Utility of Clinical Next Generation Sequencing Tests in KIT/PDGFRA/SDH Wild-Type Gastrointestinal Stromal Tumors

 ,
,  , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Genomic Sequencing
2.3. Survival Analyses and Statistics
2.4. Data Availability
2.5. Ethics
3. Results
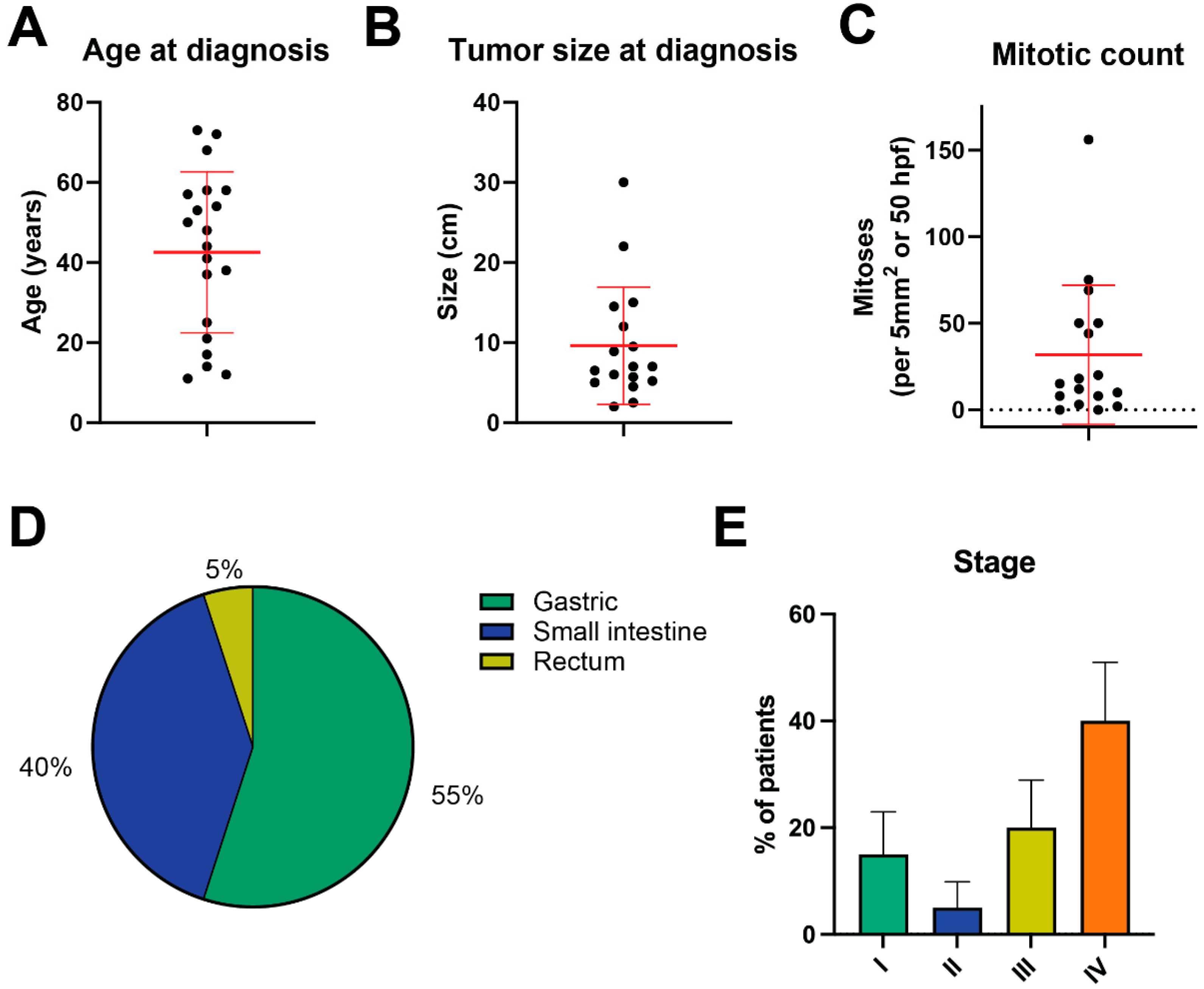
3.1. Clinical Characteristics of Triple-Negative GIST Cohort
3.2. The Genomics of Triple-Negative GIST
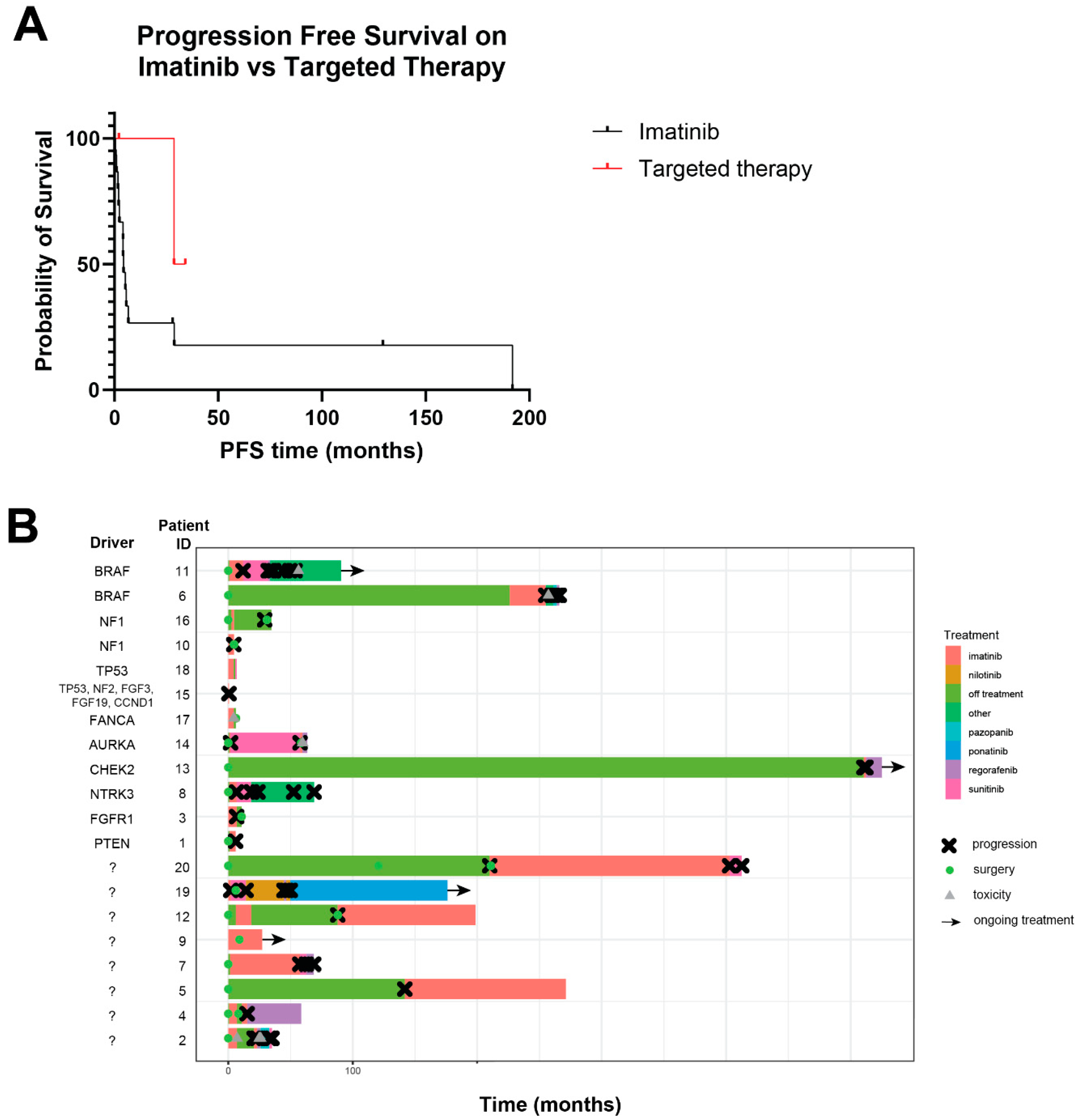
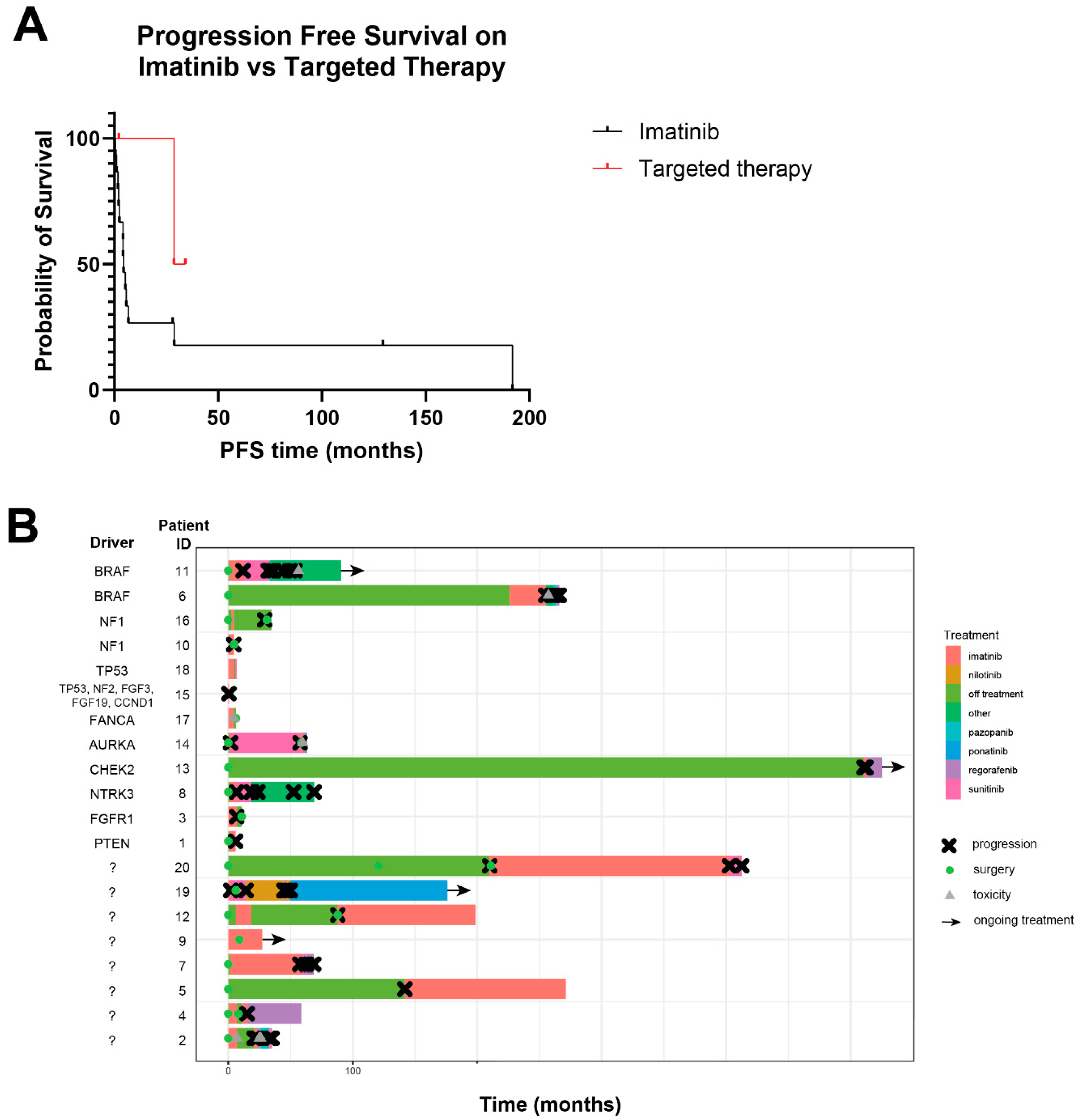
3.3. Response to Treatment
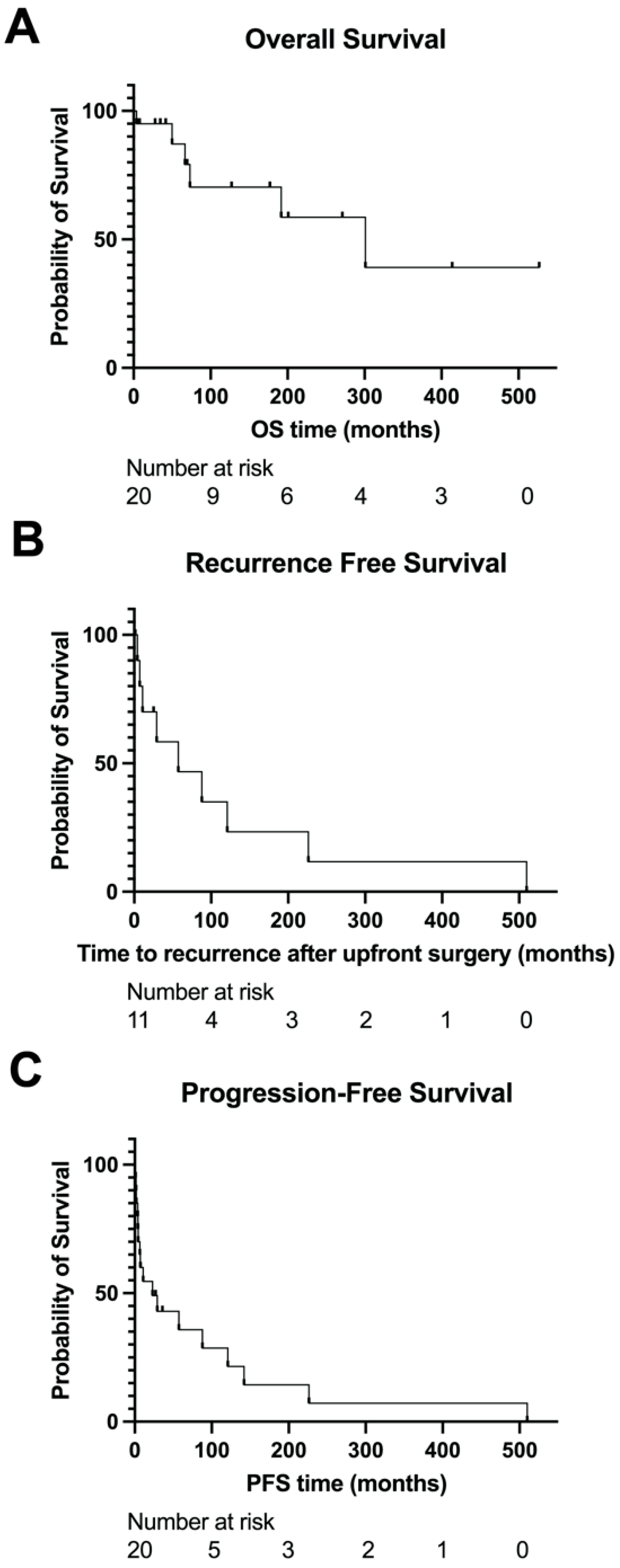
3.4. Survival Outcomes
3.5. Review of Literature on Triple-Negative GIST
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GIST | gastrointestinal stromal tumor |
| GOF | gain-of-function |
| LOF | loss-of-function |
| OS | overall survival |
| PFS | progression-free survival |
| SDH | succinate dehydrogenase |
| TKI | tyrosine kinase inhibitor |
References
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal Pacemaker Cell Tumor (GIPACT): Gastrointestinal Stromal Tumors Show Phenotypic Characteristics of the Interstitial Cells of Cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar]
- Patel, N.; Benipal, B. Incidence of Gastrointestinal Stromal Tumors in the United States from 2001–2015: A United States Cancer Statistics Analysis of 50 States. Cureus 2019, 11, e4120. [Google Scholar] [CrossRef]
- Corless, C.L.; Fletcher, J.A.; Heinrich, M.C. Biology of Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2004, 22, 3813–3825. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients with Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.M.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of Kinase Genotype and Clinical Outcome in the North American Intergroup Phase III Trial of Imatinib Mesylate for Treatment of Advanced Gastrointestinal Stromal Tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.-Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT Mutations and Dose Selection for Imatinib in Patients with Advanced Gastrointestinal Stromal Tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Pappo, A.S.; Janeway, K.A. Pediatric Gastrointestinal Stromal Tumors. Hematol. Oncol. Clin. N. Am. 2009, 23, 15–34. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Nannini, M.; Corless, C.L.; Heinrich, M.C. Quadruple Wild-Type (WT) GIST: Defining the Subset of GIST That Lacks Abnormalities of KIT, PDGFRA, SDH, or RAS Signaling Pathways. Cancer Med. 2015, 4, 101–103. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate Dehydrogenase Deficiency in Pediatric and Adult Gastrointestinal Stromal Tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in Succinate Dehydrogenase in Gastrointestinal Stromal Tumors Lacking KIT and PDGFRA Mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Mason, E.F.; Hornick, J.L. Conventional Risk Stratification Fails to Predict Progression of Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors: A Clinicopathologic Study of 76 Cases. Am. J. Surg. Pathol. 2016, 40, 1616–1621. [Google Scholar] [CrossRef]
- Gill, A.J.; Chou, A.; Vilain, R.; Clarkson, A.; Lui, M.; Jin, R.; Tobias, V.; Samra, J.; Goldstein, D.; Smith, C.; et al. Immunohistochemistry for SDHB Divides Gastrointestinal Stromal Tumors (GISTs) into 2 Distinct Types. Am. J. Surg. Pathol. 2010, 34, 636–644. [Google Scholar] [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs Cycle Intermediates and Over-Expression of HIF1alpha in Tumours Which Result from Germline FH and SDH Mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. International Carney Triad and Carney-Stratakis Syndrome Consortium Familial Gastrointestinal Stromal Tumors and Germ-Line Mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A. The Triad of Paragangliomas, Gastric Stromal Tumours and Pulmonary Chondromas (Carney Triad), and the Dyad of Paragangliomas