
HotSPOT: A Computational Tool to Design Targeted Sequencing Panels to Assess Early Photocarcinogenesis

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
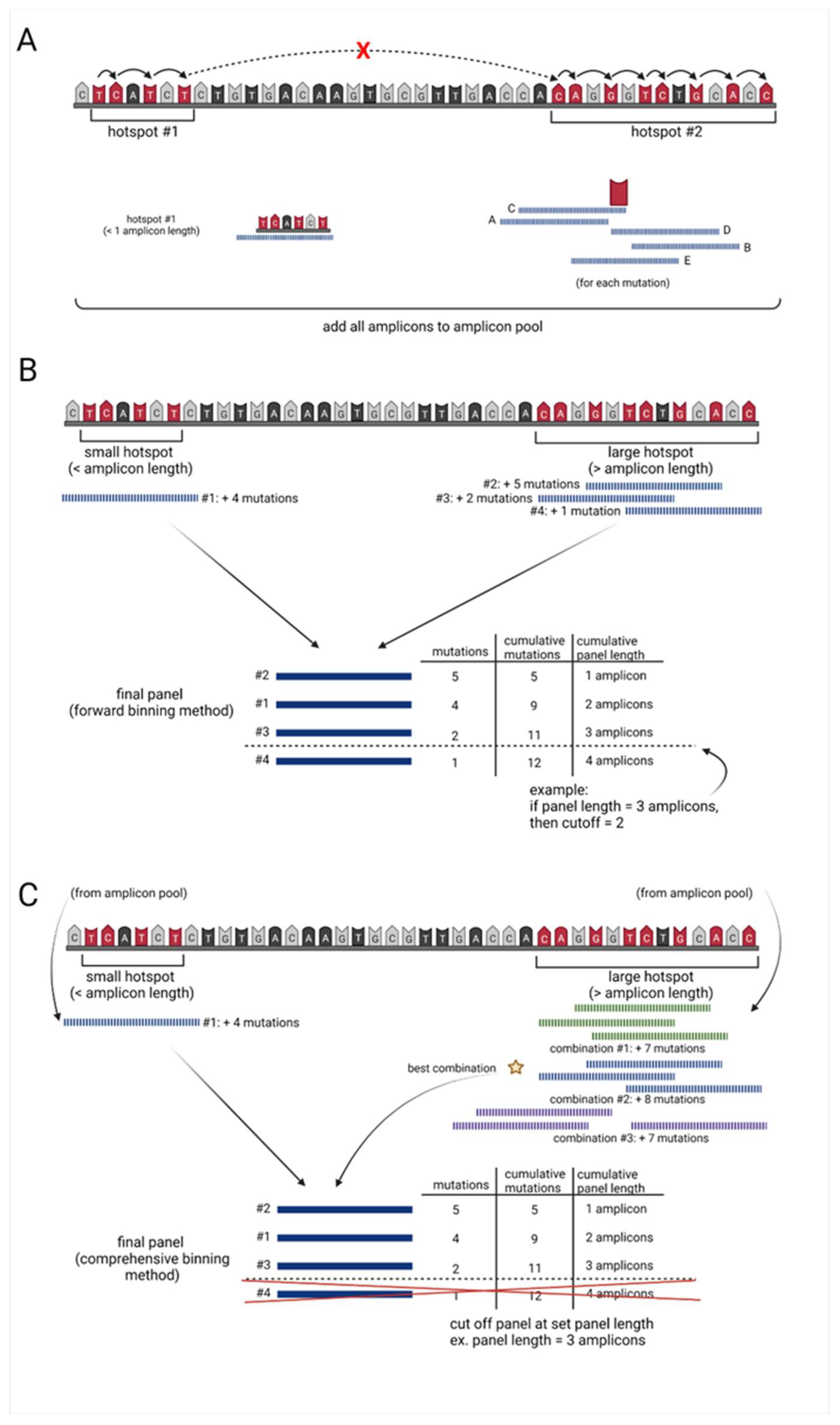
2.2. Sequencing Panel Identifier Input Data and Amplicon Generator
- 1.
- Starting at the mutation with the lowest chromosomal position (primary mutation), using a modified rank and recovery system, the algorithm searches for the closest neighboring mutation.
- 2.
- If the neighboring mutation is less than one amplicon in distance away from the primary mutation, the neighboring mutation is included within the “hotspot” region.
