
The Current State of Tumor Microenvironment-Specific Therapies for Non-Small Cell Lung Cancer

 , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
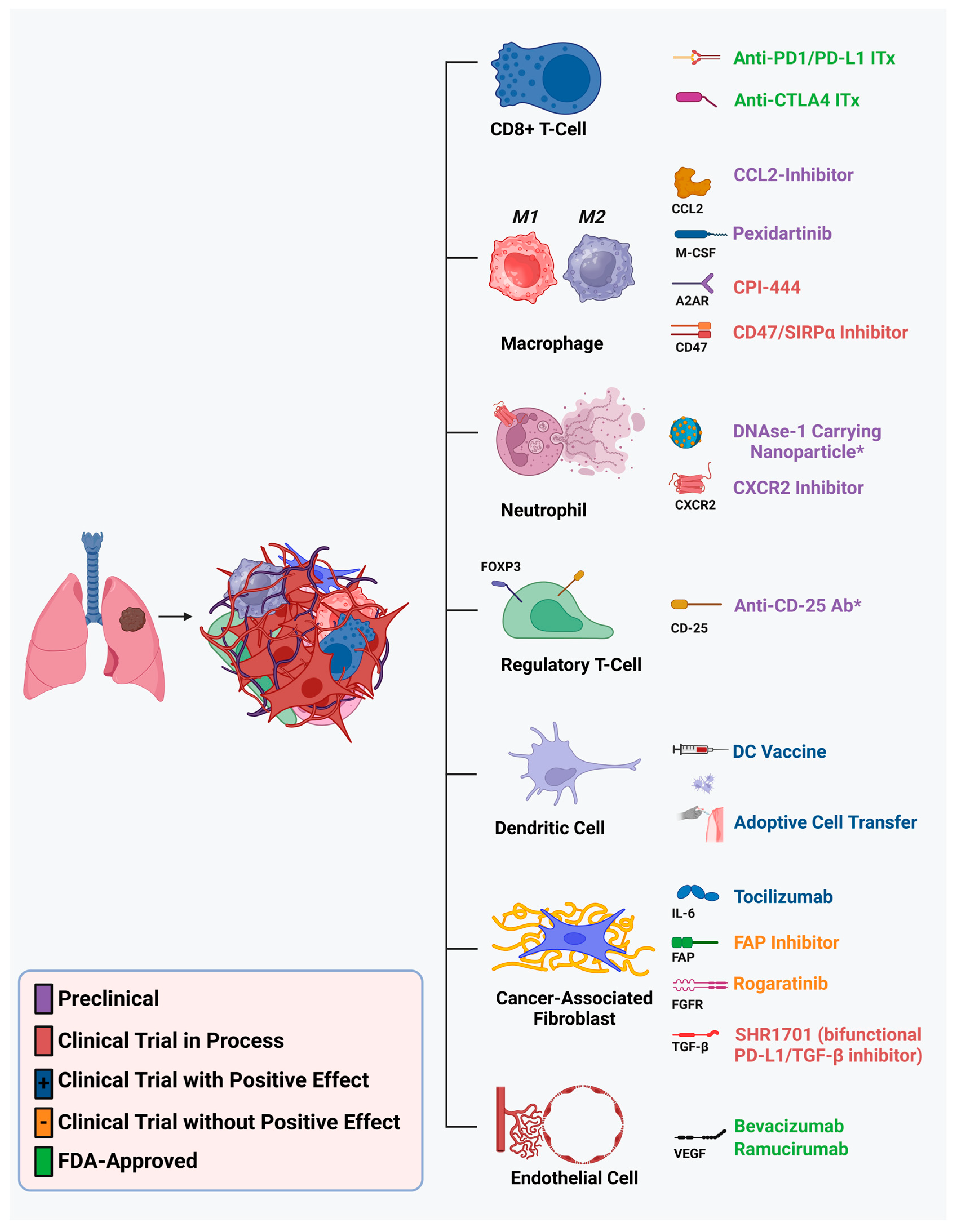
2. Single-Cell Sequencing Analyses of the Tumor Microenvironment of NSCLC
3. Emerging Therapeutic Strategies Targeting the NSCLC TME
3.1. Structural Components
3.1.1. Endothelial Cells
3.1.2. Cancer-Associated Fibroblasts
3.2. Immune Components
3.2.1. Neutrophils
3.2.2. Regulatory T-Cells
3.2.3. Dendritic Cells
3.2.4. Tumor-Associated Macrophages
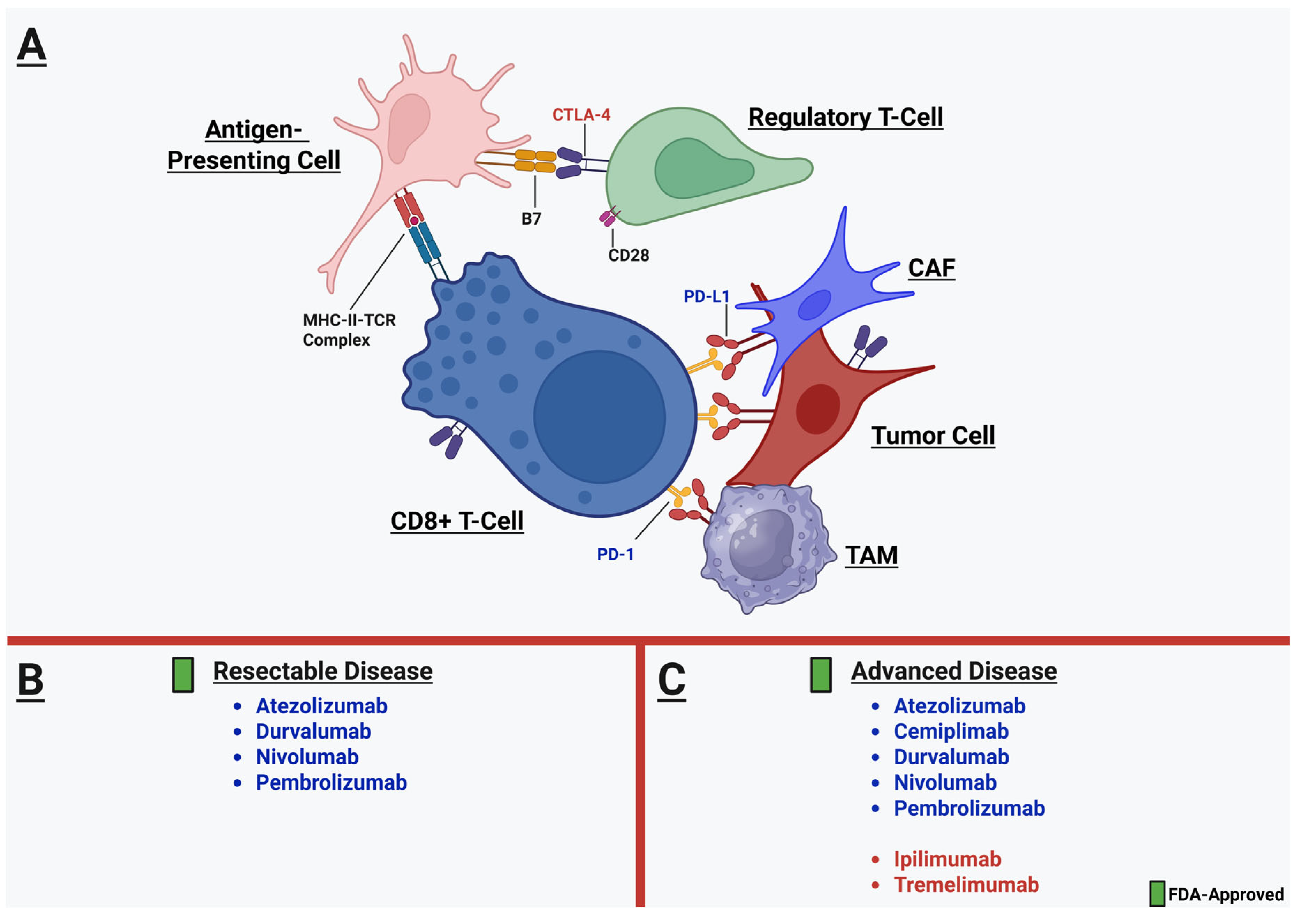
3.2.5. CD8+ T-Cells and the Immunotherapy Revolution
3.2.6. Building upon the Immunotherapy Foundation: Factors and Biomarkers Predicting Response to ICI Therapy
4. Perioperative Immunotherapy and TME-Specific Implications for the Thoracic Surgeon
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAF | Cancer-associated Fibroblast |
| CCL2 | Chemokine C-C motif ligand-2 |
| CD | Cluster of Differentiation |
| CTLA-4 | Cytotoxic T-Lymphocyte-associated Protein 4 |
| CPS | Combined Positivity Score |
| DC | Dendritic Cell |
| DFS | Disease-free Survival |
| EC | Endothelial Cell |
| ECM | Extracellular Matrix |
| EMT | Epithelial–mesenchymal Transition |
| FAP | Fibroblast Activating Protein |
| FDA | Food and Drug Administration |
| ICI | Immune Checkpoint Inhibitor |
| LUAD | Lung Adenocarcinoma |
| LUSC | Lung Squamous Cell Carcinoma |
| mOS | Median Overall Survival |
| MHC | Major Histocompatibility Complex |
| NSCLC | Non-small cell Lung Cancer |
| NKG2D | Natural Killer Group 2D |
| OS | Overall Survival |
| PARP | Poly-(ADP ribose) polymerase |
| PD/PD-L1 | Programmed death/programmed death-ligand 1 |
| PFS | Progression-free Survival |
| PMN | Polymorphonuclear Neutrophil |
| RFS | Recurrence-free Survival |
| Sc | Single-cell |
| TAM | Tumor-associated Macrophage |
| TAN | Tumor-associated Neutrophil |
| TME | Tumor Microenvironment |
| TPS | Tumor Proportion Score |
| Treg | Regulatory T-cell |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Liberini, V.; Mariniello, A.; Righi, L.; Capozza, M.; Delcuratolo, M.D.; Terreno, E.; Farsad, M.; Volante, M.; Novello, S.; Deandreis, D. NSCLC biomarkers to predict response to immunotherapy with checkpoint inhibitors (ICI): From the cells to in vivo images. Cancers 2021, 13, 4543. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Wang, H.; Liang, S.; Yu, Y.; Han, Y. Efficacy and safety of neoadjuvant immunotherapy protocols and cycles for non-small cell lung cancer: A systematic review and meta-analysis. Front. Oncol. 2024, 14, 1276549. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, J.; Qin, C.; Yan, H.; Luo, X.; Zhou, H. Advances and challenges of first-line immunotherapy for non-small cell lung cancer: A review. Medicine 2024, 103, e36861. [Google Scholar] [CrossRef]
- Yu, S.; Zhai, S.; Gong, Q.; Xiang, C.; Gong, J.; Wu, L.; Pu, X. Neoadjuvant Immunotherapy and Non–Small Cell Lung Cancer: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Am. J. Clin. Oncol. 2023, 46, 517–528. [Google Scholar] [CrossRef]
- Lahiri, A.; Maji, A.; Potdar, P.D.; Singh, N.; Parikh, P.; Bisht, B.; Mukherjee, A.; Paul, M.K. Lung cancer immunotherapy: Progress, pitfalls, and promises. Mol. Cancer 2023, 22, 40. [Google Scholar] [CrossRef]
- Cascone, T.; William Jr, W.N.; Weissferdt, A.; Leung, C.H.; Lin, H.Y.; Pataer, A.; Godoy, M.C.; Carter, B.W.; Federico, L.; Reuben, A. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: The phase 2 randomized NEOSTAR trial. Nat. Med. 2021, 27, 504–514. [Google Scholar] [CrossRef]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Heymach, J.V.; Harpole, D.; Mitsudomi, T.; Taube, J.M.; Galffy, G.; Hochmair, M.; Winder, T.; Zukov, R.; Garbaos, G.; Gao, S. Perioperative durvalumab for resectable non–small-cell lung cancer. N. Engl. J. Med. 2023, 389, 1672–1684. [Google Scholar] [CrossRef] [PubMed]
- Laza-Briviesca, R.; Cruz-Bermúdez, A.; Nadal, E.; Insa, A.; García-Campelo, M.; Huidobro, G.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A. Blood biomarkers associated to complete pathological response on NSCLC patients treated with neoadjuvant chemoimmunotherapy included in NADIM clinical trial. Clin. Transl. Med. 2021, 11, e491. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Araujo, H.A.; Do, M.T.; Qian, Y.; Sun, X.; Cobo, A.G.; Le, J.T.; Montesion, M.; Palmer, R.; Jahchan, N. CTLA4 blockade abrogates KEAP1/STK11-related resistance to PD-(L) 1 inhibitors. Nature 2024, 635, 462–471. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Elizabeth, A.Y.; Schenk, E.L.; Tan, W.; Zee, A. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Papavassiliou, K.A.; Basdra, E.K.; Papavassiliou, A.G. Advances in non-small cell lung cancer mechanomedicine: Deciphering the signaling networks that govern tumor-TME interactions. J. Exp. Clin. Cancer Res. 2024, 43, 316. [Google Scholar] [CrossRef]
