
Unveiling the Maze: Branched-Chain Amino Acids Fueling the Dynamics of Cancer Metabolism and Progression



{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
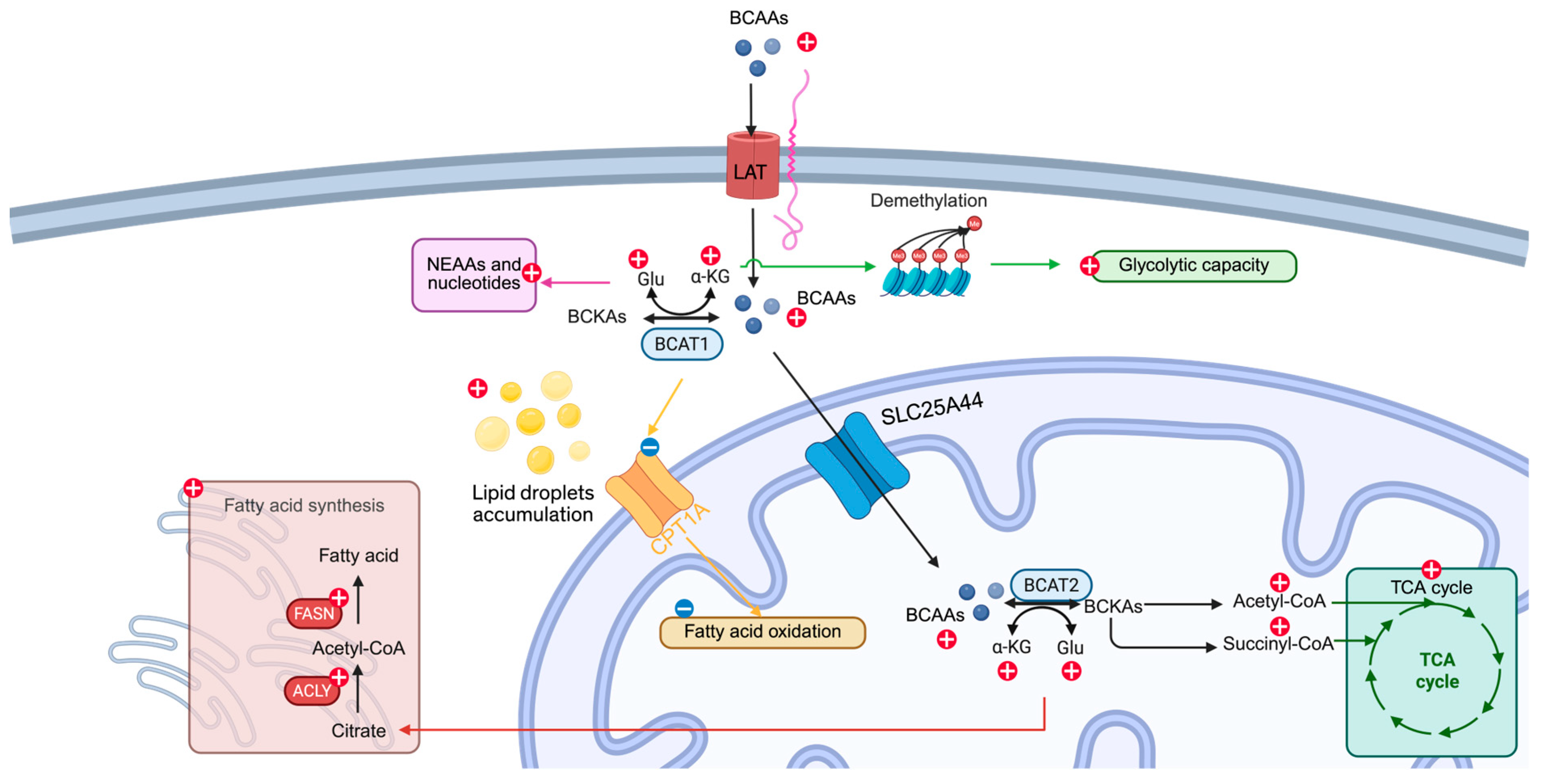
2. BCAA Metabolism
2.1. Cellular Uptake
2.2. Transamination
2.3. Oxidative Decarboxylation
3. Reprogramming BCAA Metabolism in Cancer
- Provision of Carbon and Nitrogen for BiosynthesisBCAAs supply both the carbon skeletons and amino groups required for de novo protein synthesis, supporting the rapid proliferation of malignant cells.