- This rank and recovery system is repeated, integrating mutations into the “hotspot region” until the neighboring mutation is greater than or equal to the length of one amplicon in distance, from the primary mutation (Figure 1A, Supplementary Figure S1A).
- Once neighboring mutations equal or exceed one amplicon in distance from the primary mutation, incorporation into the “hotspot region” halts incorporation.
- 3.
- For hotspots within the one amplicon range, from the lowest to highest mutation location, this area is covered by a single amplicon and added to an amplicon pool with a unique ID.
- The center of these single amplicons is then defined by the weighted distribution of mutations.
- 4.
- For all hotspots larger than one amplicon, the algorithm examines 5 potential amplicons at each covered mutation in the hotspot:
- one amplicon directly upstream of the primary mutation
- one amplicon directly downstream of the primary mutation
- one amplicon including the mutation at the end of the read and base pairs (amplicon length 1) upstream
- one amplicon including the mutation at the beginning of the read and base pairs (amplicon length 1) downstream
- one amplicon with the mutation directly in the center (Figure 1A, Supplementary Figure S1B).
- 5.
- All amplicons generated for each hotspot region of interest are assigned a unique ID and added to the amplicon pool (Figure 1A, Supplementary Figure S1C).
2.3. Forward Selection Sequencing Panel Identifier (Optimal computation time)
- 6.
- Amplicons covering hotspots less than or equal to one amplicon in length are added to the final sequencing panel dataset.
- 7.
- For amplicons covering larger hotspot regions, the algorithm uses a forward selection method to determine the optimal combination of amplicons to use in the sequencing panel:
- the algorithm first identifies the amplicon containing the highest number of mutations
- the algorithm then identifies the next amplicon, which contains the highest number of new mutations
- this process continues until all mutations are covered by at least one amplicon (Figure 1B, Supplementary Figure S1D)
- 8.
- Each of these amplicons are then added to the final sequencing panel, with their own unique IDs.
- 9.
- All amplicons in the final sequencing panel are ranked from highest to lowest based on the number of mutations they cover.
- 10.
- The algorithm then calculates the cumulative base-pair length and the cumulative mutations covered by each amplicon.
- 11.
- Dependent on the desired length of the targeted panel, a cutoff may be applied to remove all amplicons which fall below a set cumulative length (Figure 1B, Supplementary Figure S1E).
2.4. Comprehensive Selection Sequencing Panel Identifier (Optimal Mutation Capture)
- 12.
- To conserve computational power, the forward selection sequencing panel identifier is run to determine the lowest number of mutations per amplicon (mutation frequency) that need to be included in the predetermined length sequencing panel (Figure 1B, Supplementary Figure S1A–E).
- any amplicon generated by the algorithm which is less than this threshold value will be removed (Supplementary Figure S1F).
- 13.
- For the feasible exhaustive selection of amplicon combinations covering hotspot areas larger than the predefined number of amplicons in length, the algorithm breaks these large regions into multiple smaller regions.
- The amplicons covering these regions are pulled from the amplicon pool based on their unique IDs.
- 14.
- The algorithm finds both the minimum number of amplicons overlap and all positions with this value and identifies the region with the longest continuous spot of minimum value.
- The region is split at the center of this longest continuous minimum post values and continues the splitting process until all smaller regions are less than the “n” number amplicon length set by the user (Supplementary Figure S1G).
- As this set number of amplicons decreases, the computation time required also often decreases.
- 15.
- All amplicons contained in these bins are added back to the amplicon pool based on a new unique ID.
- 16.
- Amplicons covering hotspots less than or equal to one amplicon length are added to the final sequencing panel dataset.
- 17.
- To determine the optimal combination of amplicons for each region, the number of amplicons necessary for full coverage of the bin is calculated.
- 18.
- A list is generated of every possible combination of n, number of amplicons, needed. For each combination of amplicons:
- amplicons that would not meet the threshold of unique mutations are filtered out, and the number of all mutations captured by these amplicons is calculated.
- the combination of amplicons that yields the highest number of mutations is added to the final sequencing panel (Figure 1C, Supplementary Figure S1H).
- 19.
- All amplicons in the final sequencing panel are ranked from highest to lowest based on the number of mutations they cover.
- 20.
- All amplicons capturing the number of mutations equal to the cutoff are further ranked to favor amplicons that have mutations closer in location to the center of the amplicon.
- 21.
- Cumulative base-pair length and cumulative mutations covered by each amplicon are calculated.