- Zhang, L.; Ludden, C.M.; Cullen, A.J.; Tew, K.D.; de Barros, A.L.B.; Townsend, D.M. Nuclear factor kappa B expression in non-small cell lung cancer. Biomed. Pharmacother. 2023, 167, 115459. [Google Scholar] [CrossRef] [PubMed]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Xu, Y.; Tang, R.; Ren, J.; Shen, S.; Chen, Y.; Liu, B.; Hou, Y.; Wang, T. miR141–CXCL1–CXCR2 signaling–induced Treg recruitment regulates metastases and survival of non–small cell lung cancer. Mol. Cancer Ther. 2014, 13, 3152–3162. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 expression in lung cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef]
- Mansouri, S.; Heylmann, D.; Stiewe, T.; Kracht, M.; Savai, R. Cancer genome and tumor microenvironment: Reciprocal crosstalk shapes lung cancer plasticity. eLife 2022, 11, e79895. [Google Scholar] [CrossRef]
- Faruki, H.; Mayhew, G.M.; Serody, J.S.; Hayes, D.N.; Perou, C.M.; Lai-Goldman, M. Lung adenocarcinoma and squamous cell carcinoma gene expression subtypes demonstrate significant differences in tumor immune landscape. J. Thorac. Oncol. 2017, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhai, X.; Yan, W.; Zhu, H.; Yu, J. Clinical outcomes of immune checkpoint blockades and the underlying immune escape mechanisms in squamous and adenocarcinoma NSCLC. Cancer Med. 2021, 10, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Perol, M.; Banna, G.L.; Parikh, K.; Addeo, A. Oncogenic alterations in advanced NSCLC: A molecular super-highway. Biomark. Res. 2024, 12, 24. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217. [Google Scholar] [CrossRef]
- Sposito, M.; Eccher, S.; Pasqualin, L.; Scaglione, I.M.; Avancini, A.; Tregnago, D.; Trestini, I.; Insolda, J.; Bonato, A.; Ugel, S. Characterizing the immune tumor microenvironment in ALK fusion-positive lung cancer: State-of-the-art and therapeutical implications. Expert Rev. Clin. Immunol. 2024, 20, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zeng, J.; Zhang, H.; Zhu, S.; Wang, H.; He, J.; Yang, L.; Zhou, N.; Zu, L.; Xu, X. Characteristics of the immune microenvironment and their clinical significance in non-small cell lung cancer patients with ALK-rearranged mutation. Front. Immunol. 2022, 13, 974581. [Google Scholar] [CrossRef]
- Madeddu, C.; Donisi, C.; Liscia, N.; Lai, E.; Scartozzi, M.; Macciò, A. EGFR-mutated non-small cell lung cancer and resistance to immunotherapy: Role of the tumor microenvironment. Int. J. Mol. Sci. 2022, 23, 6489. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic value of smoking status in non-small-cell lung cancer patients treated with immune checkpoint inhibitors: A meta-analysis. Oncotarget 2017, 8, 93149. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Luo, D.; Yang, D.; Cao, D.; Gong, Z.; He, F.; Hou, Y.; Lin, S. Effect of smoking status on immunotherapy for lung cancer: A systematic review and meta-analysis. Front. Oncol. 2024, 14, 1422160. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Kim, J.W.; Ylaya, K.; Chung, E.J.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.-Y.; Hewitt, S.M. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, S.; Li, S.; Jiang, J.; Mei, J.; Chen, Y.; Ma, Y.; Liu, Y.; Liu, Y. Nanotechnology: A New Strategy for Lung Cancer Treatment Targeting Pro-Tumor Neutrophils. Engineering 2023, 27, 106–126. [Google Scholar] [CrossRef]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-mesenchymal transition in cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial–mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel–carboplatin alone or with bevacizumab for non–small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massutí, B.; Palmero, R.; Aix, S.P.; Carcereny, E. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): An international, multicentre, single-arm, phase 2 trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.-M.; Wang, R. Effectiveness and safety of adding bevacizumab to platinum-based chemotherapy as first-line treatment for advanced non-small-cell lung cancer: A meta-analysis. Front. Med. 2021, 8, 616380. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Zatarain-Barrón, Z.L.; Cardona, A.F.; Carmona, A.; Lopez-Mejia, M. Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 637–644. [Google Scholar] [CrossRef]
