

Current and Emerging Insights into the Causes, Immunopathogenesis, and Treatment of Cutaneous Squamous Cell Carcinoma

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Epidemiology, Clinical Features, and Diagnosis of Cutaneous Squamous Cell Carcinoma (cSCC)
2.1. Epidemiology
2.2. Clinical Features
- •
- Tumor diameter > 2 cm;
- •
- Depth of invasion > 6 mm;
- •
- Poor differentiation;
- •
- Perineural invasion (PNI);
- •
- Location on high-risk sites (e.g., ears, lips, or genitals);
- •
- Immunosuppression or a prior history of aggressive SCC [3].
2.3. Diagnosis
2.3.1. Clinical and Dermoscopy Evaluation
- •
- Scaling and white structureless areas;
- •
- Dotted or glomerular vessels;
- •
- Ulceration in more advanced tumors [2].
2.3.2. Histopathology and Grading
3. Risk Factors
3.1. Ultraviolet Radiation
3.2. Radiation Therapy
3.3. Iatrogenic Immunosuppression
3.4. Smoking
3.5. Cutaneous Squamous Cell Carcinoma Associated with Chronic Viral Infection
3.5.1. Human Papillomavirus
3.5.2. Human Immunodeficiency Virus
- Standardized incidence risk (SIR) = 3.2, 95% CI 3.2, 95% CI = 2.2–45 (for non-melanomatous skin malignancies, subtypes not indicated) [53].
- OR = 2.6, 95% CI = 1.4–4.9 [54].
- SIR = 4.64, 95% CI = 3.15–6.03 [55].
- Adjusted rate ratio = 2.6, 95% CI = 2.1–3.2 [56].
- SIR = 5.4, 95% CI = 3.07–9.52 [57].
3.6. Tattooing
4. Differential Gene Expression, Driver Mutation Profiling, and Role of Neoantigens in Cutaneous Squamous Cell Carcinoma
4.1. The Genomic Landscape
4.2. Tumor Neoantigens (Tumor-Specific Antigens)
5. Mechanisms of Immune Evasion in Cutaneous Squamous Cell Carcinoma
5.1. Mechanisms of Immune Evasion Operative in the Tumor Microenvironment
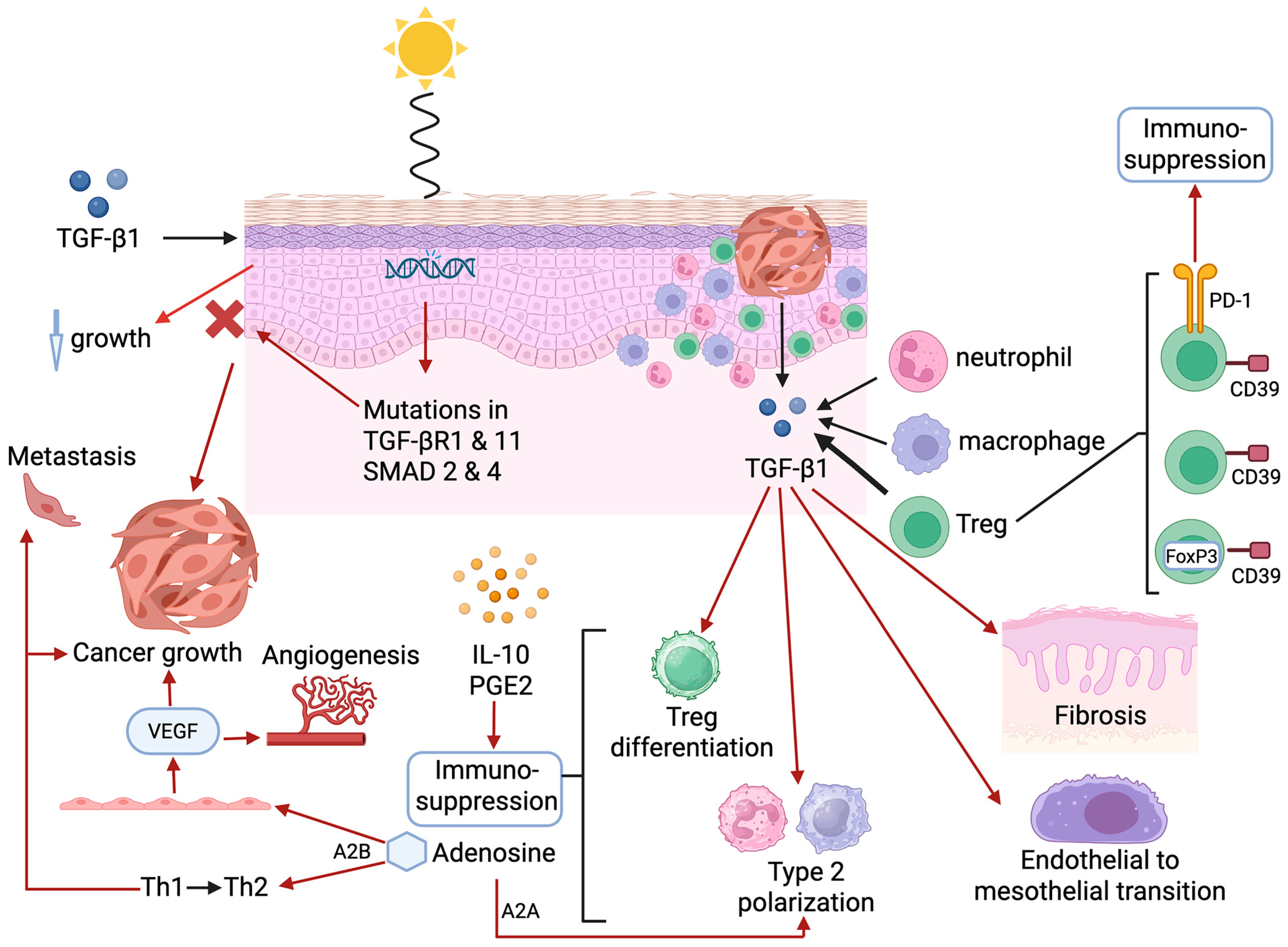
5.2. Transforming Growth Factor-β1
5.3. Adenosine
5.4. Attenuation of Expression of Highly Immunogenic Neoepitopes
5.5. Systemic Immunosuppression
6. Systemic Chemotherapy, Targeted Therapy, and Immunotherapy
6.1. Systemic Chemotherapy and Targeted Treatments
6.2. Immunotherapy
7. Potential TME-Associated and Systemic Biomarkers in Cutaneous Squamous Cell Carcinoma
Novel Potential Systemic Biomarkers
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hadian, Y.; Howell, J.Y.; Ramsey, M.L.; Buckley, C. Cutaneous Squamous Cell Carcinoma; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441939/ (accessed on 21 January 2025).
