LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Cell Culture
2.2. CRISPR/Cas9-Mediated Inactivation of LINC01021
2.3. Ectopic Expression of LINC01021
2.4. RNA Affinity Purification (RAP)
2.5. Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis
2.6. RNA isolation and qRT-PCR Analysis
2.7. Western Blot Analysis
2.8. Cell cycle Analysis by Propidium Iodide (PI) Staining
2.9. Bioinformatic Analysis of RNA-Seq Data
2.10. Statistical Analysis
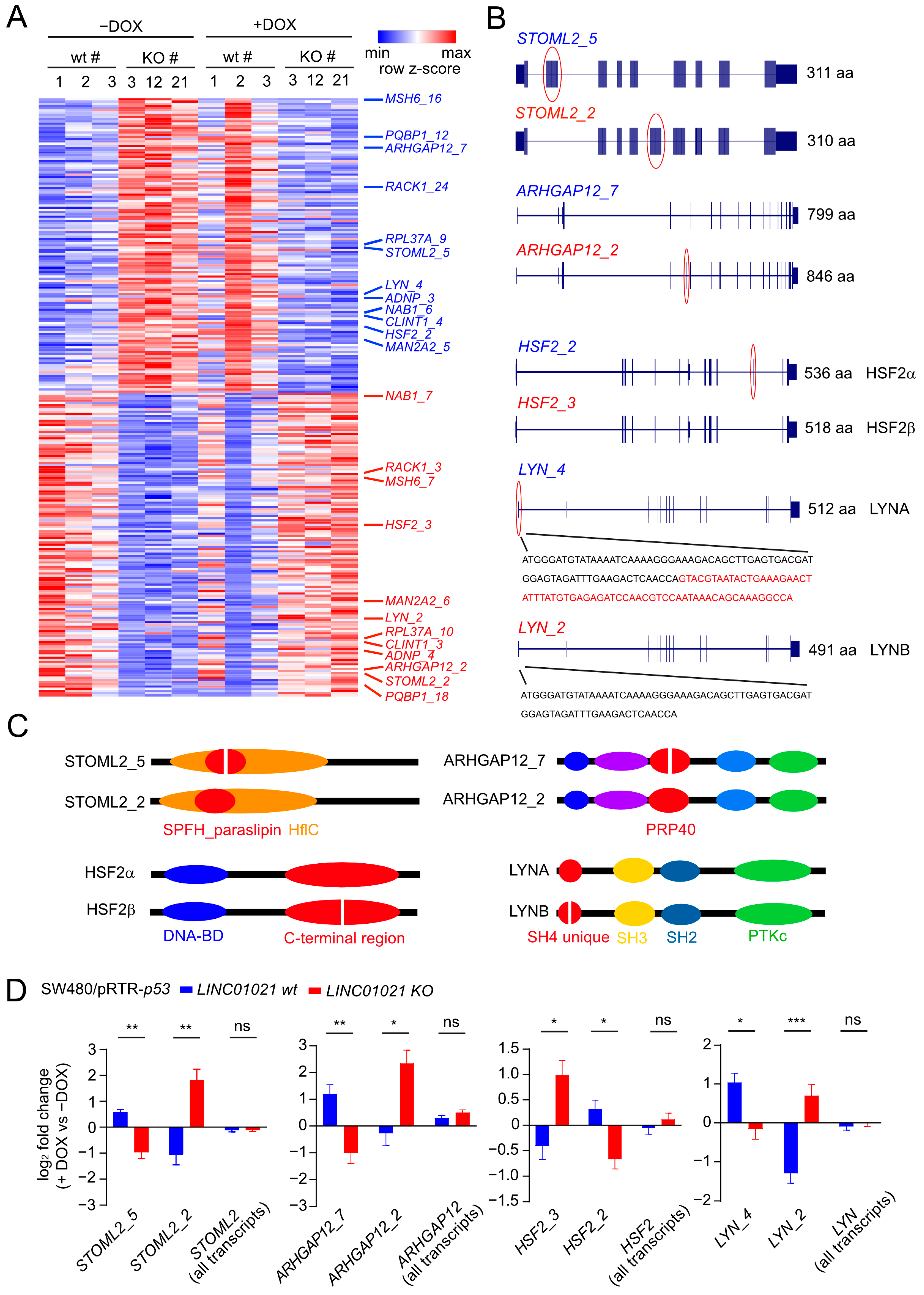
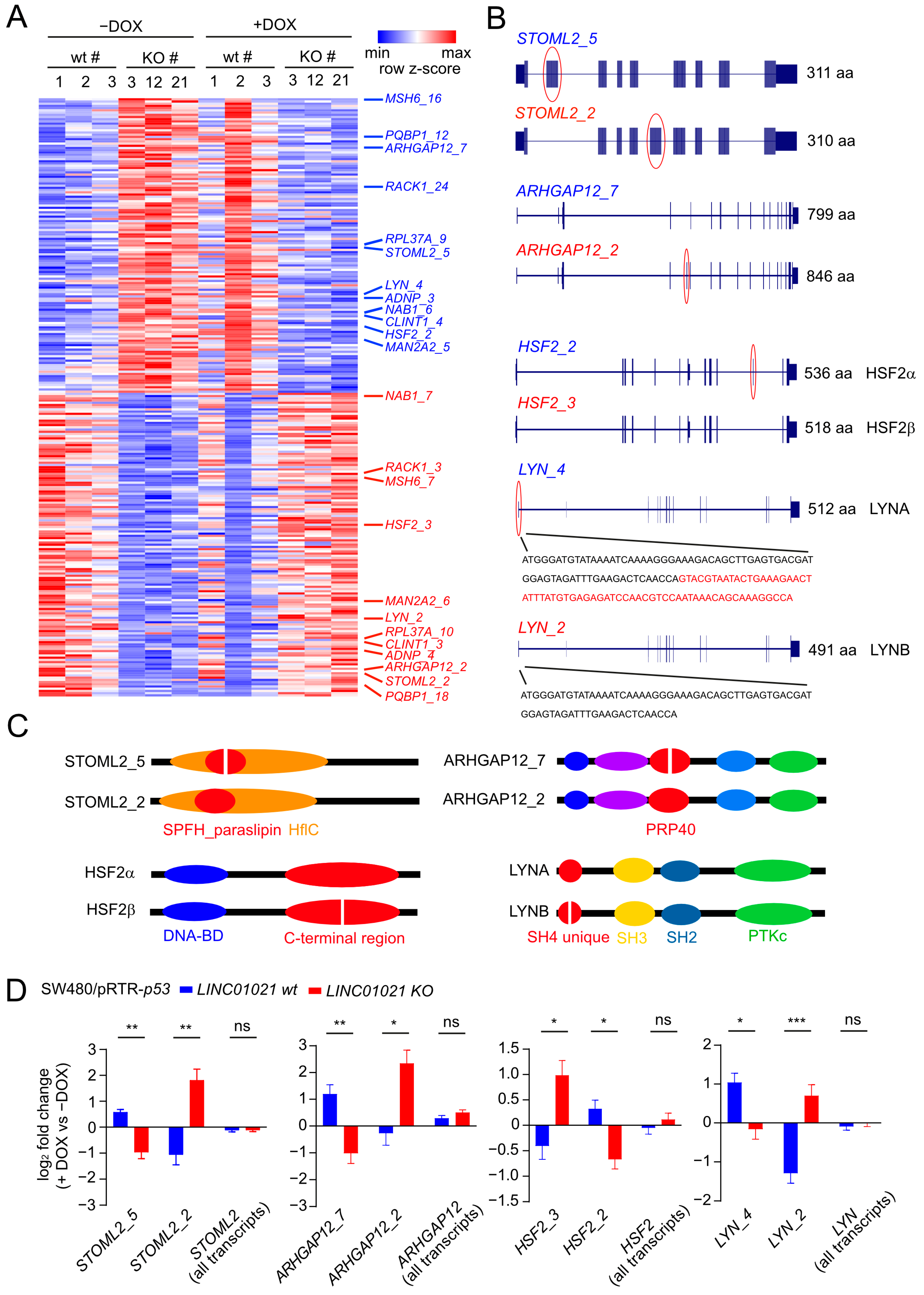
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BDS | binding site |
| cDNA | complementary DNA |
| CRC | colorectal cancer |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DOX | doxycycline |
| EMT | epithelial–mesenchymal transition |
| FACS | fluorescence-activated cell sorting |
| GSEA | gene set enrichment analysis |
| KO | knockout |
| Lnc | long noncoding |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| siRNA | Small interfering RNA |
| sgRNA | single guide RNA |
References
- Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Adv. Cancer Res. 2011, 110, 107–139. [Google Scholar] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Lal, A. Long noncoding RNAs in the p53 network. Wiley Interdiscip. Rev. RNA 2017, 8, e1410. [Google Scholar] [CrossRef]
- Grossi, E.; Sanchez, Y.; Huarte, M. Expanding the p53 regulatory network: LncRNAs take up the challenge. Biochim. Et Biophys. Acta 2016, 1859, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Xu, M.; Mo, Y.Y. Role of the lncRNA-p53 regulatory network in cancer. J. Mol. Cell Biol. 2014, 6, 181–191. [Google Scholar] [CrossRef]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigo, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Adelman, K. Emerging Roles of Non-Coding RNA Transcription. Trends Biochem. Sci. 2018, 43, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef]
- Leveille, N.; Melo, C.A.; Rooijers, K.; Diaz-Lagares, A.; Melo, S.A.; Korkmaz, G.; Lopes, R.; Akbari Moqadam, F.; Maia, A.R.; Wijchers, P.J.; et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015, 6, 6520. [Google Scholar] [CrossRef]
- Sanchez, Y.; Segura, V.; Marin-Bejar, O.; Athie, A.; Marchese, F.P.; Gonzalez, J.; Bujanda, L.; Guo, S.; Matheu, A.; Huarte, M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014, 5, 5812. [Google Scholar] [CrossRef] [PubMed]