and Gastric Stromal Sarcomas (Carney-Stratakis Syndrome): Molecular Genetics and Clinical Implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and Molecular Genetics of Patients with the Carney-Stratakis Syndrome and Germline Mutations of the Genes Coding for the Succinate Dehydrogenase Subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar] [CrossRef]
- Killian, J.K.; Miettinen, M.; Walker, R.L.; Wang, Y.; Zhu, Y.J.; Waterfall, J.J.; Noyes, N.; Retnakumar, P.; Yang, Z.; Smith, W.I.; et al. Recurrent Epimutation of SDHC in Gastrointestinal Stromal Tumors. Sci. Transl. Med. 2014, 6, 268ra177. [Google Scholar] [CrossRef]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Astolfi, A.; Indio, V.; Nannini, M.; Saponara, M.; Schipani, A.; De Leo, A.; Altimari, A.; Vincenzi, B.; Comandini, D.; Grignani, G.; et al. Targeted Deep Sequencing Uncovers Cryptic KIT Mutations in KIT/PDGFRA/SDH/RAS-P Wild-Type GIST. Front. Oncol. 2020, 10, 504. [Google Scholar] [CrossRef]
- Trent, J.C.; Gomez-Peregrina, D.; Elliott, A.; Boikos, S.A.; Florou, V. Multi-Omic Characterization of Gastrointestinal Stromal Tumor (GIST) in a Large Real-World Patient Cohort. J. Clin. Oncol. 2023, 41, 11522. [Google Scholar] [CrossRef]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF Mutations in Imatinib-Naive and Imatinib-Resistant Gastrointestinal Stromal Tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF Mutations and Phosphatidylinositol-3-Kinase Pathway Gene Alterations in Gastrointestinal Stromal Tumors (GIST). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Rossi, S.; Sbaraglia, M.; Dell’Orto, M.C.; Gasparotto, D.; Cacciatore, M.; Boscato, E.; Carraro, V.; Toffolatti, L.; Gallina, G.; Niero, M.; et al. Concomitant KIT/BRAF and PDGFRA/BRAF Mutations Are Rare Events in Gastrointestinal Stromal Tumors. Oncotarget 2016, 7, 30109–30118. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Capuzzi, S.J.; Hoang, Y.; Chien, J.; Godwin, A.K.; von Mehren, M. Somatic Loss of Function Mutations in Neurofibromin 1 and MYC Associated Factor X Genes Identified by Exome-Wide Sequencing in a Wild-Type GIST Case. BMC Cancer 2015, 15, 887. [Google Scholar] [CrossRef]