- Activation of mTORC1 SignalingLeucine acts as a direct allosteric activator of mTORC1, thereby promoting anabolic programs, cell growth, and survival under nutrient-replete conditions.
- Anaplerotic Support and Energy GenerationThrough BCAT-mediated transamination and BCKDH-driven oxidative decarboxylation, BCAAs generate acetyl-CoA and succinyl-CoA, replenishing TCA cycle intermediates and fueling ATP production.
- Maintenance of Redox HomeostasisTransamination of BCAAs yields glutamate, which is a precursor for glutathione synthesis; elevated glutathione buffers reactive oxygen species and enhances stress resistance.
- Augmentation of Nucleotide and Nonessential Amino Acid SynthesisBCAA-derived α-ketoglutarate and glutamate serve as substrates for the synthesis of nonessential amino acids and nucleotides, thus supporting DNA replication and repair.
- Epigenetic Regulation via α-KetoglutarateFluctuations in intracellular α-ketoglutarate concentrations modulate the activity of dioxygenase enzymes (e.g., TET, JmjC histone demethylases), altering gene expression profiles in favor of malignancy.
- Metabolic Plasticity and AdaptationBy toggling between glycolytic and oxidative pathways in response to microenvironmental cues, cancer cells exploit BCAA catabolism to maintain bioenergetic flexibility.
- Modulation of the Immune MicroenvironmentElevated extracellular BCAAs and their metabolites can impair T-cell activation and skew macrophage polarization, thereby fostering an immunosuppressive tumor niche.
- Cross-Talk with Lipid MetabolismBCAA-derived citrate and acetyl-CoA influence de novo lipogenesis and fatty acid oxidation, supporting membrane biogenesis and lipid-mediated signaling networks.
3.1. PDAC
3.2. HCC
3.3. Breast Cancer
3.4. NSCLC
3.5. Leukemia
3.6. Glioblastoma
3.7. Other Cancers
4. Interplay Between BCAA Metabolism and Other Metabolic Networks
4.1. Glucose Metabolism
4.2. Nonessential Amino Acids and Nucleotide Synthesis
4.3. Fatty Acid Metabolism
5. Therapeutic Strategies for Targeting BCAA Metabolism
5.1. BCAT Inhibitors
5.2. BCKDK Inhibitors
5.3. Other Potential Therapeutic Targets
5.4. Limitations and Challenges
6. Concluding Remarks
6.1. Outstanding Questions
- Mitochondrial Trafficking of BCKAs: What are the transport mechanisms and carriers responsible for shuttling BCAT-derived keto acids into the mitochondria for oxidation?
- Isoform-Specific Contributions: How does BCAT1 versus BCAT2 overexpression differentially affect tumor metabolism and growth in distinct cancer contexts?
- Context-Dependent Effects: Under which nutrient or microenvironmental conditions (e.g., glutamine deprivation, hypoxia) does BCAA catabolism most critically drive tumor progression?
- Immune and Stromal Interactions: How does tumor-intrinsic BCAA metabolism reshape the function of tumor-infiltrating immune cells and cancer-associated fibroblasts?
- Biomarker Development: Which metabolic or genetic readouts (plasma BCAA levels, BCAT/BCKDK expression) best predict responsiveness to BCAA-targeted therapies?