- Kitagawa, C.; Mori, M.; Ichiki, M.; Sukoh, N.; Kada, A.; Saito, A.M.; Ichinose, Y. Gefitinib plus bevacizumab vs. gefitinib alone for EGFR mutant non-squamous non-small cell lung cancer. In Vivo 2019, 33, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Zhao, Z.-t.; Wang, J.; Fang, L.; Qian, X.-d.; Cai, Y.; Cao, H.-q.; Wang, G.-r.; He, M.-l.; Jiang, Y.-y.; Wang, D.-g. Dual-responsive nanoparticles loading bevacizumab and gefitinib for molecular targeted therapy against non-small cell lung cancer. Acta Pharmacol. Sin. 2023, 44, 244–254. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Provencio, M.; Ortega, A.L.; Coves-Sarto, J.; Calvo, V.; Marsé-Fabregat, R.; Dómine, M.; Guirado, M.; Carcereny, E.; Fernández, N.; Álvarez, R. Atezolizumab plus bevacizumab as first-line treatment for patients with metastatic nonsquamous non–small cell lung cancer with high tumor mutation burden: A nonrandomized controlled trial. JAMA Oncol. 2023, 9, 344–353. [Google Scholar] [CrossRef]
- Sugawara, S.; Lee, J.-S.; Kang, J.-H.; Kim, H.; Inui, N.; Hida, T.; Lee, K.; Yoshida, T.; Tanaka, H.; Yang, C.-T. Nivolumab with carboplatin, paclitaxel, and bevacizumab for first-line treatment of advanced nonsquamous non-small-cell lung cancer. Ann. Oncol. 2021, 32, 1137–1147. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.-T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J.; et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef]
- Ren, Q.; Zhang, P.; Lin, H.; Feng, Y.; Chi, H.; Zhang, X.; Xia, Z.; Cai, H.; Yu, Y. A novel signature predicts prognosis and immunotherapy in lung adenocarcinoma based on cancer-associated fibroblasts. Front. Immunol. 2023, 14, 1201573. [Google Scholar] [CrossRef]
- Irvine, A.F.; Waise, S.; Green, E.W.; Stuart, B.; Thomas, G.J. Characterising cancer-associated fibroblast heterogeneity in non-small cell lung cancer: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 3727. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Kinoshita, T.; Ishii, G.; Hiraoka, N.; Hirayama, S.; Yamauchi, C.; Aokage, K.; Hishida, T.; Yoshida, J.; Nagai, K.; Ochiai, A. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci. 2013, 104, 409–415. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Cheng, W.; Diao, T.; Liu, H.; Bo, Y.; Liu, C.; Zhou, W.; Chen, M.; Zhang, Y. Cross-tissue human fibroblast atlas reveals myofibroblast subtypes with distinct roles in immune modulation. Cancer Cell 2024, 42, 1764–1783.e1710. [Google Scholar] [CrossRef]
- Cords, L.; Engler, S.; Haberecker, M.; Rüschoff, J.H.; Moch, H.; de Souza, N.; Bodenmiller, B. Cancer-associated fibroblast phenotypes are associated with patient outcome in non-small cell lung cancer. Cancer Cell 2024, 42, 396–412.e5. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, Y.; Wang, J.; Chen, X.; Han, J.; Zhen, S.; Yin, S.; Lv, W.; Yu, F.; Wang, J. circNOX4 activates an inflammatory fibroblast niche to promote tumor growth and metastasis in NSCLC via FAP/IL-6 axis. Mol. Cancer 2024, 23, 47. [Google Scholar] [CrossRef]
- Shintani, Y.; Kimura, T.; Funaki, S.; Ose, N.; Kanou, T.; Fukui, E. Therapeutic targeting of cancer-associated fibroblasts in the non-small cell lung cancer tumor microenvironment. Cancers 2023, 15, 335. [Google Scholar] [CrossRef] [PubMed]
- Addeo, A.; Rothschild, S.I.; Holer, L.; Schneider, M.; Waibel, C.; Haefliger, S.; Mark, M.; Fernandez, E.; Mach, N.; Mauti, L. Fibroblast growth factor receptor (FGFR) inhibitor rogaratinib in patients with advanced pretreated squamous-cell non-small cell lung cancer over-expressing FGFR mRNA: The SAKK 19/18 phase II study. Lung Cancer 2022, 172, 154–159. [Google Scholar] [CrossRef]
- Gao, M.; Wang, L.; Jing, F.; Zhang, F.; Tao, H.; Hu, Y. The Efficacy of Pemigatinib in Advanced NSCLC With FGFR Aberration: Case Report. Clin. Lung Cancer 2024, 25, e62–e66. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, X.-Y.; Pan, R.; Zhang, X.-T.; Si, X.-Y.; Chen, M.; Wang, M.; Zhang, L. Tocilizumab for advanced non-small cell lung cancer with concomitant inflammatory cachexia: A single-centre study. J. Clin. Oncol. 2024, 42, 16. [Google Scholar] [CrossRef]