- Younger, S.T.; Kenzelmann-Broz, D.; Jung, H.; Attardi, L.D.; Rinn, J.L. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015, 43, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- Hunten, S.; Kaller, M.; Drepper, F.; Oeljeklaus, S.; Bonfert, T.; Erhard, F.; Dueck, A.; Eichner, N.; Friedel, C.C.; Meister, G.; et al. p53-Regulated Networks of Protein, mRNA, miRNA, and lncRNA Expression Revealed by Integrated Pulsed Stable Isotope Labeling With Amino Acids in Cell Culture (pSILAC) and Next Generation Sequencing (NGS) Analyses. Mol. Cell. Proteom. MCP 2015, 14, 2609–2629. [Google Scholar] [CrossRef] [PubMed]
- Kaller, M.; Gotz, U.; Hermeking, H. Loss of p53-inducible long non-coding RNA LINC01021 increases chemosensitivity. Oncotarget 2017, 8, 102783–102800. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long Noncoding RNA PURPL Suppresses Basal p53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep. 2017, 20, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Y.; Wu, G.; Zhang, W.; Xu, S.; Wang, W. Long noncoding RNA PURPL promotes cell proliferation in liver cancer by regulating p53. Mol. Med. Rep. 2019, 19, 4998–5006. [Google Scholar] [CrossRef] [PubMed]
- Moridi, H.; Karimi, J.; Tavilani, H.; Khodadadi, I.; Emami Razavi, A.N. Overexpression of PURPL and downregulation of NONHSAT062994 as potential biomarkers in gastric cancer. Life Sci. 2019, 237, 116904. [Google Scholar] [CrossRef]
- Hartford, C.C.R.; Shrestha, R.L.; Pongor, L.; Zhao, Y.; Chen, X.; Fromont, C.; Chaudhary, R.; Li, X.L.; Pasterczyk, K.R.; Kumar, R.; et al. Context-Dependent Function of Long Noncoding RNA PURPL in Transcriptome Regulation during p53 Activation. Mol. Cell Biol. 2022, 42, e0028922. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Kaller, M.; Hermeking, H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 2013, 4, 1399–1415. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Notzold, L.; Frank, L.; Gandhi, M.; Polycarpou-Schwarz, M.; Gross, M.; Gunkel, M.; Beil, N.; Erfle, H.; Harder, N.; Rohr, K.; et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci. Rep. 2017, 7, 2265. [Google Scholar] [CrossRef]
- Singer, M.; Simon, K.; Forne, I.; Meissner, M. A central CRMP complex essential for invasion in Toxoplasma gondii. PLoS Biol. 2023, 21, e3001937. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Hassin, O.; Nataraj, N.B.; Shreberk-Shaked, M.; Aylon, Y.; Yaeger, R.; Fontemaggi, G.; Mukherjee, S.; Maddalena, M.; Avioz, A.; Iancu, O.; et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 2022, 13, 2800. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Nuytten, M.; Beke, L.; Van Eynde, A.; Ceulemans, H.; Beullens, M.; Van Hummelen, P.; Fuks, F.; Bollen, M. The transcriptional repressor NIPP1 is an essential player in EZH2-mediated gene silencing. Oncogene 2008, 27, 1449–1460. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Cacan, E. Histone Deacetylase-1-mediated Suppression of FAS in Chemoresistant Ovarian Cancer Cells. Anticancer. Res. 2016, 36, 2819–2826. [Google Scholar]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Q.; Chen, H. Genistein affects histone modifications on Dickkopf-related protein 1 (DKK1) gene in SW480 human colon cancer cell line. PLoS ONE 2012, 7, e40955. [Google Scholar] [CrossRef]
- Andrysik, Z.; Galbraith, M.D.; Guarnieri, A.L.; Zaccara, S.; Sullivan, K.D.; Pandey, A.; MacBeth, M.; Inga, A.; Espinosa, J.M. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017, 27, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed]
- Bieging-Rolett, K.T.; Kaiser, A.M.; Morgens, D.W.; Boutelle, A.M.; Seoane, J.A.; Van Nostrand, E.L.; Zhu, C.; Houlihan, S.L.; Mello, S.S.; Yee, B.A.; et al. Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol. Cell 2020, 80, 452–469. [Google Scholar] [CrossRef]
- Muys, B.R.; Anastasakis, D.G.; Claypool, D.; Pongor, L.; Li, X.L.; Grammatikakis, I.; Liu, M.; Wang, X.; Prasanth, K.V.; Aladjem, M.I.; et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes. Dev. 2021, 35, 102–116. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhong, Y.; Zhang, Y.; Yang, L.; Wu, P.; Hou, X.; Xiong, F.; Li, X.; Zhang, S.; Gong, Z.; et al. Long non-coding RNAs are involved in alternative splicing and promote cancer progression. Br. J. Cancer 2022, 126, 1113–1124. [Google Scholar] [CrossRef]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Cooke, J.; Mirza, H.; Aurrekoetxea-Rodriguez, I.; Vivanco, M.D.M.; Buckley, N.E.; Grigoriadis, A.; Smalley, M.J. Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. Cell Rep. 2018, 25, 3674–3692. [Google Scholar] [CrossRef]
- Alvelos, M.I.; Bruggemann, M.; Sutandy, F.R.; Juan-Mateu, J.; Colli, M.L.; Busch, A.; Lopes, M.; Castela, A.; Aartsma-Rus, A.; Konig, J.; et al. The RNA-binding profile of the splicing factor SRSF6 in immortalized human pancreatic beta-cells. Life Sci. Alliance 2021, 4, e202000825. [Google Scholar] [CrossRef]
- Lecomte, S.; Reverdy, L.; Le Quement, C.; Le Masson, F.; Amon, A.; Le Goff, P.; Michel, D.; Christians, E.; Le Drean, Y. Unraveling complex interplay between heat shock factor 1 and 2 splicing isoforms. PLoS ONE 2013, 8, e56085. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Monte, M.; Simonatto, M.; Peche, L.Y.; Bublik, D.R.; Gobessi, S.; Pierotti, M.A.; Rodolfo, M.; Schneider, C. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 11160–11165. [Google Scholar] [CrossRef]