6.2. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.Y.; Monleon, D.; Verhamme, P.; Staessen, J.A. Branched-Chain Amino Acids as Critical Switches in Health and Disease. Hypertension 2018, 72, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, Y.R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.W.; Hu, L.P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef]
- Li, J.T.; Li, K.Y.; Su, Y.; Shen, Y.; Lei, M.Z.; Zhang, F.; Yin, M.; Chen, Z.J.; Wen, W.Y.; Hu, W.G.; et al. Diet high in branched-chain amino acid promotes PDAC development by USP1-mediated BCAT2 stabilization. Natl. Sci. Rev. 2022, 9, nwab212. [Google Scholar] [CrossRef]
- Lei, M.Z.; Li, X.X.; Zhang, Y.; Li, J.T.; Zhang, F.; Wang, Y.P.; Yin, M.; Qu, J.; Lei, Q.Y. Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct. Target. Ther. 2020, 5, 70. [Google Scholar] [CrossRef]
- Zhu, Z.; Achreja, A.; Meurs, N.; Animasahun, O.; Owen, S.; Mittal, A.; Parikh, P.; Lo, T.W.; Franco-Barraza, J.; Shi, J.; et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2020, 2, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Noguchi, R.; Kaji, K.; Ikenaka, Y.; Shirai, Y.; Namisaki, T.; Kitade, M.; Tsujimoto, T.; Kawaratani, H.; Fukui, H. Attenuation of insulin-resistance-based hepatocarcinogenesis and angiogenesis by combined treatment with branched-chain amino acids and angiotensin-converting enzyme inhibitor in obese diabetic rats. J. Gastroenterol. 2010, 45, 443–450. [Google Scholar] [CrossRef]
- Iwasa, J.; Shimizu, M.; Shiraki, M.; Shirakami, Y.; Sakai, H.; Terakura, Y.; Takai, K.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Dietary supplementation with branched-chain amino acids suppresses diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Sci. 2010, 101, 460–467. [Google Scholar] [CrossRef]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S.; et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol. Res. 2006, 35, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-chain amino acids prevent insulin-induced hepatic tumor cell proliferation by inducing apoptosis through mTORC1 and mTORC2-dependent mechanisms. J. Cell Physiol. 2012, 227, 2097–2105. [Google Scholar] [CrossRef]
- Wubetu, G.Y.; Utsunomiya, T.; Ishikawa, D.; Ikemoto, T.; Yamada, S.; Morine, Y.; Iwahashi, S.; Saito, Y.; Arakawa, Y.; Imura, S.; et al. Branched chain amino acid suppressed insulin-initiated proliferation of human cancer cells through induction of autophagy. Anticancer Res. 2014, 34, 4789–4796. [Google Scholar]
- Lee, I.J.; Seong, J.; Bae, J.I.; You, S.H.; Rhee, Y.; Lee, J.H. Effect of Oral Supplementation with Branched-chain Amino Acid (BCAA) during Radiotherapy in Patients with Hepatocellular Carcinoma: A Double-Blind Randomized Study. Cancer Res. Treat. 2011, 43, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, Y.; Xu, H.; Yi, F. Network meta-analysis of adjuvant treatments for patients with hepatocellular carcinoma after curative resection. BMC Gastroenterol. 2023, 23, 320. [Google Scholar] [CrossRef]
- Sideris, G.A.; Tsaramanidis, S.; Vyllioti, A.T.; Njuguna, N. The Role of Branched-Chain Amino Acid Supplementation in Combination with Locoregional Treatments for Hepatocellular Carcinoma: Systematic Review and Meta-Analysis. Cancers 2023, 15, 926. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ong, Q.; Liao, Y.; Ding, Z.; Tan, A.Q.L.; Lim, L.T.R.; Tan, H.M.; Lim, S.L.; Lee, Q.Y.; Han, W. Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling. Int. J. Mol. Sci. 2023, 24, 9171. [Google Scholar] [CrossRef]