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous squamous cell carcinoma: From pathophysiology to novel therapeutic approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Winge, M.C.G.; Kellman, L.N.; Guo, K.; Tang, J.Y.; Swetter, S.M.; Aasi, S.Z.; Sarin, K.Y.; Chang, A.L.S.; Khavari, P.A. Advances in cutaneous squamous cell carcinoma. Nat. Rev. Cancer 2023, 23, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Geidel, G.; Heidrich, I.; Kött, J.; Schneider, S.W.; Pantel, K.; Gebhardt, C. Emerging precision diagnostics in advanced cutaneous squamous cell carcinoma. NPJ Precis. Oncol. 2022, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Oncology (Editorial). Climate change and skin cancer: Urgent call for action. Lancet Oncol. 2023, 24, 823. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Townes, T.; Na’ara, S. Current advances and challenges in the management of cutaneous squamous cell carcinoma in immunosuppressed patients. Cancers 2024, 16, 3118. [Google Scholar] [CrossRef] [PubMed]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S.; Schmults, C.D.; Ruiz, E.S. A Review of cutaneous squamous cell carcinoma epidemiology, diagnosis and management. Int. J. Cancer Manag. 2018, 11, e60846. [Google Scholar] [CrossRef]
- Sinz, C.; Tschandl, P.; Rosendahl, C.; Akay, B.N.; Argenziano, G.; Blum, A.; Braun, R.P.; Cabo, H.; Gourhant, J.Y.; Kreusch, J.; et al. Accuracy of dermatoscopy for the diagnosis of nonpigmented cancers of the skin. J. Am. Acad. Dermatol. 2017, 77, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Vonk, J.; de Wit, J.G.; Voskuil, F.J.; Witjes, M.J.H. Improving oral cavity cancer diagnosis and treatment with fluorescence molecular imaging. Oral Dis. 2021, 27, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Atak, M.F.; Farabi, B.; Navarrete-Dechent, C.; Rubinstein, G.; Rajadhyaksha, M.; Jain, M. Confocal microscopy for diagnosis and management of cutaneous malignancies: Clinical impacts and innovation. Diagnostics 2023, 13, 854. [Google Scholar] [CrossRef] [PubMed]
- Cinotti, E.; Brunetti, T.; Cartocci, A.; Tognetti, L.; Suppa, M.; Malvehy, J.; Perez-Anker, J.; Puig, S.; Perrot, J.L.; Rubegni, P. Diagnostic accuracy of line-field confocal optical coherence tomography for the diagnosis of skin carcinomas. Diagnostics 2023, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Guida, S.; Mirra, M.; Pampena, R.; Ciardo, S.; Bassoli, S.; Casari, A.; Rongioletti, F.; Spadafora, M.; Chester, J.; et al. Dermatoscopy and reflectance confocal microscopy for basal cell carcinoma diagnosis and diagnosis prediction score: A prospective and multicenter study on 1005 lesions. J. Am. Acad. Dermatol. 2024, 90, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Danescu, S.; Negrutiu, M.; Focsan, M.; Baican, A. An overview of cutaneous squamous cell carcinoma imaging diagnosis methods. Front. Med. 2024, 11, 1388835. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.; Kubicki, S.; Nguyen, Q.B.; Aboul-Fettouh, N.; Wilmas, K.M.; Chen, O.M.; Doan, H.Q.; Silapunt, S.; Migden, M.R. Advances in cutaneous squamous cell carcinoma management. Cancers 2022, 14, 3653. [Google Scholar] [CrossRef] [PubMed]
- Urbach, F. Ultraviolet radiation and skin cancer of humans. J. Photochem. Photobiol. B 1997, 40, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Norval, M. Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem. Photobiol. Sci. 2018, 17, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Prime, S.S.; Darski, P.; Hunter, K.D.; Cirillo, N.; Parkinson, E.K. A Review of the repair of DNA double strand breaks in the development of oral cancer. Int. J. Mol. Sci. 2024, 25, 4092. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Mullenders, L.H.F. Solar UV damage to cellular DNA: From mechanisms to biological effects. Photochem. Photobiol. Sci. 2018, 17, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Azzimonti, B.; Zavattaro, E.; Provasi, M.; Vidali, M.; Conca, A.; Catalano, E.; Rimondini, L.; Colombo, E.; Valente, G. Intense Foxp3+ CD25+ regulatory T-cell infiltration is associated with high-grade cutaneous squamous cell carcinoma and counterbalanced by CD8+/Foxp3+ CD25+ ratio. Br. J. Dermatol. 2015, 172, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; August, S.; Albibas, A.; Behar, R.; Cho, S.Y.; Polak, M.E.; Theaker, J.; MacLeod, A.S.; French, R.R.; Glennie, M.J.; et al. OX40+ Regulatory T cells in cutaneous squamous cell carcinoma suppress effector T-cell responses and associate with metastatic potential. Clin. Cancer Res. 2016, 22, 4236–4248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Wijaya, W.A.; Liang, Z.; Chen, J. Efficacy and prognostic factors of adjuvant radiotherapy for cutaneous squamous cell carcinoma: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Lichter, M.D.; Karagas, M.R.; Mott, L.A.; Spencer, S.K.; Stukel, T.A.; Greenberg, E.R. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch. Dermatol. 2000, 136, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Karagas, M.R.; McDonald, J.A.; Greenberg, E.R.; Stukel, T.A.; Weiss, J.E.; Baron, J.A.; Stevens, M.M. Risk of basal cell and squamous cell skin cancers after ionizing radiation therapy. For The Skin Cancer Prevention Study Group. J. Natl. Cancer Inst. 1996, 88, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, K.; Shibuya, H.; Yoshimura, R.; Okada, N. The risk of radiation-induced cancer in patients with squamous cell carcinoma of the head and neck and its results of treatment. Br. J. Radiol. 2005, 78, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Zavdy, O.; Coreanu, T.; Bar-On, D.Y.; Ritter, A.; Bachar, G.; Shpitzer, T.; Kurman, N.; Mansour, M.; Ad-El, D.; Rozovski, U.; et al. Cutaneous squamous cell carcinoma in immunocompromised patients–A comparison between different immunomodulating conditions. Cancers 2023, 15, 1764. [Google Scholar] [CrossRef] [PubMed]
