S.T.; Behnam, B.; Abbassi, M.; Asgari, H.; Nazmara, Z.; Shirinbayan, P.; Joghataei, M.; Koruji, M. Tsga10 expression correlates with sperm profiles in the adult formalin-exposed mice. Andrologia 2016, 48, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas Project. The Human Protein Atlas: The Open Access Resource for Human Proteins. 2024. Available online: https://www.proteinatlas.org (accessed on 29 April 2025).
- Jahani, M.; Shahlaei, M.; Norooznezhad, F.; Miraghaee, S.S.; Hosseinzadeh, L.; Moasefi, N.; Khodarahmi, R.; Farokhi, A.; Mahnam, A.; Mansouri, K. TSGA10 Overexpression Decreases Metastatic and Metabolic Activity by Inhibiting HIF-1 in Breast Cancer Cells. Arch. Med. Res. 2020, 51, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, W.; Luo, C.; Zhang, W. Exploring a four-gene risk model based on doxorubicin resistance-associated lncRNAs in hepatocellular carcinoma. Front. Pharmacol. 2022, 13, 1015842. [Google Scholar] [CrossRef]
- Asghari-Givehchi, S.; Hossein-Modarressi, M. Identification and expression analysis of zebrafish testis-specific gene 10 (tsga10). Int. J. Dev. Biol. 2019, 63, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Amoorahim, M.; Valipour, E.; Hoseinkhani, Z.; Mahnam, A.; Rezazadeh, D.; Ansari, M.; Shahlaei, M.; Gamizgy, Y.H.; Moradi, S.; Mansouri, K. TSGA10 overexpression inhibits angiogenesis of HUVECs: A HIF-2α biased perspective. Microvasc. Res. 2020, 128, 103952. [Google Scholar] [CrossRef]
- Wei, W.; Gong, Y.; Guo, X.; Liu, M.; Zhou, Y.; Li, Z.; Zhou, L.; Wang, Z.; Gui, J. Gonadal transcriptomes reveal sex-biased expression genes associated with sex determination and differentiation in red-tail catfish (Hemibagrus wyckioides). BMC Genom. 2023, 24, 183. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, J.; Fu, Z.; Dong, L.; Tang, Y.; Xu, C.; Wang, H.; Zhang, T.; Wu, Y.; Dong, C.; et al. Hypoxia-induced microRNA-10b-3p promotes esophageal squamous cell carcinoma growth and metastasis by targeting TSGA10. Aging 2019, 11, 10374–10384. [Google Scholar] [CrossRef]
- Behnam, B. Investigation of TSGA10 Gene Expression, Localization, and Protein Interaction in Human and Mouse Spermatogenesis. Ph.D. Thesis, University College London, London, UK, 2005. [Google Scholar]
- Van Haute, L.; Lee, S.Y.; McCann, B.J.; Powell, C.A.; Bansal, D.; Vasiliauskaitė, L.; Garone, C.; Shin, S.; Kim, J.O.; Frye, M.; et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8720–8733. [Google Scholar] [CrossRef]
- Ye, Y.; Wei, X.; Sha, Y.; Li, N.; Yan, X.; Cheng, L.; Qiao, D.; Zhou, W.; Wu, R.; Liu, Q.; et al. Loss-of-function mutation in TSGA10 causes acephalic spermatozoa phenotype in human. Mol. Genet. Genom. Med. 2020, 8, e1284. [Google Scholar] [CrossRef]
- Asgari, R.; Bakhtiari, M.; Rezazadeh, D.; Yarani, R.; Esmaeili, F.; Mansouri, K. TSGA10 as a Potential Key Factor in the Process of Spermatid Differentiation/Maturation: Deciphering Its Association with Autophagy Pathway. Reprod. Sci. 2021, 28, 3228–3240. [Google Scholar] [CrossRef]
- Rich, P.R. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003, 31, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Esser, L.; Zhou, F.; Yu, C.A.; Xia, D. Crystal structure of bacterial cytochrome bc1 in complex with azoxystrobin reveals a conformational switch of the Rieske iron-sulfur protein subunit. J. Biol. Chem. 2019, 294, 12007–12019. [Google Scholar] [CrossRef] [PubMed]
- Behnam, B.; Fazilaty, H.; Ghadyani, M.; Fadavi, P.; Taghizadeh-Hesary, F. Ciliated, Mitochondria-Rich Postmitotic Cells are Immune-privileged, and Mimic Immunosuppressive Microenvironment of Tumor-Initiating Stem Cells: From Molecular Anatomy to Molecular Pathway. Front. Biosci. (Landmark Ed.) 2023, 28, 261. [Google Scholar] [CrossRef]
- Tabassum, N.; Kheya, I.S.; Asaduzzaman, S.A.I.; Maniha, S.M.; Fayz, A.H.; Zakaria, A.; Noor, R. A Review on the Possible Leakage of Electrons through the Electron Transport Chain within Mitochondria. J. Biomed. Environ. Sci. 2020, 1, 105–113. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Conte, L.; Zara, V. The Rieske Iron-Sulfur Protein: Import and Assembly into the Cytochrome bc(1) Complex of Yeast Mitochondria. Bioinorg. Chem. Appl. 2011, 2011, 363941. [Google Scholar] [CrossRef]
- Zerbes, R.M.; Colina-Tenorio, L.; Bohnert, M.; von der Malsburg, K.; Peikert, C.D.; Mehnert, C.S.; Perschil, I.; Klar, R.F.U.; de Boer, R.; Kram, A.; et al. Coordination of cytochrome bc1 complex assembly at MICOS. EMBO Rep. 2025, 26, 353–384. [Google Scholar] [CrossRef]