- Ericksen, R.E.; Lim, S.L.; McDonnell, E.; Shuen, W.H.; Vadiveloo, M.; White, P.J.; Ding, Z.; Kwok, R.; Lee, P.; Radda, G.K.; et al. Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019, 29, 1151–1165.e1156. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.; Cai, Y.; Lu, K.; Zhong, X.; Xing, S.; Song, W.; Zhang, Y.; Ye, L.; Zhu, X.; et al. Branched-chain amino acid catabolism breaks glutamine addiction to sustain hepatocellular carcinoma progression. Cell Rep. 2022, 41, 111691. [Google Scholar] [CrossRef]
- Biswas, D.; Slade, L.; Duffley, L.; Mueller, N.; Dao, K.T.; Mercer, A.; Pakkiriswami, S.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. Inhibiting BCKDK in triple negative breast cancer suppresses protein translation, impairs mitochondrial function, and potentiates doxorubicin cytotoxicity. Cell Death Discov. 2021, 7, 241. [Google Scholar] [CrossRef]
- Abdul Kader, S.; Dib, S.; Achkar, I.W.; Thareja, G.; Suhre, K.; Rafii, A.; Halama, A. Defining the landscape of metabolic dysregulations in cancer metastasis. Clin. Exp. Metastasis 2022, 39, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem. Biophys. Res. Commun. 2017, 486, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, G.; Zhang, Y.; Zhuge, R.; Qin, S.; Qian, J.; Chen, R.; Kwan Wong, Y.; Tang, H.; Wang, P.; et al. Small-molecule targeting BCAT1-mediated BCAA metabolism inhibits the activation of SHOC2-RAS-ERK to induce apoptosis of Triple-negative breast cancer cells. J. Adv. Res. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.L.; Abed, M.N.; Mohamed, G.; Price, J.C.; Abdullah, M.I.; Richardson, A. Inhibition of branched-chain alpha-keto acid dehydrogenase kinase augments the sensitivity of ovarian and breast cancer cells to paclitaxel. Br. J. Cancer 2023, 128, 896–906. [Google Scholar] [CrossRef]
- Chi, R.; Yao, C.; Chen, S.; Liu, Y.; He, Y.; Zhang, J.; Ellies, L.G.; Wu, X.; Zhao, Q.; Zhou, C.; et al. Elevated BCAA Suppresses the Development and Metastasis of Breast Cancer. Front. Oncol. 2022, 12, 887257. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Jiang, W.; Zuo, D.; Wang, X.; Jin, Y.; Qiao, L.; An, H.; Yang, L.; Dumoulin, D.W.; et al. BCKDK alters the metabolism of non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 4459–4476. [Google Scholar] [CrossRef]
- Mao, L.; Wang, L.; Lyu, Y.; Zhuang, Q.; Li, Z.; Zhang, J.; Gu, Z.; Lu, S.; Wang, X.; Guan, Y.; et al. Branch Chain Amino Acid Metabolism Promotes Brain Metastasis of NSCLC through EMT Occurrence by Regulating ALKBH5 activity. Int. J. Biol. Sci. 2024, 20, 3285–3301. [Google Scholar] [CrossRef]
- Xue, M.; Xiao, J.; Jiang, W.; Wang, Y.; Zuo, D.; An, H.; Ren, L. Loss of BCAA catabolism enhances Rab1A-mTORC1 signaling activity and promotes tumor proliferation in NSCLC. Transl. Oncol. 2023, 34, 101696. [Google Scholar] [CrossRef]