- Struckmeier, A.K.; Gosau, M.; Smeets, R. Cutaneous squamous cell carcinoma in solid organ transplant recipients: Current therapeutic and screening strategies. Transplant Rev. 2024, 38, 100882. [Google Scholar] [CrossRef] [PubMed]
- Barsouk, A.; Aluru, J.S.; Rawla, P.; Saginala, K.; Barsouk, A. Epidemiology, risk factors, and prevention of head and neck squamous cell carcinoma. Med. Sci. 2023, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Vidal, M.; Whitaker-Menezes, D.; Martos-Rus, C.; Tassone, P.; Snyder, C.M.; Tuluc, M.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Cigarette smoke induces metabolic reprogramming of the tumor stroma in head and neck squamous cell carcinoma. Mol. Cancer Res. 2019, 17, 1893–1909. [Google Scholar] [CrossRef] [PubMed]
- De Hertog, S.A.; Wensveen, C.A.; Bastiaens, M.T.; Kielich, C.J.; Berkhout, M.J.; Westendorp, R.G.; Vermeer, B.J.; Bouwes Bavinck, J.N.; Leiden Skin Cancer Study. Relation between smoking and skin cancer. J. Clin. Oncol. 2001, 19, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Arafa, A.; Mostafa, A.; Navarini, A.A.; Dong, J.Y. The association between smoking and risk of skin cancer: A meta-analysis of cohort studies. Cancer Causes Control 2020, 31, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; George, C.D.; Jiang, C.; Asgari, M.M.; Nijsten, T.; Pardo, L.M.; Choquet, H. Association between lifetime smoking and cutaneous squamous cell carcinoma: A 2-sample Mendelian randomization study. JAAD Int. 2023, 14, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Batra, V.; Patkar, A.A.; Berrettini, W.H.; Weinstein, S.P.; Leone, F.T. The genetic determinants of smoking. Chest 2003, 123, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Saunders, G.R.B.; Wang, X.; Chen, F.; Jang, S.K.; Liu, M.; Wang, C.; Gao, S.; Jiang, Y.; Khunsriraksakul, C.; Otto, J.M.; et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature 2022, 612, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Uotila, I.; Siiskonen, H.; Haimakainen, S.; Harvima, I. Tobacco smoking is associated with cutaneous squamous cell carcinoma but not with basal cell carcinoma or melanoma in adult subjects at risk of skin cancer: A cross-sectional study. Tob. Induc. Dis. 2024, 22. [Google Scholar] [CrossRef] [PubMed]
- Odenbro, A.; Bellocco, R.; Boffetta, P.; Lindelöf, B.; Adami, J. Tobacco smoking, snuff dipping and the risk of cutaneous squamous cell carcinoma: A nationwide cohort study in Sweden. Br. J. Cancer 2005, 92, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- McBride, P.; Olsen, C.M.; Green, A.C. Tobacco smoking and cutaneous squamous cell carcinoma: A 16-year longitudinal population-based study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Theron, A.J.; Anderson, R. Activation of neutrophil membrane-associated oxidative metabolism by ultraviolet radiation. J. Investig. Dermatol. 1993, 101, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C. Cases of warty tumours in cicatrices. Med. Chir. Trans. 1835, 19, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T.; Butel, J.S. The history of tumor virology. Cancer Res. 2008, 68, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A.; Vogt, P.K. 100 years of Rous sarcoma virus. J. Exp. Med. 2011, 208, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aldabagh, B.; Yu, J.; Arron, S.T. Role of human papillomavirus in cutaneous squamous cell carcinoma: A meta-analysis. J. Am. Acad. Dermatol. 2014, 70, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Strickley, J.D.; Messerschmidt, J.L.; Awad, M.E.; Li, T.; Hasegawa, T.; Ha, D.T.; Nabeta, H.W.; Bevins, P.A.; Ngo, K.H.; Asgari, M.M.; et al. Immunity to commensal papillomaviruses protects against skin cancer. Nature 2019, 575, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Son, H.G.; Ha, D.T.; Xia, Y.; Li, T.; Blandin, J.; Oka, T.; Azin, M.; Conrad, D.N.; Zhou, C.; Zeng, Y.; et al. Commensal papillomavirus immunity preserves the homeostasis of highly mutated normal skin. Cancer Cell 2025, 43, 36–48.e10. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. HPV and skin carcinogenesis. Papillomavirus Res. 2019, 7, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Yugawa, T.; Handa, K.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell Biol. 2007, 27, 3732–3742. [Google Scholar] [CrossRef] [PubMed]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef] [PubMed]