- Ramundo, V.; Palazzo, M.L.; Aldieri, E. TGF-β as predictive marker and pharmacological target in lung cancer approach. Cancers 2023, 15, 2295. [Google Scholar] [CrossRef]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.-J.; Lee, S.H.; Park, M.S.; Yim, H.W. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Sato, R.; Imamura, K.; Semba, T.; Tomita, Y.; Saeki, S.; Ikeda, K.; Komohara, Y.; Suzuki, M.; Sakagami, T.; Saya, H. TGFβ signaling activated by cancer-associated fibroblasts determines the histological signature of lung adenocarcinoma. Cancer Res. 2021, 81, 4751–4765. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.-Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Faivre, S.J.; Santoro, A.; Gane, E.; Kelley, R.K.; Hourmand, I.O.; Assenat, E.; Gueorguieva, I.; Cleverly, A.; Desaiah, D.; Lahn, M.M. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J. Clin. Oncol. 2016, 34, 5. [Google Scholar] [CrossRef]
- Horvath, L.; Puschmann, C.; Scheiber, A.; Martowicz, A.; Sturm, G.; Trajanoski, Z.; Wolf, D.; Pircher, A.; Salcher, S. Beyond binary: Bridging neutrophil diversity to new therapeutic approaches in NSCLC. Trends Cancer 2024, 10, 457–474. [Google Scholar] [CrossRef]
- Kargl, J.; Busch, S.E.; Yang, G.H.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Mitchell, K.G.; Diao, L.; Karpinets, T.; Negrao, M.V.; Tran, H.T.; Parra, E.R.; Corsini, E.M.; Reuben, A.; Federico, L.; Bernatchez, C.; et al. Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: A descriptive analysis of a prospectively immunoprofiled cohort. J. Immunother. Cancer 2020, 8, e000405. [Google Scholar] [CrossRef]
- Leach, J.; Morton, J.P.; Sansom, O.J. Neutrophils: Homing in on the myeloid mechanisms of metastasis. Mol Immunol 2019, 110, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef]
- Yao, Y.; Yuan, D.; Liu, H.; Gu, X.; Song, Y. Pretreatment neutrophil to lymphocyte ratio is associated with response to therapy and prognosis of advanced non-small cell lung cancer patients treated with first-line platinum-based chemotherapy. Cancer Immunol. Immunother. 2013, 62, 471–479. [Google Scholar] [CrossRef]
- Chen, J.; Hou, S.; Liang, Q.; He, W.; Li, R.; Wang, H.; Zhu, Y.; Zhang, B.; Chen, L.; Dai, X. Localized degradation of neutrophil extracellular traps by photoregulated enzyme delivery for cancer immunotherapy and metastasis suppression. ACS Nano 2022, 16, 2585–2597. [Google Scholar] [CrossRef]
- Cheng, Y.; Mo, F.; Li, Q.; Han, X.; Shi, H.; Chen, S.; Wei, Y.; Wei, X. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol. Cancer 2021, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Chiec, L.; Mohindra, N.A.; Munshi, H.G. Regulatory T-cells as an emerging barrier to immune checkpoint inhibition in lung cancer. Front. Oncol. 2021, 11, 684098. [Google Scholar] [CrossRef]
- Liang, J.; Bi, G.; Shan, G.; Jin, X.; Bian, Y.; Wang, Q. Tumor-Associated Regulatory T Cells in Non-Small-Cell Lung Cancer: Current Advances and Future Perspectives. J. Immunol. Res. 2022, 2022, 4355386. [Google Scholar] [CrossRef]
- Devi-Marulkar, P.; Fastenackels, S.; Karapentiantz, P.; Goc, J.; Germain, C.; Kaplon, H.; Knockaert, S.; Olive, D.; Panouillot, M.; Validire, P. Regulatory T cells infiltrate the tumor-induced tertiary lymphoïd structures and are associated with poor clinical outcome in NSCLC. Commun. Biol. 2022, 5, 1416. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Ueyama, A.; Funaki, S.; Jinushi, K.; Higuchi, N.; Morihara, H.; Hirata, M.; Nagira, Y.; Saito, T.; Kawashima, A. In situ analysis of CCR8+ regulatory T cells in lung cancer: Suppression of GzmB+ CD8+ T cells and prognostic marker implications. BMC Cancer 2024, 24, 627. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wei, J.; Xu, J. Inducers, attractors and modulators of CD4+ Treg cells in non-small-cell lung cancer. Frontiers in immunology 2020, 11. [Google Scholar]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Horvath, L.; Thienpont, B.; Zhao, L.; Wolf, D.; Pircher, A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)-novel approaches and future outlook. Molecular Cancer 2020, 19, 1–5. [Google Scholar]
- Petersen, R.P.; Campa, M.J.; Sperlazza, J.; Conlon, D.; Joshi, M.B.; Harpole, D.H., Jr.; Patz, E.F., Jr. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 2006, 107, 2866–2872. [Google Scholar] [CrossRef]
- Song, D.; Liu, X.; Dong, C.; Wang, Q.; Sha, C.; Liu, C.; Ning, Z.; Han, J.; Liu, H.; Zong, M. Two novel human anti-CD25 antibodies with antitumor activity inversely related to their affinity and in vitro activity. Sci. Rep. 2021, 11, 22966. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef]
- López, L.; Morosi, L.G.; La Terza, F.; Bourdely, P.; Rospo, G.; Amadio, R.; Piperno, G.M.; Russo, V.; Volponi, C.; Vodret, S.; et al. Dendritic cell-targeted therapy expands CD8 T cell responses to bona-fide neoantigens in lung tumors. Nat. Commun. 2024, 15, 2280. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.J.; Salehi-Rad, R.; Tran, L.M.; Oh, M.S.; Dumitras, C.; Crosson, W.P.; Li, R.; Patel, T.S.; Man, S.; Yean, C.E. CXCL9/10-engineered dendritic cells promote T cell activation and enhance immune checkpoint blockade for lung cancer. Cell Rep. Med. 2024, 5, 101479. [Google Scholar] [CrossRef]