- Zhang, T.; Pan, Z.; Gao, J.; Wu, Q.; Bai, G.; Li, Y.; Tong, L.; Feng, F.; Lai, M.; Liu, Y.; et al. Branched-chain amino acid transaminase 1 confers EGFR-TKI resistance through epigenetic glycolytic activation. Signal Transduct. Target. Ther. 2024, 9, 216. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Velasco-Hernandez, T.; Mess, J.; Lalioti, M.E.; Romero-Mulero, M.C.; Obier, N.; Karantzelis, N.; Rettkowski, J.; Schonberger, K.; Karabacz, N.; et al. GPRC5C drives branched-chain amino acid metabolism in leukemogenesis. Blood Adv. 2023, 7, 7525–7538. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, F.; Zhang, Y.; Li, X.; Chen, C.; Zhou, M.; Yu, Z.; Liu, Y.; Zhao, Y.; Hao, X.; et al. PPM1K Regulates Hematopoiesis and Leukemogenesis through CDC20-Mediated Ubiquitination of MEIS1 and p21. Cell Rep. 2018, 23, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Kochi, Y.; Semba, Y.; Ohishi, M.; Irifune, H.; Hatakeyama, K.; Kunisaki, Y.; Sugio, T.; Sakoda, T.; et al. Human acute leukemia uses branched-chain amino acid catabolism to maintain stemness through regulating PRC2 function. Blood Adv. 2023, 7, 3592–3603. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef]
- Tosello, V.; Di Martino, L.; Papathanassiu, A.E.; Santa, S.D.; Pizzi, M.; Mussolin, L.; Liu, J.; Van Vlierberghe, P.; Piovan, E. BCAT1 is a NOTCH1 target and sustains the oncogenic function of NOTCH1. Haematologica 2025, 110, 350–367. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Wang, Y.; Huang, S.; Mao, S.; Ling, Q.; Li, C.; Li, F.; Yu, M.; Huang, X.; Huang, J.; et al. High expression of BCAT1 sensitizes AML cells to PARP inhibitor by suppressing DNA damage response. J. Mol. Med. 2024, 102, 415–433. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Han, L.; Dong, L.; Leung, K.; Zhao, Z.; Li, Y.; Gao, L.; Chen, Z.; Xue, J.; Qing, Y.; Li, W.; et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell 2023, 30, 52–68.e13. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef]
- Tosello, V.; Rompietti, C.; Papathanassiu, A.E.; Arrigoni, G.; Piovan, E. BCAT1 Associates with DNA Repair Proteins KU70 and KU80 and Contributes to Regulate DNA Repair in T-Cell Acute Lymphoblastic Leukemia (T-ALL). Int. J. Mol. Sci. 2024, 25, 3571. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, D.; He, X.; Chen, C.; Xie, L.; Liu, L.; Yu, Z.; Zhang, Y.; Zheng, J.; Huang, D. BCAT1 contributes to the development of TKI-resistant CML. Cell. Oncol. 2024, 48, 411–424. [Google Scholar] [CrossRef]
- Li, Z.; Gu, Z.; Wang, L.; Guan, Y.; Lyu, Y.; Zhang, J.; Wang, Y.; Wang, X.; Xiong, J.; Liu, Y. Nuclear Translocation of LDHA Promotes the Catabolism of BCAAs to Sustain GBM Cell Proliferation through the TxN Antioxidant Pathway. Int. J. Mol. Sci. 2023, 24, 9365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Sun, G.F.; He, K.Y.; Zhang, Z.; Han, X.H.; Qu, X.H.; Wan, D.F.; Yao, D.; Tou, F.F.; Han, X.J.; et al. Targeted inhibition of branched-chain amino acid metabolism drives apoptosis of glioblastoma by facilitating ubiquitin degradation of Mfn2 and oxidative stress. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167220. [Google Scholar] [CrossRef]
- Tonjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Tang, L.; Shi, Y.; Li, W.; Zou, L.; Zhang, L.; Cheng, Y.; Yuan, Z.; Zhu, F.; et al. Cross-talk between BCKDK-mediated phosphorylation and STUB1-dependent ubiquitination degradation of BCAT1 promotes GBM progression. Cancer Lett. 2024, 591, 216849. [Google Scholar] [CrossRef]
- Suh, E.H.; Hackett, E.P.; Wynn, R.M.; Chuang, D.T.; Zhang, B.; Luo, W.; Sherry, A.D.; Park, J.M. In vivo assessment of increased oxidation of branched-chain amino acids in glioblastoma. Sci. Rep. 2019, 9, 340. [Google Scholar] [CrossRef]