- Blakely, W.J.; Hatterschide, J.; White, E.A. HPV18 E7 inhibits LATS1 kinase and activates YAP1 by degrading PTPN14. mBio 2024, 15, e0181124. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Polesel, J.; Rickenbach, M.; Dal Maso, L.; Keiser, O.; Kofler, A.; Rapiti, E.; Levi, F.; Jundt, G.; Fisch, T.; et al. Cancer risk in the Swiss HIV. Cohort Study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J. Natl. Cancer Inst. 2005, 97, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.; Urban, M.I.; O’Connell, D.; Yu, X.Q.; Beral, V.; Newton, R.; Ruff, P.; Donde, B.; Hale, M.; Patel, M.; et al. The spectrum of human immunodeficiency virus-associated cancers in a South African black population: Results from a case-control study, 1995–2004. Int. J. Cancer. 2008, 122, 2260–2265. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.K.; Chang, P.-Y.; Martínez-Maza, O.; Zhang, Z.-F.; Jacobson, L.; Detels, R. Risk factors for squamous cell skin cancer in HIV. Infect. Agents Cancer. 2012, 7 (Suppl. S1), O26. [Google Scholar] [CrossRef]
- Silverberg, M.J.; Leyden, W.; Warton, E.M.; Quesenberry, C.P., Jr.; Engels, E.A.; Asgari, M.M. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J. Natl. Cancer Inst. 2013, 105, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Omland, S.H.; Ahlström, M.G.; Gerstoft, J.; Pedersen, G.; Mohey, R.; Pedersen, C.; Kronborg, G.; Larsen, C.S.; Kvinesdal, B.; Gniadecki, R.; et al. Risk of skin cancer in patients with HIV: A Danish nationwide cohort study. J. Am. Acad. Dermatol. 2018, 79, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, J.P.; Leslie, K.S. Skin cancer risk in people living with HIV: A call for action. Lancet HIV 2024, 11, e60–e62. [Google Scholar] [CrossRef] [PubMed]
- Lineberry, C. The Worldwide History of Tattoos. Smithsonian Magazine. Updated by Anderson, S., Updated: 18 October 2023. 2007. Available online: https://www.smithsonianmag.com/history/tattoos-worldwide-history-144038580/ (accessed on 28 April 2025).
- Junqueira, A.L.; Wanat, K.A.; Farah, R.S. Squamous neoplasms arising within tattoos: Clinical presentation, histopathology and management. Clin. Exp. Dermatol. 2017, 42, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Rahbarinejad, Y.; Guio-Aguilar, P.; Vu, A.N.; Lo, M.; McTigue, C.; Nirenberg, A.; Rozen, W.M. Pathogenesis, Diagnosis and management of squamous cell carcinoma and pseudoepithelial hyperplasia secondary to red ink tattoo: A case series and review. J. Clin. Med. 2023, 12, 2424. [Google Scholar] [CrossRef] [PubMed]
- Lebhar, J.; Jacobs, J.; Rundle, C.; Kaplan, S.J.; Mosca, P.J. Skin cancers arising within tattoos: A systematic review. JAAD Int. 2024, 16, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Liljedahl, E.R.; Nielsen, K.; Engfeldt, M.; Jöud, A.; Nielsen, C. Does tattoo exposure increase the risk of skin cancer? A population-based case-control study. In Proceedings of the ISEE 2024: 36th Annual Conference of the International Society of Environmental Epidemiology, Santiago, Chile, 25–28 August 2024; ISEE Conference Abstracts. Volume 1. [Google Scholar] [CrossRef]
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Phillips, D.H.; Hanawalt, P.C. Heterogeneous DNA damage and repair in the mammalian genome. Cancer Res. 1987, 47, 6426–6436. [Google Scholar] [PubMed]
- Wei, W.; Chen, Y.; Xu, J.; Zhou, Y.; Bai, X.; Yang, M.; Zhu, J. Identification of biomarker for cutaneous squamous cell carcinoma using microarray data analysis. J. Cancer 2018, 9, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.D.; Xu, D.; Deng, Y.Y.; Wu, W.J.; Zhang, J.; Huang, L.; He, L. Identification of key genes in cutaneous squamous cell carcinoma: A transcriptome sequencing and bioinformatics profiling study. Ann. Transl. Med. 2021, 9, 1497. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Shain, A.H. The landscape of driver mutations in cutaneous squamous cell carcinoma. NPJ Genom. Med. 2021, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Hernando, B.; Dietzen, M.; Parra, G.; Gil-Barrachina, M.; Pitarch, G.; Mahiques, L.; Valcuende-Cavero, F.; McGranahan, N.; Martinez-Cadenas, C. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 2021, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Droll, S.; Bao, X. Oh, the mutations you’ll acquire! A systematic overview of cutaneous squamous cell carcinoma. Cell Physiol. Biochem. 2021, 55, 89–119. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.; Lammerts, M.U.P.A.; Genders, R.E.; Bouwes Bavinck, J.N. Update of advanced cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on tumor neoantigens and their utility: Why it is good to be different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.S.; Kang, P.; Natri, H.M.; Phung, T.N.; Wilson, M.A.; Buetow, K.H.; Hastings, K.T. Neoantigen fitness model predicts lower immune recognition of cutaneous squamous cell carcinomas than actinic keratoses. Front. Immunol. 2019, 10, 2799. [Google Scholar] [CrossRef] [PubMed]
- Hastings, K.; Adams, A.; Macy, A.; Borden, E.; Herrmann, L.; Brambley, C.; Ma, T.; Li, X.; Hughes, A.; Roe, D.; et al. Characteristics of tumor-rejecting neoantigens identified in a physiologically relevant cutaneous squamous cell carcinoma model. J. Immunol. 2024, 212 (Suppl. S1), 0547_4385. [Google Scholar] [CrossRef]