- Ingels, J.; De Cock, L.; Stevens, D.; Mayer, R.L.; Théry, F.; Sanchez, G.S.; Vermijlen, D.; Weening, K.; De Smet, S.; Lootens, N.; et al. Neoantigen-targeted dendritic cell vaccination in lung cancer patients induces long-lived T cells exhibiting the full differentiation spectrum. Cell Rep. Med. 2024, 5, 101516. [Google Scholar] [CrossRef]
- Vounckx, M.; Tijtgat, J.; Stevens, L.; Dirven, I.; Ilsen, B.; Vandenbroucke, F.; Raeymaeckers, S.; Vekens, K.; Forsyth, R.; Geeraerts, X. A randomized phase II clinical trial of stereotactic body radiation therapy (SBRT) and systemic pembrolizumab with or without intratumoral avelumab/ipilimumab plus CD1c (BDCA-1)+/CD141 (BDCA-3)+ myeloid dendritic cells in solid tumors. Cancer Immunol. Immunother. 2024, 73, 167. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Pan, Q.-Z.; Weng, D.-S.; Xie, C.-M.; Zhao, J.-J.; Chen, M.-S.; Peng, R.-Q.; Li, D.-D.; Wang, Y.; Tang, Y. Safety and activity of PD-1 blockade-activated DC-CIK cells in patients with advanced solid tumors. Oncoimmunology 2018, 7, e1417721. [Google Scholar] [CrossRef]
- Kimura, H.; Matsui, Y.; Ishikawa, A.; Nakajima, T.; Iizasa, T. Randomized controlled phase III trial of adjuvant chemoimmunotherapy with activated cytotoxic T cells and dendritic cells from regional lymph nodes of patients with lung cancer. Cancer Immunol. Immunother. 2018, 67, 1231–1238. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. npj Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Park, J.V.; Chandra, R.; Cai, L.; Ganguly, D.; Li, H.; Toombs, J.E.; Girard, L.; Brekken, R.A.; Minna, J.D. Tumor cells modulate macrophage phenotype in a novel in vitro co-culture model of the NSCLC tumor microenvironment. J. Thorac. Oncol. 2022, 17, 1178–1191. [Google Scholar] [CrossRef]
- Sedighzadeh, S.S.; Khoshbin, A.P.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. A narrative review of tumor-associated macrophages in lung cancer: Regulation of macrophage polarization and therapeutic implications. Transl. Lung Cancer Res. 2021, 10, 1889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiang, X.; Zou, Y.; Yuan, L.; Wang, X. Pexidartinib synergize PD-1 antibody through inhibiting treg infiltration by reducing TAM-derived CCL22 in lung adenocarcinoma. Front. Pharmacol. 2023, 14, 1092767. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Zhen, S.; Zhang, L.; Zhao, Q.; Yang, L.; Zhang, Y. A2AR-mediated CXCL5 upregulation on macrophages promotes NSCLC progression via NETosis. Cancer Immunol. Immunother. 2024, 73, 108. [Google Scholar] [CrossRef]
- Zhang, X.-w.; Qin, X.; Qin, C.Y.; Yin, Y.-l.; Chen, Y.; Zhu, H.-l. Expression of monocyte chemoattractant protein-1 and CC chemokine receptor 2 in non-small cell lung cancer and its significance. Cancer Immunol. Immunother. 2013, 62, 563–570. [Google Scholar] [CrossRef]
- An, J.; Xue, Y.; Long, M.; Zhang, G.; Zhang, J.; Su, H. Targeting CCR2 with its antagonist suppresses viability, motility and invasion by downregulating MMP-9 expression in non-small cell lung cancer cells. Oncotarget 2017, 8, 39230. [Google Scholar]
- Chen, X.; Zhou, J.; Wang, Y.; Wang, X.; Chen, K.; Chen, Q.; Huang, D.; Jiang, R. PIM1/NF-κB/CCL2 blockade enhances anti-PD-1 therapy response by modulating macrophage infiltration and polarization in tumor microenvironment of NSCLC. Oncogene 2024, 43, 2517–2530. [Google Scholar] [CrossRef]
- Lau, A.P.; Khavkine Binstock, S.S.; Thu, K.L. CD47: The Next Frontier in Immune Checkpoint Blockade for Non-Small Cell Lung Cancer. Cancers 2023, 15, 5229. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M. Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef]
- Cui, Z.; Xu, D.; Zhang, F.; Sun, J.; Song, L.; Ye, W.; Zeng, J.; Zhou, M.; Ruan, Z.; Zhang, L. CD47 blockade enhances therapeutic efficacy of cisplatin against lung carcinoma in a murine model. Exp. Cell Res. 2021, 405, 112677. [Google Scholar] [CrossRef]
- Huang, P.-W.; Chang, J.W.-C. Immune checkpoint inhibitors win the 2018 Nobel Prize. Biomed. J. 2019, 42, 299–306. [Google Scholar] [CrossRef]
- FDA. Pembrolizumab. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125514s012lbl.pdf (accessed on 5 May 2025).
- FDA. Atezolizumab. 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761034s028lbl.pdf (accessed on 5 May 2025).
- FDA. Nivolumab. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125554s095lbl.pdf (accessed on 5 May 2025).