- Sun, Y.; Mu, G.; Zhang, X.; Wu, Y.; Wang, S.; Wang, X.; Xue, Z.; Wang, C.; Liu, J.; Li, W.; et al. Metabolic modulation of histone acetylation mediated by HMGCL activates the FOXM1/beta-catenin pathway in glioblastoma. Neuro Oncol. 2024, 26, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Li, N.; Lu, X.C.; Xu, M.; Liu, Y.; Li, K.; Zhang, Y.; Hu, K.; Qi, Y.T.; Yao, J.; et al. Enhanced BCAT1 activity and BCAA metabolism promotes RhoC activity in cancer progression. Nat. Metab. 2023, 5, 1159–1173. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, J.; Wang, M.; Yi, X.; Guo, S.; Wang, H.; Zhang, H.; Wang, H.; Yang, Y.; Zhang, B.; et al. BCKDHA contributes to melanoma progression by promoting the expressions of lipogenic enzymes FASN and ACLY. Exp. Dermatol. 2023, 32, 1633–1643. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, J.; Wang, H.; Yi, X.; Wang, H.; Zhang, H.; Guo, S.; Yang, Y.; Zhang, B.; Du, J.; et al. BCAT2 promotes melanoma progression by activating lipogenesis via the epigenetic regulation of FASN and ACLY expressions. Cell Mol. Life Sci. 2023, 80, 315. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.C.; Sun, R.M.; Yang, Y.; Zhou, H.Y.; Meng, Z.W.; Chi, R.; Xia, L.L.; Ji, P.; Chen, Y.Y.; Zhang, G.Q.; et al. Accumulation of branched-chain amino acids reprograms glucose metabolism in CD8(+) T cells with enhanced effector function and anti-tumor response. Cell Rep. 2023, 42, 112186. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.S.; Poschet, G.; Nonnenmacher, Y.; Becker, H.M.; Sapcariu, S.; Gaupel, A.C.; Schlotter, M.; Wu, Y.; Kneisel, N.; Seiffert, M.; et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017, 18, 2172–2185. [Google Scholar] [CrossRef]
- Zou, L.; Wang, W.; Huang, W.; Ni, X.; Li, W.; Cheng, Y.; Tian, Q.; Liu, L.; Zhu, F.; Duan, Q. FYN-mediated phosphorylation of BCKDK at Y151 promotes GBM proliferation by increasing the oncogenic metabolite N-acetyl-L-alanine. Heliyon 2024, 10, e33663. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef]
- Gotvaldova, K.; Spackova, J.; Novotny, J.; Baslarova, K.; Jezek, P.; Rossmeislova, L.; Gojda, J.; Smolkova, K. BCAA metabolism in pancreatic cancer affects lipid balance by regulating fatty acid import into mitochondria. Cancer Metab. 2024, 12, 10. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Yan, G.; Tong, Y.; Guo, W.; Li, S.; Qian, Y.; Li, Q.; Shu, Y.; Zhang, L.; et al. CPT1A loss disrupts BCAA metabolism to confer therapeutic vulnerability in TP53-mutated liver cancer. Cancer Lett. 2024, 595, 217006. [Google Scholar] [CrossRef]
- Wang, J.; Han, L.; Liu, Z.; Zhang, W.; Zhang, L.; Jing, J.; Gao, A. Targeting IGF2BP1 alleviated benzene hematotoxicity by reprogramming BCAA metabolism and fatty acid oxidation. Chem. Biol. Interact. 2024, 398, 111107. [Google Scholar] [CrossRef] [PubMed]
- Fala, M.; Ros, S.; Sawle, A.; Rao, J.U.; Tsyben, A.; Tronci, L.; Frezza, C.; Mair, R.; Brindle, K.M. The role of branched-chain aminotransferase 1 in driving glioblastoma cell proliferation and invasion varies with tumor subtype. Neurooncol. Adv. 2023, 5, vdad120. [Google Scholar] [CrossRef]
- Luo, W.; Pan, Z.; Zhu, X.; Li, Y.; Li, Y.; Zhang, Y.; Pan, J.; Ding, J.; Xie, H.; Zhao, G. Design, Synthesis and Biological Activity Study of gamma-Aminobutyric Acid (GABA) Derivatives Containing Bridged Bicyclic Skeletons as BCAT1 Inhibitors. Molecules 2025, 30, 904. [Google Scholar] [CrossRef]