- Pham, T.V.; Boichard, A.; Goodman, A.; Riviere, P.; Yeerna, H.; Tamayo, P.; Kurzrock, R. Role of ultraviolet mutational signature versus tumor mutation burden in predicting response to immunotherapy. Mol. Oncol. 2020, 14, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.S.; Azin, M.; Demehri, S. Cutaneous squamous cell carcinoma: The frontier of cancer immunoprevention. Annu. Rev. Pathol. 2022, 17, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Fritz, M.; Que, S.K.T. Cutaneous squamous cell carcinoma: An updated review. Cancers 2024, 16, 1800. [Google Scholar] [CrossRef] [PubMed]
- Tuong, Z.K.; Lewandowski, A.; Bridge, J.A.; Cruz, J.L.G.; Yamada, M.; Lambie, D.; Lewandowski, R.; Steptoe, R.J.; Leggatt, G.R.; Simpson, F.; et al. Cytokine/chemokine profiles in squamous cell carcinoma correlate with precancerous and cancerous disease stage. Sci. Rep. 2019, 9, 17754. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, E.; Toia, F.; Oieni, S.; Buccheri, S.; Turdo, A.; Mangiapane, L.R.; Campisi, G.; Caputo, V.; Todaro, M.; Stassi, G.; et al. Squamous cell tumors recruit γδ T cells producing either IL17 or IFNγ depending on the tumor stage. Cancer Immunol. Res. 2017, 5, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.; Stafford, H.; Buell, J.; Ramesh, U.; Amit, M.; Nagarajan, P.; Migden, M.; Yaniv, D. Review of the tumor microenvironment in basal and squamous cell carcinoma. Cancers 2023, 15, 2453. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, X.J. TGFβ signaling in photoaging and UV-induced skin cancer. J. Investig. Dermatol. 2021, 141, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Rentero, M.; Gutiérrez-Pérez, M.; Fernández-Guarino, M.; Mascaraque, M.; Portillo-Esnaola, M.; Gilaberte, Y.; Carrasco, E.; Juarranz, Á. TGFβ1 secreted by cancer-associated fibroblasts as an inductor of resistance to photodynamic therapy in squamous cell carcinoma cells. Cancers 2021, 13, 5613. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-β1–A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer. 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Chen, W. TGF-β regulation of T cells. Annu. Rev. Immunol. 2023, 41, 483–512. [Google Scholar] [CrossRef] [PubMed]
- Oliver, M.A.; Davis, X.D.; Bohannon, J.K. TGFβ macrophage reprogramming: A new dimension of macrophage plasticity. J. Leukoc. Biol. 2024, 115, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Extracellular adenosine-mediated modulation of regulatory T cells. Front. Immunol. 2014, 5, 304. [Google Scholar] [CrossRef] [PubMed]
- Baratelli, F.; Lee, J.M.; Hazra, S.; Lin, Y.; Walser, T.C.; Schaue, D.; Pak, P.S.; Elashoff, D.; Reckamp, K.; Zhang, L.; et al. PGE(2) contributes to TGF-beta induced T regulatory cell function in human non-small cell lung cancer. Am. J. Transl. Res. 2010, 2, 356–367. [Google Scholar] [PubMed]
- Nestor, M.; Berman, B.; Lu, P.; Molyneaux, M. Safety and efficacy of TGF-β1/COX-2 silencing therapeutic in adults with cutaneous squamous cell carcinoma in situ. J. Drugs Dermatol. 2022, 21, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, M.; Berman, B.; Xu, J.; Evans, D.M.; Lu, P.Y. 15580 Effect of TGF-β1/COX-2 small interfering RNA combination product (STP705) on cell viability and tumor growth in a human squamous carcinoma xenograft tumor model in nude mice. J. Am. Acad. Dermatol. 2020, 83, AB156. [Google Scholar] [CrossRef]
- Kim, W.; Ye, Z.; Simonenko, V.; Shahi, A.; Malikzay, A.; Long, S.Z.; Xu, J.J.; Lu, A.; Horng, J.H.; Wu, C.R.; et al. Codelivery of TGFβ and Cox2 siRNA inhibits HCC by promoting T-cell penetration into the tumor and improves response to Immune Checkpoint Inhibitors. NAR Cancer 2024, 6, zcad059. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Napolitano, F.; Maresca, D.C.; Scala, M.; Amato, A.; Belli, S.; Ascione, C.M.; Vallefuoco, A.; Attanasio, G.; Somma, F.; et al. Early assessment of IL8 and PD1+ Treg predicts response and guides treatment monitoring in cemiplimab-treated cutaneous squamous cell carcinoma. J. Immunother. Cancer 2025, 13, e010421. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R.C.; Leone, R.D.; Naing, A.; Fong, L.; Bao, R.; Luke, J.J. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004089. [Google Scholar] [CrossRef] [PubMed]
- Mandapathil, M.; Szczepanski, M.; Harasymczuk, M.; Ren, J.; Cheng, D.; Jackson, E.K.; Gorelik, E.; Johnson, J.; Lang, S.; Whiteside, T.L. CD26 expression and adenosine deaminase activity in regulatory T cells (Treg) and CD4(+) T effector cells in patients with head and neck squamous cell carcinoma. Oncoimmunology 2012, 1, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Devi, V.J.; Radhika, A.; Biju, P.G. Adenosine receptor activation promotes macrophage class switching from LPS-induced acute inflammatory M1 to anti-inflammatory M2 phenotype. Immunobiology 2023, 228, 152362. [Google Scholar] [CrossRef] [PubMed]
- Lovászi, M.; Németh, Z.H.; Pacher, P.; Gause, W.C.; Wagener, G.; Haskó, G. A2A adenosine receptor activation prevents neutrophil aging and promotes polarization from N1 towards N2 phenotype. Purinergic Signal 2022, 18, 345–358. [Google Scholar] [CrossRef] [PubMed]