- Klimova, T.; Chandel, N. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, M.; Jeong, Y.Y.; Margolis, D.J.; Cai, Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy 2021, 17, 4182–4201. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef]
- Wang, T.; Xu, Y. Tumor hypoxia, HIF-1α, and microRNA-210: An emerging hypoxia pathway in cancer. Cell Cycle 2010, 9, 1346–1353. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Choi, D.; Jeong, J.; Chung, S.; Chang, Y.; Na, K.; Lee, K. Preservation of Hypoxia-Inducible Factor-1 Induced By ERK Phosphorylation Is Involved in Hypothermic Protection of Renal Ischemia-Reperfusion Injury. Transplantation 2014, 98, 354. Available online: https://journals.lww.com/transplantjournal/fulltext/2014/07151/preservation_of_hypoxia_inducible_factor_1_induced.1145.aspx (accessed on 17 May 2025). [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Yamashita, S.I.; Kanki, T. Mitophagy Responds to the Environmental Temperature and Regulates Mitochondrial Mass in Adipose Tissues. Adv. Exp. Med. Biol. 2024, 1461, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Yamada, T. Adipose tissue thermogenesis by calcium futile cycling. J. Biochem. 2022, 172, 197–203. [Google Scholar] [CrossRef]
- Lee, J.H.; Rao, M.V.; Yang, D.S.; Stavrides, P.; Im, E.; Pensalfini, A.; Huo, C.; Sarkar, P.; Yoshimori, T.; Nixon, R.A. Transgenic expression of a ratiometric autophagy probe specifically in neurons enables the interrogation of brain autophagy in vivo. Autophagy 2019, 15, 543–557. [Google Scholar] [CrossRef]
- Aitken, R.J.; Lewis, S.E.M. DNA damage in testicular germ cells and spermatozoa. When and how is it induced? How should we measure it? What does it mean? Andrology 2023, 11, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Moustakli, E.; Zikopoulos, A.; Skentou, C.; Bouba, I.; Tsirka, G.; Stavros, S.; Vrachnis, D.; Vrachnis, N.; Potiris, A.; Georgiou, I.; et al. Sperm Mitochondrial Content and Mitochondrial DNA to Nuclear DNA Ratio Are Associated with Body Mass Index and Progressive Motility. Biomedicines 2023, 11, 3014. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, J.; Petricca, S.; Iorio, R.; Toniato, E.; Flati, V. Mitochondrial and metabolic alterations in cancer cells. Eur. J. Cell Biol. 2022, 101, 151225. [Google Scholar] [CrossRef]
- García-Aguilar, A.; Cuezva, J.M. A Review of the Inhibition of the Mitochondrial ATP Synthase by IF1 in vivo: Reprogramming Energy Metabolism and Inducing Mitohormesis. Front. Physiol. 2018, 9, 1322. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Ding, H.; Jiang, M.; Yin, H.; Gollasch, M.; Huang, Y. Perivascular adipose tissue: Fine-tuner of vascular redox status and inflammation. Redox Biol. 2023, 62, 102683. [Google Scholar] [CrossRef]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.L. Energy metabolism in health and diseases. Signal Transduct. Target. Ther. 2025, 10, 69. [Google Scholar] [CrossRef]
- Chen, X.S.; Li, L.Y.; Guan, Y.D.; Yang, J.-M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef]
- Taghizadeh-Hesary, F. Is Chronic Ice Water Ingestion a Risk Factor for Gastric Cancer Development? An Evidence-Based Hypothesis Focusing on East Asian Populations. Oncol. Ther. 2024, 12, 629–646. [Google Scholar] [CrossRef]
- Behnam, B.; Mobahat, M.; Fazilaty, H.; Wolfe, J.; Omran, H. TSGA10 is a Centrosomal Protein, Interacts with ODF2 and Localizes to Basal Body. J. Cell Sci. Ther. 2015, 6, 217. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amini, A.; Taghizadeh-Hesary, F.; Bracht, J.; Behnam, B. TSGA10 as a Model of a Thermal Metabolic Regulator: Implications for Cancer Biology. Cancers 2025, 17, 1756. https://doi.org/10.3390/cancers17111756
Amini A, Taghizadeh-Hesary F, Bracht J, Behnam B. TSGA10 as a Model of a Thermal Metabolic Regulator: Implications for Cancer Biology. Cancers. 2025; 17(11):1756. https://doi.org/10.3390/cancers17111756
Chicago/Turabian StyleAmini, Ali, Farzad Taghizadeh-Hesary, John Bracht, and Babak Behnam. 2025. "TSGA10 as a Model of a Thermal Metabolic Regulator: Implications for Cancer Biology" Cancers 17, no. 11: 1756. https://doi.org/10.3390/cancers17111756
APA StyleAmini, A., Taghizadeh-Hesary, F., Bracht, J., & Behnam, B. (2025). TSGA10 as a Model of a Thermal Metabolic Regulator: Implications for Cancer Biology. Cancers, 17(11), 1756. https://doi.org/10.3390/cancers17111756