- FDA. Cemiplimab. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761097s014lbl.pdf (accessed on 5 May 2025).
- FDA. Ipilimumab. 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125377s115lbl.pdf (accessed on 5 May 2025).
- FDA. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761270s000lbl.pdf (accessed on 5 May 2025).
- FDA. Durvalumab. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761069s035lbl.pdf (accessed on 5 May 2025).
- Riely, G.J.; Wood, D.E.; Ettinger, D.S.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R. Non–Small Cell Lung Cancer, Version 4.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2024, 22, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Azarov, I.; Helmlinger, G.; Kosinsky, Y.; Peskov, K. Elaborating on anti CTLA-4 mechanisms of action using an agent-based modeling approach. Front. Appl. Math. Stat. 2022, 8, 993581. [Google Scholar] [CrossRef]
- Zhang, H.; Dutta, P.; Liu, J.; Sabri, N.; Song, Y.; Li, W.X.; Li, J. Tumour cell-intrinsic CTLA 4 regulates PD-L1 expression in non-small cell lung cancer. J. Cell. Mol. Med. 2019, 23, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Kwiecien, I.; Stelmaszczyk-Emmel, A.; Polubiec-Kownacka, M.; Dziedzic, D.; Domagala-Kulawik, J. Elevated regulatory T cells, surface and intracellular CTLA-4 expression and interleukin-17 in the lung cancer microenvironment in humans. Cancer Immunol. Immunother. 2017, 66, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, E.-E.; Kilvaer, T.K.; Rakaee, M.; Richardsen, E.; Hald, S.M.; Andersen, S.; Busund, L.-T.; Bremnes, R.M.; Donnem, T. CTLA-4 expression in the non-small cell lung cancer patient tumor microenvironment: Diverging prognostic impact in primary tumors and lymph node metastases. Cancer Immunol. Immunother. 2017, 66, 1449–1461. [Google Scholar] [CrossRef]
- Govindan, R.; Szczesna, A.; Ahn, M.-J.; Schneider, C.-P.; Gonzalez Mella, P.F.; Barlesi, F.; Han, B.; Ganea, D.E.; Von Pawel, J.; Vladimirov, V. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non–small-cell lung cancer. J. Clin. Oncol. 2017, 35, 3449–3457. [Google Scholar] [CrossRef]
- Puri, S.; Shafique, M. Combination checkpoint inhibitors for treatment of non-small-cell lung cancer: An update on dual anti-CTLA-4 and anti-PD-1/PD-L1 therapies. Drugs Context 2020, 9, 2019-9-2. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Shinchi, Y.; Ishizuka, S.; Komohara, Y.; Matsubara, E.; Mito, R.; Pan, C.; Yoshii, D.; Yonemitsu, K.; Fujiwara, Y.; Ikeda, K. The expression of PD-1 ligand 1 on macrophages and its clinical impacts and mechanisms in lung adenocarcinoma. Cancer Immunol. Immunother. 2022, 71, 2645–2661. [Google Scholar] [CrossRef]
- Palliyage, G.H.; Samart, P.; Bobbala, S.; Rojanasakul, L.W.; Coyle, J.; Martin, K.; Callery, P.S.; Rojanasakul, Y. Chemotherapy-induced PDL-1 expression in cancer-associated fibroblasts promotes chemoresistance in NSCLC. Lung Cancer 2023, 181, 107258. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Mooradian, M.J.; Cai, L.; Wang, A.; Qiao, Y.; Chander, P.; Whitaker, R.M. Durvalumab After Chemoradiotherapy in Patients With Unresectable Stage III Non–Small Cell Lung Cancer. JAMA Netw. Open 2024, 7, e247542. [Google Scholar] [CrossRef]
- Shaverdashvili, K.; Reyes, V.; Wang, H.; Mehta, D.; Marsh, C.; Waas, J.K.; VanderWeele, R.A.; Peracha, S.M.; Liang, H.; Socinski, M.A. A phase II clinical trial evaluating the safety and efficacy of durvalumab as first line therapy in advanced and metastatic non-small cell lung cancer patients with Eastern Cooperative Oncology Group performance status of 2. eClinicalMedicine 2023, 66, 102317. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef]
- Saad, M.B.; Hong, L.; Aminu, M.; Vokes, N.I.; Chen, P.; Salehjahromi, M.; Qin, K.; Sujit, S.J.; Lu, X.; Young, E. Predicting benefit from immune checkpoint inhibitors in patients with non-small-cell lung cancer by CT-based ensemble deep learning: A retrospective study. Lancet Digit. Health 2023, 5, e404–e420. [Google Scholar] [CrossRef] [PubMed]
- Blons, H.; Garinet, S.; Laurent-Puig, P.; Oudart, J.-B. Molecular markers and prediction of response to immunotherapy in non-small cell lung cancer, an update. J. Thorac. Dis. 2019, 11, S25. [Google Scholar] [CrossRef]
- Patel, K.H.; Alpert, N.; Tuminello, S.; Taioli, E. Personal and clinical characteristics associated with immunotherapy effectiveness in stage IV non-small cell lung cancer. Transl. Lung Cancer Res. 2023, 12, 1210. [Google Scholar] [CrossRef]
- Zhao, W.; Jiang, W.; Wang, H.; He, J.; Su, C.; Yu, Q. Impact of smoking history on response to immunotherapy in non-small-cell lung cancer: A systematic review and meta-analysis. Front. Oncol. 2021, 11, 703143. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Qin, L.; Yu, G. The progresses of relevant factors on the efficacy of immune checkpoint inhibitors in the non-small cell lung cancer patients. Cancer Treat. Res. Commun. 2023, 37, 100758. [Google Scholar] [CrossRef]
- Raphael, J.; Batra, A.; Boldt, G.; Shah, P.S.; Blanchette, P.; Rodrigues, G.; Vincent, M.D. Predictors of survival benefit from immune checkpoint inhibitors in patients with advanced non–small-cell lung cancer: A systematic review and meta-analysis. Clin. Lung Cancer 2020, 21, 106–113.e105. [Google Scholar] [CrossRef]
- Ulas, E.B.; Hashemi, S.M.; Houda, I.; Kaynak, A.; Veltman, J.D.; Fransen, M.F.; Radonic, T.; Bahce, I. Predictive value of combined positive score and tumor proportion score for immunotherapy response in advanced NSCLC. JTO Clin. Res. Rep. 2023, 4, 100532. [Google Scholar] [CrossRef]
- Aggarwal, C.; Ben-Shachar, R.; Gao, Y.; Hyun, S.W.; Rivers, Z.; Epstein, C.; Kaneva, K.; Sangli, C.; Nimeiri, H.; Patel, J. Assessment of tumor mutational burden and outcomes in patients with diverse advanced cancers treated with immunotherapy. JAMA Netw. Open 2023, 6, e2311181. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Wu, L.; Su, D.; Mao, W.; Fang, B. Immunotherapy for non-small cell lung cancers: Biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer 2018, 18, 1082. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N. Molecular determinants of response to anti–programmed cell death (PD)-1 and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022, 8, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non–small cell lung carcinoma: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- To, K.K.; Fong, W.; Cho, W.C. Immunotherapy in treating EGFR-mutant lung cancer: Current challenges and new strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef]