- El-Naccache, D.W.; Chen, F.; Palma, M.J.; Lemenze, A.; Fischer, M.A.; Wu, W.; Mishra, P.K.; Eltzschig, H.K.; Robson, S.C.; Di Virgilio, F.; et al. Adenosine metabolized from extracellular ATP promotes type 2 immunity through triggering A2BAR signaling in intestinal epithelial cells. Cell Rep. 2022, 40, 111150. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Goldstein, A.E.; Ryzhov, S.; Zeng, D.; Belardinelli, L.; Voyno-Yasenetskaya, T.; Biaggioni, I. Differential expression of adenosine receptors in human endothelial cells: Role of A2B receptors in angiogenic factor regulation. Circ. Res. 2002, 90, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Chandrakiran, C.; Jogy, T.; Patil, S.B. Serum adenosine deaminase levels and human papillomavirus as prognostic and predictive factors for laryngeal and pharyngeal carcinomas. Indian J. Otolaryngol. Head Neck Surg. 2019, 71, 522–527. [Google Scholar] [CrossRef]
- Huo, X.X.; Wang, S.J.; Song, H.; Li, M.D.; Yu, H.; Wang, M.; Gong, H.X.; Qiu, X.T.; Zhu, Y.F.; Zhang, J.Y. Roles of major RNA adenosine modifications in head and neck squamous cell carcinoma. Front. Pharmacol. 2021, 12, 779779. [Google Scholar] [CrossRef] [PubMed]
- Whitley, M.J.; Suwanpradid, J.; Lai, C.; Jiang, S.W.; Cook, J.L.; Zelac, D.E.; Rudolph, R.; Corcoran, D.L.; Degan, S.; Spasojevic, I.; et al. ENTPD1 (CD39) expression inhibits UVR-induced DNA damage repair through purinergic signaling and is associated with metastasis in human cutaneous squamous cell carcinoma. J. Invest. Dermatol. 2021, 141, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Kgokolo, M.C.M.; Malinga, N.Z.; Steel, H.C.; Meyer, P.W.A.; Smit, T.; Anderson, R.; Rapoport, B.L. Transforming growth factor-β1 and soluble co-inhibitory immune checkpoints as putative drivers of immune suppression in patients with basal cell carcinoma. Transl. Oncol. 2024, 42, 101867. [Google Scholar] [CrossRef] [PubMed]
- Rollison, D.E.; Messina, J.L.; Cherpelis, B.S.; Fenske, N.A.; Schell, M.J.; Adeegbe, D.O.; Zhao, Y.; Amorrortu, R.P.; Akuffo, A.A.; Hesterberg, R.S.; et al. Circulating immunosuppressive regulatory T cells predict risk of incident cutaneous squamous cell carcinoma. Front. Med. 2021, 8, 735585. [Google Scholar] [CrossRef] [PubMed]
- Queirolo, P.; Cinquini, M.; Argenziano, G.; Bassetto, F.; Bossi, P.; Boutros, A.; Clemente, C.; de Giorgi, V.; Del Vecchio, M.; Patuzzo, R.; et al. Guidelines for the diagnosis and treatment of cutaneous squamous cell carcinoma: A GRADE approach for evidence evaluation and recommendations by the Italian Association of Medical Oncology. ESMO Open 2024, 9, 103005. [Google Scholar] [CrossRef] [PubMed]
- Chapalain, M.; Baroudjian, B.; Dupont, A.; Lhote, R.; Lambert, J.; Bagot, M.; Lebbe, C.; Basset-Seguin, N. Stage IV cutaneous squamous cell carcinoma: Treatment outcomes in a series of 42 patients. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.; Tamimi, A.; Sorkheh, F.; Asl, S.M.; Ghafari, A.; Karimi, A.G.; Erabi, G.; Pourmontaseri, H.; Deravi, N. Monoclonal antibodies for the treatment of squamous cell carcinoma: A literature review. Cancer Rep. 2023, 6, e1802. [Google Scholar] [CrossRef] [PubMed]
- Montaudié, H.; Viotti, J.; Combemale, P.; Dutriaux, C.; Dupin, N.; Robert, C.; Mortier, L.; Kaphan, R.; Duval-Modeste, A.B.; Dalle, S.; et al. Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: A retrospective, multicentre study. Oncotarget 2020, 11, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Lebbe, C.; van Akkooi, A.; Bataille, V.; Bastholt, L.; Dreno, B.; Dummer, R.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for invasive cutaneous squamous cell carcinoma: Part 2. Treatment-Update 2023. Eur. J. Cancer. 2023, 193, 113252. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The treatment of inoperable sarcoma with the ‘mixed toxins of erysipelas and bacillus prodigiosus. Immediate and final results in one hundred and forty cases. JAMA 1898, XXXI, 456–465. [Google Scholar] [CrossRef]
- Ichim, C.V. Revisiting immunosurveillance and immunostimulation: Implications for cancer immunotherapy. J. Transl. Med. 2005, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC cancer). BMC Cancer 2018, 18, 1086. [Google Scholar] [CrossRef] [PubMed]
- Clingan, P.; Ladwa, R.; Brungs, D.; Harris, D.L.; McGrath, M.; Arnold, S.; Coward, J.; Fourie, S.; Kurochkin, A.; Malan, D.R.; et al. Efficacy and safety of cosibelimab, an anti-PD-L1 antibody, in metastatic cutaneous squamous cell carcinoma. J. Immunother. Cancer. 2023, 11, e007637. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.G.M.; Guminski, A.; Bowyer, S.; Migden, M.R.; Schmults, C.D.; Khushalani, N.I.; Chang, A.L.S.; Grob, J.J.; Lewis, K.D.; Ansstas, G.; et al. A phase 2 open-label study of cemiplimab in patients with advanced cutaneous squamous cell carcinoma (EMPOWER-CSCC-1): Final long-term analysis of groups 1, 2, and 3, and primary analysis of fixed-dose treatment group 6. J. Am. Acad. Dermatol. 2025, 92, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Baggi, A.; Quaglino, P.; Rubatto, M.; Depenni, R.; Guida, M.; Ascierto, P.A.; Trojaniello, C.; Queirolo, P.; Saponara, M.; Peris, K.; et al. Real world data of cemiplimab in locally advanced and metastatic cutaneous squamous cell carcinoma. Eur. J. Cancer. 2021, 157, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Couselo, E.; Hughes, B.G.M.; Mortier, L.; Grob, J.J.; Gutzmer, R.; Roshdy, O.; Gonzalez Mendoza, R.L.; Schachter, J.; Arance, A.M.; Grange, F.; et al. Pembrolizumab (pembro) for locally advanced (LA) or recurrent/metastatic (R/M) cutaneous squamous cell carcinoma (cSCC): Long-term results of the phase 2 KEYNOTE-629 study. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9554. [Google Scholar] [CrossRef]