- Depending on the desired length of the targeted panel, a cutoff may be applied to remove all amplicons which fall below a set cumulative length (Figure 1C, Supplementary Figure S1).
2.5. Calculation of Mutation Capture Efficacy
2.6. Computational Development of hotSPOT Algorithm and RShiny Web Application
2.7. Statistical Analysis
2.8. Calculation of Optimal Sample Size for hotSPOT Panel Design
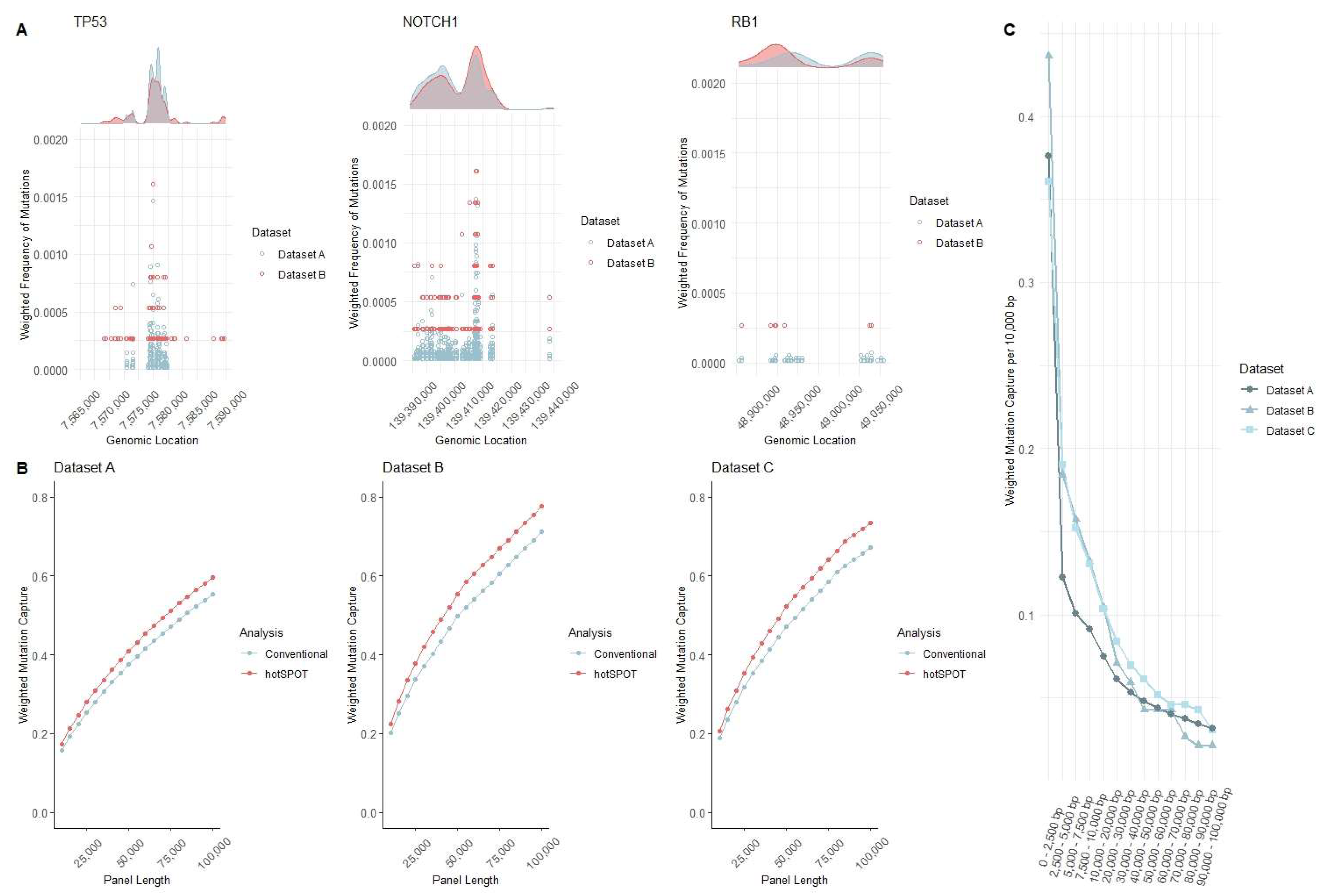
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martincorena, I. Somatic mutation and clonal expansions in human tissues. Genome Med. 2019, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Hernando, B.; Dietzen, M.; Parra, G.; Gil-Barrachina, M.; Pitarch, G.; Mahiques, L.; Valcuende-Cavero, F.; McGranahan, N.; Martinez-Cadenas, C. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 2021, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.C.; King, C.; Bryant, C.; Hall, M.W.J.; Sood, R.; Ong, S.H.; Earp, E.; Fernandez-Antoran, D.; Koeppel, J.; Dentro, S.C.; et al. Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site. Cancer Discov. 2021, 11, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, M.; Fouque, L.; Pelletier, M.; Chabert, R.; Pinacolo, S.; Restellini, L.; Cucumel, K. Blue light: Friend or foe? J. Photochem. Photobiol. B 2020, 212, 112026. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Gordenin, D.A. Clusters of Multiple Mutations: Incidence and Molecular Mechanisms. Annu. Rev. Genet. 2015, 49, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Christensen, S.R.; Fitzgerald, M.E.; Graham, J.; Hutson, N.D.; Zhang, C.; Huang, Z.; Hu, Q.; Zhan, F.; Xie, J.; et al. Ultradeep sequencing differentiates patterns of skin clonal mutations associated with sun-exposure status and skin cancer burden. Sci. Adv. 2021, 7, eabd7703. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]
- Cario, C.L.; Chen, E.; Leong, L.; Emami, N.C.; Lopez, K.; Tenggara, I.; Simko, J.P.; Friedlander, T.W.; Li, P.S.; Paris, P.L.; et al. A machine learning approach to optimizing cell-free DNA sequencing panels: With an application to prostate cancer. BMC Cancer 2020, 20, 820. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ledesma, E.; Flores, D.; Trevino, V. Computational methods for detecting cancer hotspots. Comput. Struct. Biotechnol. J. 2020, 18, 3567–3576. [Google Scholar] [CrossRef] [PubMed]