- Solassol, I.; Pinguet, F.; Quantin, X. FDA-and EMA-approved tyrosine kinase inhibitors in advanced EGFR-mutated non-small cell lung cancer: Safety, tolerability, plasma concentration monitoring, and management. Biomolecules 2019, 9, 668. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.-L.; Papadimitrakopoulou, V.A.; Su, W.-C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in combination with erlotinib or gefitinib as first-line therapy for advanced NSCLC with sensitizing EGFR mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef]
- Chan, D.W.-K.; Choi, H.C.-W.; Lee, V.H.-F. Treatment-related adverse events of combination EGFR tyrosine kinase inhibitor and immune checkpoint inhibitor in EGFR-mutant advanced non-small cell lung cancer: A systematic review and meta-analysis. Cancers 2022, 14, 2157. [Google Scholar] [CrossRef] [PubMed]
- Tu, E.; McGlinchey, K.; Wang, J.; Martin, P.; Ching, S.L.; Floc’h, N.; Kurasawa, J.; Starrett, J.H.; Lazdun, Y.; Wetzel, L. Anti–PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. JCI Insight 2022, 7, e142843. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Negrao, M.V.; Reuben, A.; Federico, L.; Diao, L.; McGrail, D.; Nilsson, M.; Robichaux, J.; Munoz, I.G.; Patel, S. Characterization of the immune landscape of EGFR-mutant NSCLC identifies CD73/adenosine pathway as a potential therapeutic target. J. Thorac. Oncol. 2021, 16, 583–600. [Google Scholar] [CrossRef]
- Amanam, I.; Mambetsariev, I.; Gupta, R.; Achuthan, S.; Wang, Y.; Pharaon, R.; Massarelli, E.; Koczywas, M.; Reckamp, K.; Salgia, R. Role of immunotherapy and co-mutations on KRAS-mutant non-small cell lung cancer survival. J. Thorac. Dis. 2020, 12, 5086. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zeng, D.; Ou, Q.; Liu, S.; Li, A.; Chen, Y.; Lin, D.; Gao, Q.; Zhou, H.; Liao, W. Association of survival and immune-related biomarkers with immunotherapy in patients with non–small cell lung cancer: A meta-analysis and individual patient–level analysis. JAMA Netw. Open 2019, 2, e196879. [Google Scholar] [CrossRef]
- Wu, L.; Cheng, D.; Yang, X.; Zhao, W.; Fang, C.; Chen, R.; Ji, M. M2-TAMs promote immunoresistance in lung adenocarcinoma by enhancing METTL3-mediated m6A methylation. Ann. Transl. Med. 2022, 10, 1380. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Jiang, P.; Zhang, Y.; Li, Y.; Jia, X.; Wang, Q.; Jiao, M.; Jiang, L.; Shen, Y.; Guo, H. First-line immune-based combination therapies for advanced non-small cell lung cancer: A Bayesian network meta-analysis. Cancer Med. 2021, 10, 9139–9155. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Peters, S. Immunotherapy-based combinations in metastatic NSCLC. Cancer Treat. Rev. 2023, 116, 102545. [Google Scholar] [CrossRef]
- Yang, J.-H.; Luft, A.; Jiménez, E.D.L.M.; Lee, J.; Koralewski, P.; Karadurmus, N.; Sugawara, S.; Livi, L.; Basappa, N.; Quantin, X. 120O Pembrolizumab (Pembro) with or without lenvatinib (Lenva) in first-line metastatic NSCLC with PD-L1 TPS ≥ 1%(LEAP-007): A phase III, randomized, double-blind study. Ann. Oncol. 2021, 32, S1429–S1430. [Google Scholar] [CrossRef]
- Besse, B.; Pons-Tostivint, E.; Park, K.; Hartl, S.; Forde, P.M.; Hochmair, M.J.; Awad, M.M.; Thomas, M.; Goss, G.; Wheatley-Price, P. Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: A phase 2 umbrella trial. Nat. Med. 2024, 30, 716–729. [Google Scholar] [CrossRef]
- Xing, S.; Hu, K.; Wang, Y. Tumor immune microenvironment and immunotherapy in non-small cell lung cancer: Update and new challenges. Aging Dis. 2022, 13, 1615. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- van der Woude, L.L.; Gorris, M.A.; Wortel, I.M.; Creemers, J.H.; Verrijp, K.; Monkhorst, K.; Grünberg, K.; van den Heuvel, M.M.; Textor, J.; Figdor, C.G. Tumor microenvironment shows an immunological abscopal effect in patients with NSCLC treated with pembrolizumab-radiotherapy combination. J. Immunother. Cancer 2022, 10, e005248. [Google Scholar] [CrossRef]
- Yang, H.; Lei, Z.; He, J.; Zhang, L.; Lai, T.; Zhou, L.; Wang, N.; Tang, Z.; Sui, J.; Wu, Y. Single-cell RNA sequencing reveals recruitment of the M2-like CCL8high macrophages in Lewis lung carcinoma-bearing mice following hypofractionated radiotherapy. J. Transl. Med. 2024, 22, 306. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; DeCamp, M. NCCN guidelines® Insights: Non–small cell lung cancer, version 2.2023: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2023, 21, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Petrella, F.; Rizzo, S.; Attili, I.; Passaro, A.; Zilli, T.; Martucci, F.; Bonomo, L.; Del Grande, F.; Casiraghi, M.; De Marinis, F. Stage III non-small-cell lung cancer: An overview of treatment options. Curr. Oncol. 2023, 30, 3160–3175. [Google Scholar] [CrossRef]
- Pignon, J.-P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.-Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef]
- Cascone, T.; Hamdi, H.; Zhang, F.; Poteete, A.; Li, L.; Hudgens, C.W.; Williams, L.J.; Wu, Q.; Gudikote, J.; Peng, W. Superior efficacy of neoadjuvant compared to adjuvant immune checkpoint blockade in non-small cell lung cancer. Cancer Res. 2018, 78, 1719. [Google Scholar] [CrossRef]
- Wakelee, H.; Liberman, M.; Kato, T.; Tsuboi, M.; Lee, S.-H.; Gao, S.; Chen, K.-N.; Dooms, C.; Majem, M.; Eigendorff, E. Perioperative pembrolizumab for early-stage non–small-cell lung cancer. N. Engl. J. Med. 2023, 389, 491–503. [Google Scholar] [CrossRef]
- Rusch, V.W.; Nicholas, A.; Patterson, G.A.; Waqar, S.N.; Toloza, E.M.; Haura, E.B.; Raz, D.J.; Reckamp, K.L.; Merritt, R.E.; Owen, D.H. Surgical results of the Lung Cancer Mutation Consortium 3 trial: A phase II multicenter single-arm study to investigate the efficacy and safety of atezolizumab as neoadjuvant therapy in patients with stages IB-select IIIB resectable non–small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2023, 165, 828–839.e5. [Google Scholar]
- Felip, E.; Altorki, N.; Zhou, C.; Csőszi, T.; Vynnycheno, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet 2021, 398, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Paz-Ares, L.; Marreaud, S.; Dafni, U.; Oselin, K.; Havel, L.; Esteban, E.; Isla, D.; Martinez-Marti, A.; Faehling, M. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB–IIIA non-small-cell lung cancer (PEARLS/KEYNOTE-091): An interim analysis of a randomised, triple-blind, phase 3 trial. Lancet Oncol. 2022, 23, 1274–1286. [Google Scholar] [CrossRef]
- FDA. FDA Approves Neoadjuvant/Adjuvant Pembrolizumab for Resectable Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neoadjuvant-adjuvant-pembrolizumab-resectable-non-small-cell-lung-cancer (accessed on 5 May 2025).