- Hughes, B.G.M.; Munoz-Couselo, E.; Mortier, L.; Bratland, Å.; Gutzmer, R.; Roshdy, O.; González Mendoza, R.; Schachter, J.; Arance, A.; Grange, F.; et al. Pembrolizumab for locally advanced and recurrent/metastatic cutaneous squamous cell carcinoma (KEYNOTE-629 study): An open-label, nonrandomized, multicenter, phase II trial. Ann. Oncol. 2021, 32, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Amatore, F.; Sridharan, S.; Karunamurthy, A.; Wang, H.; Patel, R.; Pugliano-Mauro, M.; Kim, S.; Asif Choudry, M.H.; Pingpank, J.F., Jr.; Holtzman, M.P.; et al. Pathologic response rates to neoadjuvant pembrolizumab in locally advanced (LA) resectable cutaneous squamous cell carcinoma (cSCC). J. Clin. Oncol. 2024, 42 (Suppl. S16), 9591. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. Cutaneous Squamous Cell Carcinoma and Other Cutaneous Carcinomas. In The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM; Edge, S., Ed.; Chapter 29; Springer: New York, NY, USA, 2010; pp. 301–314. ISBN 978-0-387-88440-0. [Google Scholar]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Willenbrink, T.J.; Jambusaria-Pahlajani, A.; Arron, S.; Seckin, D.; Harwood, C.A.; Proby, C.M. Treatment approaches in immunosuppressed patients with advanced cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Montano, E.; Bhatia, N.; Ostojić, J. Biomarkers in cutaneous keratinocyte carcinomas. Dermatol. Ther. 2024, 14, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Slater, N.A.; Sayed, C.J.; Googe, P.B. PD-L1 and LAG-3 expression in advanced cutaneous squamous cell carcinomas. J. Cutan. Pathol. 2020, 47, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Dany, M.; Doudican, N.; Carucci, J. The Novel Checkpoint Target Lymphocyte-Activation Gene 3 Is Highly Expressed in Cutaneous Squamous Cell Carcinoma. Dermatol. Surg. 2023, 49, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Piao, M.S.; Choi, J.Y.; Yun, S.J.; Lee, J.B.; Lee, S.C. Up-regulation of cyclooxygenase 2 and matrix metalloproteinases-2 and -9 in cutaneous squamous cell carcinoma: Active role of inflammation and tissue remodeling in carcinogenesis. Ann. Dermatol. 2013, 25, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kivisaari, A.K.; Kallajoki, M.; Ala-aho, R.; McGrath, J.A.; Bauer, J.W.; Königová, R.; Medvecz, M.; Beckert, W.; Grénman, R.; Kähäri, V.M. Matrix metalloproteinase-7 activates heparin-binding epidermal growth factor-like growth factor in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 163, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H.; Yan, Q.; Gao, S.; Gao, J.; Wang, Z.; Sun, Y. Serum matrix metalloproteinase-13 as a diagnostic biomarker for cutaneous squamous cell carcinoma. BMC Cancer 2021, 21, 816. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Viiklepp, K.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Tumour-cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br. J. Dermatol. 2020, 182, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Stravodimou, A.; Tzelepi, V.; Papadaki, H.; Mouzaki, A.; Georgiou, S.; Melachrinou, M.; Kourea, E.P. Evaluation of T-lymphocyte subpopulations in actinic keratosis, in situ and invasive squamous cell carcinoma of the skin. J. Cutan. Pathol. 2018, 45, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Mousa, A.M.; Enk, A.H.; Hassel, J.C.; Reschke, R. Immune Checkpoints and Cellular Landscape of the Tumor Microenvironment in Non-Melanoma Skin Cancer (NMSC). Cells 2024, 13, 1615. [Google Scholar] [CrossRef] [PubMed]
- Amôr, N.G.; Santos, P.S.D.S.; Campanelli, A.P. The tumor microenvironment in SCC: Mechanisms and therapeutic opportunities. Front. Cell Dev. Biol. 2021, 9, 636544. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Brito, K.; Geiger, J.L.; Sussman, T.A.; Campbell, S.; Silver, N.L.; McGrail, D.; Ku, J.; Lamarre, E.; Yilmaz, E.; et al. Plasma circulating tumor DNA in patients with cutaneous squamous cell carcinoma: Initial experience. J. Clin. Oncol. 2024, 42 (Suppl. S16), e15034. [Google Scholar] [CrossRef]
- Lee, I.T.; Shen, C.H.; Tsai, F.C.; Chen, C.B.; Ma, K.S. Cancer-derived extracellular vesicles as biomarkers for cutaneous squamous cell carcinoma: A systematic review. Cancers 2022, 14, 5098. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Natarelli, N.; Boby, A.; Aflatooni, S.; Tran, J.T.; Diaz, M.J.; Taneja, K.; Forouzandeh, M. Regulatory miRNAs and lncRNAs in skin cancer: A narrative review. Life 2023, 13, 1696. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, J.; Wang, S.; Zhou, A.; Zhao, G.; Li, P. Roles of small extracellular vesicles in the development, diagnosis and possible treatment strategies for hepatocellular carcinoma (Review). Int. J. Oncol. 2022, 61, 91. [Google Scholar] [CrossRef] [PubMed]
- Lak, N.S.M.; van der Kooi, E.J.; Enciso-Martinez, A.; Lozano-Andrés, E.; Otto, C.; Wauben, M.H.M.; Tytgat, G.A.M. Extracellular vesicles: A new source of biomarkers in pediatric solid tumors? A systematic review. Front. Oncol. 2022, 12, 887210. [Google Scholar] [CrossRef] [PubMed]
- Hinestrosa, J.P.; Kurzrock, R.; Lewis, J.M.; Schork, N.J.; Schroeder, G.; Kamat, A.M.; Lowy, A.M.; Eskander, R.N.; Perrera, O.; Searson, D.; et al. Early-stage multi-cancer detection using an extracellular vesicle protein-based blood test. Commun. Med. 2022, 2, 29. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Risk Factor | Mechanism |