- Mastall, M.; Roth, P.; Bink, A.; Fischer Maranta, A.; Laubli, H.; Hottinger, A.F.; Hundsberger, T.; Migliorini, D.; Ochsenbein, A.; Seystahl, K.; et al. A phase Ib/II randomized, open-label drug repurposing trial of glutamate signaling inhibitors in combination with chemoradiotherapy in patients with newly diagnosed glioblastoma: The GLUGLIO trial protocol. BMC Cancer 2024, 24, 82. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.M.; Ancellin, N.; Beaufils, B.; Bingham, R.P.; Borthwick, J.A.; Boullay, A.B.; Boursier, E.; Carter, P.S.; Chung, C.W.; Churcher, I.; et al. The Discovery of in Vivo Active Mitochondrial Branched-Chain Aminotransferase (BCATm) Inhibitors by Hybridizing Fragment and HTS Hits. J. Med. Chem. 2015, 58, 7140–7163. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, S.; Ho, M.Y.; Ng, K.K.; Cheng, K.K. Branched-chain amino acid metabolism: Pathophysiological mechanism and therapeutic intervention in metabolic diseases. Obes. Rev. 2025, 26, e13856. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Sun, G.F.; Pan, X.A.; Qu, X.H.; Yang, P.; Chen, Z.P.; Han, X.J.; Wang, T. BCATc inhibitor 2 ameliorated mitochondrial dysfunction and apoptosis in oleic acid-induced non-alcoholic fatty liver disease model. Front. Pharmacol. 2022, 13, 1025551. [Google Scholar] [CrossRef]
- Burrage, L.C.; Nagamani, S.C.; Campeau, P.M.; Lee, B.H. Branched-chain amino acid metabolism: From rare Mendelian diseases to more common disorders. Hum. Mol. Genet. 2014, 23, R1–R8. [Google Scholar] [CrossRef]
- Abdualkader, A.M.; Karwi, Q.G.; Lopaschuk, G.D.; Al Batran, R. The role of branched-chain amino acids and their downstream metabolites in mediating insulin resistance. J. Pharm. Pharm. Sci. 2024, 27, 13040. [Google Scholar] [CrossRef]
- Liu, S.; Kormos, B.L.; Knafels, J.D.; Sahasrabudhe, P.V.; Rosado, A.; Sommese, R.F.; Reyes, A.R.; Ward, J.; Roth Flach, R.J.; Wang, X.; et al. Structural studies identify angiotensin II receptor blocker-like compounds as branched-chain ketoacid dehydrogenase kinase inhibitors. J. Biol. Chem. 2023, 299, 102959. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, X.; Huang, P.; Li, C.; Han, L.; Han, Y.; Gan, R.; Xin, B.; Tu, Y.; Zhou, S.; et al. BCKDK modification enhances the anticancer efficacy of CAR-T cells by reprogramming branched chain amino acid metabolism. Mol. Ther. 2024, 32, 3128–3144. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Shah, P.; Makhanasa, D.; Khan, M.W. Unveiling the Maze: Branched-Chain Amino Acids Fueling the Dynamics of Cancer Metabolism and Progression. Cancers 2025, 17, 1751. https://doi.org/10.3390/cancers17111751
Xu K, Shah P, Makhanasa D, Khan MW. Unveiling the Maze: Branched-Chain Amino Acids Fueling the Dynamics of Cancer Metabolism and Progression. Cancers. 2025; 17(11):1751. https://doi.org/10.3390/cancers17111751
Chicago/Turabian StyleXu, Kai, Pratham Shah, Dhruvi Makhanasa, and Md. Wasim Khan. 2025. "Unveiling the Maze: Branched-Chain Amino Acids Fueling the Dynamics of Cancer Metabolism and Progression" Cancers 17, no. 11: 1751. https://doi.org/10.3390/cancers17111751
APA StyleXu, K., Shah, P., Makhanasa, D., & Khan, M. W. (2025). Unveiling the Maze: Branched-Chain Amino Acids Fueling the Dynamics of Cancer Metabolism and Progression. Cancers, 17(11), 1751. https://doi.org/10.3390/cancers17111751