- Luthra, R.; Patel, K.P.; Routbort, M.J.; Broaddus, R.R.; Yau, J.; Simien, C.; Chen, W.; Hatfield, D.Z.; Medeiros, L.J.; Singh, R.R. A Targeted High-Throughput Next-Generation Sequencing Panel for Clinical Screening of Mutations, Gene Amplifications, and Fusions in Solid Tumors. J. Mol. Diagn. 2017, 19, 255–264. [Google Scholar] [CrossRef]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical Validation of a Next-Generation Sequencing Screen for Mutational Hotspots in 46 Cancer-Related Genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef]
- Chen, K.; Meric-Bernstam, F.; Zhao, H.; Zhang, Q.; Ezzeddine, N.; Tang, L.-Y.; Qi, Y.; Mao, Y.; Chen, T.; Chong, Z.; et al. Clinical Actionability Enhanced through Deep Targeted Sequencing of Solid Tumors. Clin. Chem. 2015, 61, 544–553. [Google Scholar] [CrossRef]
- Hong, D.S.; Bauer, T.M.; Lee, J.J.; Dowlati, A.; Brose, M.S.; Farago, A.F.; Taylor, M.; Shaw, A.T.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in Adult Patients with Solid Tumours: A Multi-Centre, Open-Label, Phase I Dose-Escalation Study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef]
- Yang, D.Y.; Wang, X.; Yuan, W.J.; Chen, Z.H. Metastatic Pattern and Prognosis of Gastrointestinal Stromal Tumor (GIST): A SEER-Based Analysis. Clin. Transl. Oncol. 2019, 21, 1654–1662. [Google Scholar] [CrossRef]
- Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. KRAS and BRAF Mutations Predict Primary Resistance to Imatinib in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2012, 18, 1769–1776. [Google Scholar] [CrossRef]