- FDA. FDA Approves Neoadjuvant Nivolumab and Platinum-Doublet Chemotherapy for Early-Stage Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neoadjuvant-nivolumab-and-platinum-doublet-chemotherapy-early-stage-non-small-cell-lung (accessed on 5 May 2025).
- FDA. FDA Approves Neoadjuvant/Adjuvant Durvalumab for Resectable Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neoadjuvantadjuvant-durvalumab-resectable-non-small-cell-lung-cancer (accessed on 5 May 2025).
- FDA. FDA Approves Atezolizumab as Adjuvant Treatment for Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer (accessed on 5 May 2025).
- Sorin, M.; Prosty, C.; Ghaleb, L.; Nie, K.; Katergi, K.; Shahzad, M.H.; Dubé, L.-R.; Atallah, A.; Swaby, A.; Dankner, M. Neoadjuvant chemoimmunotherapy for NSCLC: A systematic review and meta-analysis. JAMA Oncol. 2024, 10, 621–633. [Google Scholar] [CrossRef]
- Schuler, M.; Cuppens, K.; Plönes, T.; Wiesweg, M.; Du Pont, B.; Hegedus, B.; Köster, J.; Mairinger, F.; Darwiche, K.; Paschen, A. Neoadjuvant nivolumab with or without relatlimab in resectable non-small-cell lung cancer: A randomized phase 2 trial. Nat. Med. 2024, 30, 1602–1611. [Google Scholar] [CrossRef]
- Predina, J.D.; Kapoor, V.; Judy, B.F.; Cheng, G.; Fridlender, Z.G.; Albelda, S.M.; Singhal, S. Cytoreduction surgery reduces systemic myeloid suppressor cell populations and restores intratumoral immunotherapy effectiveness. J. Hematol. Oncol. 2012, 5, 34. [Google Scholar] [CrossRef]
- Delaunay, M.; Prévot, G.; Collot, S.; Guilleminault, L.; Didier, A.; Mazières, J. Management of pulmonary toxicity associated with immune checkpoint inhibitors. Eur. Respir. Rev. 2019, 28, 190012. [Google Scholar] [CrossRef]
- Qiu, B.; Cai, K.; Chen, C.; Chen, J.; Chen, K.-N.; Chen, Q.-X.; Cheng, C.; Dai, T.-Y.; Fan, J.; Fan, Z. Expert consensus on perioperative immunotherapy for local advanced non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 3713–3736. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.J.; Yang, S.C.; Park, B.J.; Adusumilli, P.S.; Rusch, V.W.; Isbell, J.M.; Downey, R.J.; Brahmer, J.R.; Battafarano, R.; Bush, E. Initial results of pulmonary resection after neoadjuvant nivolumab in patients with resectable non–small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2019, 158, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Obeid, M.; Spain, L.; Carbonnel, F.; Wang, Y.; Robert, C.; Lyon, A.; Wick, W.; Kostine, M.; Peters, S. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Terashima, Y.; Toda, E.; Itakura, M.; Otsuji, M.; Yoshinaga, S.; Okumura, K.; Shand, F.H.; Komohara, Y.; Takeda, M.; Kokubo, K. Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties. Nat. Commun. 2020, 11, 609. [Google Scholar] [CrossRef]
- Okabe, Y.; Toda, E.; Urushiyama, H.; Terashima, Y.; Kunugi, S.; Kajimoto, Y.; Terasaki, M.; Matsushima, K.; Saito, A.; Yamauchi, Y. Antifibrotic effect of disulfiram on bleomycin-induced lung fibrosis in mice and its impact on macrophage infiltration. Sci. Rep. 2024, 14, 23653. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Key Knowledge Gaps |
|---|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, R.; Ehab, J.; Hauptmann, E.; Gunturu, N.S.; Karalis, J.D.; Kent, D.O.; Heid, C.A.; Reznik, S.I.; Sarkaria, I.S.; Huang, H.; et al. The Current State of Tumor Microenvironment-Specific Therapies for Non-Small Cell Lung Cancer. Cancers 2025, 17, 1732. https://doi.org/10.3390/cancers17111732
Chandra R, Ehab J, Hauptmann E, Gunturu NS, Karalis JD, Kent DO, Heid CA, Reznik SI, Sarkaria IS, Huang H, et al. The Current State of Tumor Microenvironment-Specific Therapies for Non-Small Cell Lung Cancer. Cancers. 2025; 17(11):1732. https://doi.org/10.3390/cancers17111732
Chicago/Turabian StyleChandra, Raghav, Jasmina Ehab, Edward Hauptmann, Naga Swati Gunturu, John D. Karalis, Daniel O. Kent, Christopher A. Heid, Scott I. Reznik, Inderpal S. Sarkaria, Huocong Huang, and et al. 2025. "The Current State of Tumor Microenvironment-Specific Therapies for Non-Small Cell Lung Cancer" Cancers 17, no. 11: 1732. https://doi.org/10.3390/cancers17111732
APA StyleChandra, R., Ehab, J., Hauptmann, E., Gunturu, N. S., Karalis, J. D., Kent, D. O., Heid, C. A., Reznik, S. I., Sarkaria, I. S., Huang, H., Brekken, R. A., & Minna, J. D. (2025). The Current State of Tumor Microenvironment-Specific Therapies for Non-Small Cell Lung Cancer. Cancers, 17(11), 1732. https://doi.org/10.3390/cancers17111732