- Trevino, V. HotSpotAnnotations-a database for hotspot mutations and annotations in cancer. Database 2020, 2020, baaa025. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hua, Y.; Gu, S.; Lv, L.; Li, X.; Chen, P.; Dai, P.; Hu, Y.; Liu, A.; Li, J. kataegis: An R package for identification and visualization of the genomic localized hypermutation regions using high-throughput sequencing. BMC Genom. 2021, 22, 440. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xue, H. Genetic-variant hotspots and hotspot clusters in the human genome facilitating adaptation while increasing instability. Hum. Genom. 2021, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Woodman, S.E. HotSpotter: Efficient visualization of driver mutations. BMC Genom. 2014, 15, 1044. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing, R version 4.1.1; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Hadley Wickham, R.F.; Henry, L.; Müller, L. dplyr: A Grammar of Data Manipulation, R package version 1.0.7; CRAN team: Vienna, Austria, 2021. [Google Scholar]
- Brown, C. hash: Full Feature Implementation of Hash/Associated Arrays/Dictionaries, R package version 2.2.6.1; CRAN team: Vienna, Austria, 2019. [Google Scholar]
- Ren, K. rlist: A Toolbox for Non-Tabular Data Manipulation, R package version 0.4.6.2; CRAN team: Vienna, Austria, 2021. [Google Scholar]
- Bengtsson, H.R. utils: Various Programming Utilities, R package version 2.10.1; CRAN team: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; CRAN team: Vienna, Austria, 2016. [Google Scholar]
- Dean Attali, C.B. ggExtra: Add Marginal Histograms to ‘ggplot2’, and More ‘ggplot2’ Enhancements, R package version 0.9; CRAN team: Vienna, Austria, 2019. [Google Scholar]
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots, R package version 0.4.0; CRAN team: Vienna, Austria, 2020. [Google Scholar]
- Chang, W.; Cheng, J.; Allaire, J.; Sievert, C.; Schloerke, B.; Xie, Y.; Allen, J.; McPherson, J.; Dipert, A.; Borges, B. shiny: Web Application Framework for R; CRAN team: Vienna, Austria, 2021. [Google Scholar]
- Chang, W.; Ribeiro, B.B. shinydashboard: Create Dashboards with ‘Shiny’; CRAN team: Vienna, Austria, 2021. [Google Scholar]
- Sali, A.; Attali, D. shinycssloaders: Add Loading Animations to a ‘shiny’ Output While It’s Recalculating; CRAN team: Vienna, Austria, 2020. [Google Scholar]
- Lilovski, N. dashboardthemes: Customise the Appearance of ‘shinydashboard’ Applications Using Themes; CRAN team: Vienna, Austria, 2021. [Google Scholar]
- Xie, Y.; Cheng, J.; Tan, X. DT: A Wrapper of the JavaScript Library ‘DataTables’; CRAN team: Vienna, Austria, 2022. [Google Scholar]
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall/CRC: Boca Raton, FL, USA, 2020. [Google Scholar]
- Juul, R.I.; Nielsen, M.M.; Juul, M.; Feuerbach, L.; Pedersen, J.S. The landscape and driver potential of site-specific hotspots across cancer genomes. NPJ Genom. Med. 2021, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Nesta, A.V.; Tafur, D.; Beck, C.R. Hotspots of Human Mutation. Trends. Genet. 2021, 37, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Han, L.; Mehta, S.; Knight, J.; Zhao, D.; Palmatier, M.A.; Kornacker, K.; Brash, D.E. Genomic sites hypersensitive to ultraviolet radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 24196–24205. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Dataset | Source | Samples Sequenced | Average Sequencing Depth | Original Sequencing Panel Size | Sample Type | Panel Design |