|---|---|
| Potentially avoidable occupational and recreational excessive exposure to ultraviolet radiation | Most prominent risk factor due to potent pro-mutagenic and pro-inflammatory/immunosuppressive activities of UVR [1,2,3,4,5,19,21,22,23,24]. |
| Radiation therapy | A cornerstone treatment of various types of malignancy, including cSCC, in which the potential risk is clearly outweighed by clinical benefit [25,26,28]. |
| Iatrogenic immunosuppression | Promotes considerable augmentation of the intrinsic immunosuppressive activity of solid malignancies [4,29,30]. |
| Smoking | Appears to augment the pro-mutagenic, immunosuppressive effects of UVR, but conflicting reports exist [33,34,35,38]. |
| Human papillomavirus infection | A double-edged sword in which cell-mediated immune responses to commensal skin HPV augment the survival of UVR-mutated keratinocytes. Progression of cSCC is, however, augmented by oncogenic strains of the virus in the setting of defective cell-mediated immunity [46,47,48,50,51,52]. |
| Human immunodeficiency virus | Progression of cSCC driven by defective cell-mediated immunity [53,54,55,56,57,58]. |
| Tattooing | Possible augmentative pro-tumorigenic activity, particularly in the setting of red-ink tattoos acting as a potential dermatotoxin [60,61,62,63]. |
| Mediators/Site | Mechanisms of Immune Evasion |
|---|---|
| TME | Influx of various types of immunosuppressive cells expressing ligands interactive with CCR5 (macrophages/monocytes, neutrophils, Tregs, Th2 cells, and invariant Ɣδ T cells) [79,80,81]. |
| Transforming growth factor-β1 |
|
| Adenosine | Potent broad-spectrum immunosuppressive and pro-mutagenic nucleoside produced by CD39-expressing infiltrating T cells [95,96,97,98,99,102]. |
| Down-regulation of expression of highly immunogenic neoepitopes | Results in attenuation of tumor recognition by CD8+ cytotoxic T cells [76]. |
| Circulation | Increased numbers of pro-tumorigenic CCR4hi Tregs [104]. |
| Biomarker | Significance in cSCC |
|---|---|
| TME-associated: | |
| Immune checkpoint molecules | |
| PD-1 | Expressed in locally advanced and metastatic cancer [124,131]. |
| LAG-3 | Expressed by a subset of TILs, specifically CD8+ T cells [124,125]. |
| Matrix metalloproteinases | |
| MMP-2 | Upregulated expression in cSCC [126]. |
| MMP-7 | Promotes the growth of cSCC by shedding HB-EGF, leading to the activation of EGFR [127]. |
| MMP-9 | Upregulated expression in cSCC [126]. |
| MMP-13 | Potential early detection of invasiveness and monitoring progression of cSCC [128]. |
| Complement components | |
| C1r and C1s detected by increased levels of mRNA | Elevated levels in cSCC cells linked to inhibition of ERK1/2 and PI3K signaling pathways, promoting apoptosis of cSCC cells, and suppressing vascularization and growth of cSCC xenografts in vivo [129]. |
| TILs | |
| Tregs | Larger Treg population in pre-metastatic lesions and a reduction in Treg number in invasive cSCC [130]. |
| CD8+ T cells | Lower numbers of CD8+ T cells due to TGF-β1 inducing the expression of T cell exhaustion markers such as PD-1, TIM-3, and CTLA-4 [132]. |
| Systemic | |
| Peripheral Tregs | Greater proportion of CCR4hi peripheral Tregs predictive of incident cSCC, particularly in individuals highly exposed to UV radiation [104]. |
| CTC | Can be quantified and analyzed for immune-marker expression [4]. |
| ctDNA/cfDNA | Selective tumor mutations can be detected [133]. |
| EVs | Comprise proteins, RNA, DNA, and lipids that are selectively incorporated from their cells of origin [134,135].Post-translational modification of desmoglein 2, a component of the desmosomal cell-to-cell adhesion structure may assist with characterization and treatment of cSCC [134]. |
| miRNAs | Influence cellular processes of apoptosis, invasion, proliferation, or migration by means of post-transcriptional regulation of gene expression [4,136]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, R.; Mkhize, N.M.; Kgokolo, M.M.C.; Steel, H.C.; Rossouw, T.M.; Anderson, L.; Rapoport, B.L. Current and Emerging Insights into the Causes, Immunopathogenesis, and Treatment of Cutaneous Squamous Cell Carcinoma. Cancers 2025, 17, 1702. https://doi.org/10.3390/cancers17101702
Anderson R, Mkhize NM, Kgokolo MMC, Steel HC, Rossouw TM, Anderson L, Rapoport BL. Current and Emerging Insights into the Causes, Immunopathogenesis, and Treatment of Cutaneous Squamous Cell Carcinoma. Cancers. 2025; 17(10):1702. https://doi.org/10.3390/cancers17101702
Chicago/Turabian StyleAnderson, Ronald, Nomzamo M. Mkhize, Mahlatse M. C. Kgokolo, Helen C. Steel, Theresa M. Rossouw, Lindsay Anderson, and Bernardo L. Rapoport. 2025. "Current and Emerging Insights into the Causes, Immunopathogenesis, and Treatment of Cutaneous Squamous Cell Carcinoma" Cancers 17, no. 10: 1702. https://doi.org/10.3390/cancers17101702
APA StyleAnderson, R., Mkhize, N. M., Kgokolo, M. M. C., Steel, H. C., Rossouw, T. M., Anderson, L., & Rapoport, B. L. (2025). Current and Emerging Insights into the Causes, Immunopathogenesis, and Treatment of Cutaneous Squamous Cell Carcinoma. Cancers, 17(10), 1702. https://doi.org/10.3390/cancers17101702