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF Mutant Gastrointestinal Stromal Tumor: First Report of Regression with BRAF Inhibitor Dabrafenib (GSK2118436) and Whole Exomic Sequencing for Analysis of Acquired Resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef]
- Zheng, S.; Huang, K.; Pan, Y.; Zhou, Y.; Pan, S.; Li, X.; Jia, J.; Zheng, X.; Tao, D. KIT and BRAF Heterogeneous Mutations in Gastrointestinal Stromal Tumors after Secondary Imatinib Resistance. Gastric Cancer 2015, 18, 796–802. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF Mutations Are Alternative Early Molecular Events in a Subset of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.-F.; Bringuier, P.-P.; Scoazec, J.-Y.; Coindre, J.-M. BRAF Mutation Status in Gastrointestinal Stromal Tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef]
- Nannini, M.; Astolfi, A.; Urbini, M.; Indio, V.; Santini, D.; Heinrich, M.C.; Corless, C.L.; Ceccarelli, C.; Saponara, M.; Mandrioli, A.; et al. Integrated Genomic Study of Quadruple-WT GIST (KIT/PDGFRA/SDH/RAS Pathway Wild-Type GIST). BMC Cancer 2014, 14, 685. [Google Scholar] [CrossRef]
- Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int. J. Mol. Sci. 2020, 21, 3313. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.-M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 Actionable Alterations in “Wild-Type” Gastrointestinal Stromal Tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef]
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast Growth Factor Receptors (FGFRs) and Noncanonical Partners in Cancer Signaling. Cells 2021, 10, 1201. [Google Scholar] [CrossRef]
- Urbini, M.; Indio, V.; Tarantino, G.; Ravegnini, G.; Angelini, S.; Nannini, M.; Saponara, M.; Santini, D.; Ceccarelli, C.; Fiorentino, M.; et al. Gain of FGF4 Is a Frequent Event in KIT/PDGFRA/SDH/RAS-P WT GIST. Genes Chromosomes Cancer 2019, 58, 636–642. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered Chromosomal Topology Drives Oncogenic Programs in SDH-Deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-Term Follow-up Results of the Multicenter Phase II Trial of Regorafenib in Patients with Metastatic and/or Unresectable GI Stromal Tumor after Failure of Standard Tyrosine Kinase Inhibitor Therapy. Ann. Oncol. 2016, 27, 1794–1799. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome Sequencing Identifies ETV6-NTRK3 as a Gene Fusion Involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Wang, S.; Sun, R.Z.; Han, Q.; Wang, S.Y.; Wang, E.H.; Yang, L. Genomic Study of Chinese Quadruple-Negative GISTs Using next-Generation Sequencing Technology. Appl. Immunohistochem. Mol. Morphol. 2021, 29, 34–41. [Google Scholar] [CrossRef]
- Call, J.W.; Wang, Y.; Montoya, D.; Scherzer, N.J.; Heinrich, M.C. Survival in Advanced GIST Has Improved over Time and Correlates with Increased Access to Post-Imatinib Tyrosine Kinase Inhibitors: Results from Life Raft Group Registry. Clin. Sarcoma Res. 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Li, S.-H.; Yu, S.-C.; Chou, F.-F.; Tzeng, C.-C.; Hu, T.-H.; Uen, Y.-H.; Tian, Y.-F.; Wang, Y.-H.; Fang, F.-M.; et al. Homozygous Deletion of MTAP Gene as a Poor Prognosticator in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2009, 15, 6963–6972. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Alhalabi, O.; Chen, J.; Zhang, Y.; Lu, Y.; Wang, Q.; Ramachandran, S.; Tidwell, R.S.; Han, G.; Yan, X.; Meng, J.; et al. MTAP Deficiency Creates an Exploitable Target for Antifolate Therapy in 9p21-Loss Cancers. Nat. Commun. 2022, 13, 1797. [Google Scholar] [CrossRef]
- Liu, T.-T.; Li, C.-F.; Tan, K.-T.; Jan, Y.-H.; Lee, P.-H.; Huang, C.-H.; Yu, S.-C.; Tsao, C.-F.; Wang, J.-C.; Huang, H.-Y. Characterization of Aberrations in DNA Damage Repair Pathways in Gastrointestinal Stromal Tumors: The Clinicopathologic Relevance of γH2AX and 53BP1 in Correlation with Heterozygous Deletions of CHEK2, BRCA2, and RB1. Cancers 2022, 14, 1787. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age at Diagnosis | Primary Site | Stage at Diagnosis (AJCC 8th) | Site of Recurrence/Metastasis | Imatinib Received in Which Setting? | PFS on Imatinib (Months) | Time to Relapse from First Therapy (Months) | Driver | Sequencing Platform |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 41 | Gastric | IIIB | Local | Adjuvant | NA | 4.1 | PTEN deletion | Neotype, Boston Gene |
| 2 | 68 | Small intestine | IV | Pelvis | Adjuvant | NA | 2.4 | ? | MDA |
| 3 | 38 | Small intestine | IA | NA | Neoadjuvant | 6.7 | NA | FGFR1 K654E | MDA |
| 4 | 21 | Gastric | IB | Local, liver | Neoadjuvant and adjuvant | 4.1 | 7.1 | ? | Boston Gene, Foundation One, MDA |
| 5 | 17 | Gastric | IV | Liver | Recurrent/metastatic | 129.4 | 141.8 | ? | MDA |
| 6 | 12 | Gastric | ? | Right psoas, retroperitoneum | Recurrent/metastatic | 28.8 | 226.2 | BRAF V600E | MDA |
| 7 | 57 | Small intestine | IB | Liver | Adjuvant | NA | 57.4 | ? | MDA |
| 8 | 44 | Gastric | IV | Local, liver, spleen | Recurrent/metastatic | 5.7 | 6.6 | ETV6-NTRK3 fusion | Foundation One, MDA |
| 9 | 37 | Gastric | IV | Omentum | Neoadjuvant | 28 | NA | ? | OSI |
| 10 | 72 | Small intestine | IV | Pancreas | Neoadjuvant | 5.2 | 4.4 | NF1 deletion | MDA |
| 11 | 58 | Small intestine | IIIB | Bladder, abdomen | Adjuvant | NA | 10.8 | BRAF V600E | MDA |
| 12 | 48 | Small intestine | IIIA | Local | Adjuvant | NA | 88 | ? | MDA |
| 13 | 14 | Gastric | ? | Liver, lung | Recurrent/metastatic | 1.7 | 509.9 | CHEK2 frameshift | Foundation One |
| 14 | 73 | Small intestine | IV | Omentum, lymph nodes | Recurrent/metastatic | 1 | 1 | AURKA-CSTF1 fusion | Foundation One |
| 15 | 50 | Gastric | ? | Local, liver, pancreas, periportal region, duodenum, omentum | Neoadjuvant | 0.5 | 2.2 | NF2 splice site mutation, FGF3/FGF19 amplification, CCND1 amplification, TP53 frameshift | MDA |
| 16 | 58 | Gastric | II | Local | Adjuvant | NA | 29.2 | NF1 splice site mutation | MDA |