|---|---|---|---|---|---|---|---|
| “Dataset A” | Test/Training Datasets | Fowler et al. [3] | 1261 | 690x | 0.39 Mb | Normal Epidermis (Head, forearm, leg, trunk, abdomen) | 74 Cancer- related genes |
| “Dataset B” | Validation Dataset #1 | Martincorena et al. [4] | 234 | 374x | 0.67 Mb | Normal Epidermis (Eyelid) | 74 Cancer- related genes |
| “Dataset C” | Validation Dataset #2 | Hernando et al. [2] | 123 | 923x | 0.32 Mb | Normal Epidermis (Back, chest, leg, upper arm, neck, face, hands) | 46 genes frequently mutated in skin cancer |
| Test/Training Datasets | Pickering et al. [6] | 39 | 115x | - | Tumor | WES | |
| Validation Dataset | Inman et al. [9] | 36 | 54x | - | Tumor | WES |
| ALGORITHM | FGFR3 | FLG2 | MLL2 | MUC17 | ||||
|---|---|---|---|---|---|---|---|---|
| Time | Count | Time | Count | Time | Count | Time | Count | |
| hotSPOT “Forward” | 0.696 | 45 | 0.864 | 14 | 0.888 | 27 | 1.238 | 45 |
| hotSPOT “Comprehensive” | 3.594 | 45 | 6.232 | 14 | 3.344 | 27 | 8.642 | 45 |
| Exhaustive | 34.455 | 45 | 298.415 | 14 | 10.668 | 27 | 569.354 | 45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grant, S.R.; Rosario, S.R.; Patentreger, A.D.; Shary, N.; Fitzgerald, M.E.; Singh, P.K.; Foster, B.A.; Huss, W.J.; Wei, L.; Paragh, G. HotSPOT: A Computational Tool to Design Targeted Sequencing Panels to Assess Early Photocarcinogenesis. Cancers 2023, 15, 1612. https://doi.org/10.3390/cancers15051612
Grant SR, Rosario SR, Patentreger AD, Shary N, Fitzgerald ME, Singh PK, Foster BA, Huss WJ, Wei L, Paragh G. HotSPOT: A Computational Tool to Design Targeted Sequencing Panels to Assess Early Photocarcinogenesis. Cancers. 2023; 15(5):1612. https://doi.org/10.3390/cancers15051612
Chicago/Turabian StyleGrant, Sydney R., Spencer R. Rosario, Andrew D. Patentreger, Nico Shary, Megan E. Fitzgerald, Prashant K. Singh, Barbara A. Foster, Wendy J. Huss, Lei Wei, and Gyorgy Paragh. 2023. "HotSPOT: A Computational Tool to Design Targeted Sequencing Panels to Assess Early Photocarcinogenesis" Cancers 15, no. 5: 1612. https://doi.org/10.3390/cancers15051612
APA StyleGrant, S. R., Rosario, S. R., Patentreger, A. D., Shary, N., Fitzgerald, M. E., Singh, P. K., Foster, B. A., Huss, W. J., Wei, L., & Paragh, G. (2023). HotSPOT: A Computational Tool to Design Targeted Sequencing Panels to Assess Early Photocarcinogenesis. Cancers, 15(5), 1612. https://doi.org/10.3390/cancers15051612