- Choi, H.K.; Choi, Y.; Park, E.S.; Park, S.Y.; Lee, S.H.; Seo, J.; Jeong, M.H.; Jeong, J.W.; Jeong, J.H.; Lee, P.C.; et al. Programmed cell death 5 mediates HDAC3 decay to promote genotoxic stress response. Nat. Commun. 2015, 6, 7390. [Google Scholar] [CrossRef]
- Duan, Y.; Jia, Y.; Wang, J.; Liu, T.; Cheng, Z.; Sang, M.; Lv, W.; Qin, J.; Liu, L. Long noncoding RNA DGCR5 involves in tumorigenesis of esophageal squamous cell carcinoma via SRSF1-mediated alternative splicing of Mcl-1. Cell Death Dis. 2021, 12, 587. [Google Scholar] [CrossRef]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef]
- Lv, Y.F.; Yan, G.N.; Meng, G.; Zhang, X.; Guo, Q.N. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci. Rep. 2015, 5, 12999. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, S.; Fan, J.; Jin, Y.; Tian, B.; Zheng, X.; Fu, H. Nuclear retention of the lncRNA SNHG1 by doxorubicin attenuates hnRNPC-p53 protein interactions. EMBO Rep. 2017, 18, 536–548. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Liu, X.; Xu, Y.; Zhai, R.; Zhang, J.; Wang, M.; Wang, M.; Liu, L. lncRNA CYTOR promotes aberrant glycolysis and mitochondrial respiration via HNRNPC-mediated ZEB1 stabilization in oral squamous cell carcinoma. Cell Death Dis. 2022, 13, 703. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Yu, J.; Yao, X.; Yang, S.; Li, W.; Xu, L.; Zhao, L. LncRNA CRNDE attenuates chemoresistance in gastric cancer via SRSF6-regulated alternative splicing of PICALM. Mol. Cancer 2021, 20, 6. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaller, M.; Forné, I.; Imhof, A.; Hermeking, H. LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes. Cancers 2024, 16, 1639. https://doi.org/10.3390/cancers16091639
Kaller M, Forné I, Imhof A, Hermeking H. LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes. Cancers. 2024; 16(9):1639. https://doi.org/10.3390/cancers16091639
Chicago/Turabian StyleKaller, Markus, Ignasi Forné, Axel Imhof, and Heiko Hermeking. 2024. "LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes" Cancers 16, no. 9: 1639. https://doi.org/10.3390/cancers16091639
APA StyleKaller, M., Forné, I., Imhof, A., & Hermeking, H. (2024). LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes. Cancers, 16(9), 1639. https://doi.org/10.3390/cancers16091639