| 17 | 54 | Rectum | IIIA | Peritoneum, ovary | Neoadjuvant | 4.4 | 4.4 | FANCA loss | Boston Gene |
| 18 | 53 | Small intestine | IV | Liver | Recurrent/metastatic | 4.2 | 4.2 | TP53 missense and deletion | Caris, Boston Gene |
| 19 | 25 | Gastric | IV | Liver | Recurrent/metastatic | 2.0 | 2.0 | ? | OSI panel |
| 20 | 11 | Gastric | ? | Liver, omentum | Recurrent/metastatic | 191.8 | 120.7 | ? | Endeavor, Boston Gene |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denu, R.A.; Joseph, C.P.; Urquiola, E.S.; Byrd, P.S.; Yang, R.K.; Ratan, R.; Zarzour, M.A.; Conley, A.P.; Araujo, D.M.; Ravi, V.; et al. Utility of Clinical Next Generation Sequencing Tests in KIT/PDGFRA/SDH Wild-Type Gastrointestinal Stromal Tumors. Cancers 2024, 16, 1707. https://doi.org/10.3390/cancers16091707
Denu RA, Joseph CP, Urquiola ES, Byrd PS, Yang RK, Ratan R, Zarzour MA, Conley AP, Araujo DM, Ravi V, et al. Utility of Clinical Next Generation Sequencing Tests in KIT/PDGFRA/SDH Wild-Type Gastrointestinal Stromal Tumors. Cancers. 2024; 16(9):1707. https://doi.org/10.3390/cancers16091707
Chicago/Turabian StyleDenu, Ryan A., Cissimol P. Joseph, Elizabeth S. Urquiola, Precious S. Byrd, Richard K. Yang, Ravin Ratan, Maria Alejandra Zarzour, Anthony P. Conley, Dejka M. Araujo, Vinod Ravi, and et al. 2024. "Utility of Clinical Next Generation Sequencing Tests in KIT/PDGFRA/SDH Wild-Type Gastrointestinal Stromal Tumors" Cancers 16, no. 9: 1707. https://doi.org/10.3390/cancers16091707
APA StyleDenu, R. A., Joseph, C. P., Urquiola, E. S., Byrd, P. S., Yang, R. K., Ratan, R., Zarzour, M. A., Conley, A. P., Araujo, D. M., Ravi, V., Nassif Haddad, E. F., Nakazawa, M. S., Patel, S., Wang, W.-L., Lazar, A. J., & Somaiah, N. (2024). Utility of Clinical Next Generation Sequencing Tests in KIT/PDGFRA/SDH Wild-Type Gastrointestinal Stromal Tumors. Cancers, 16(9), 1707. https://doi.org/10.3390/cancers16091707