
T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies



Abstract
Simple Summary
Abstract
1. Introduction
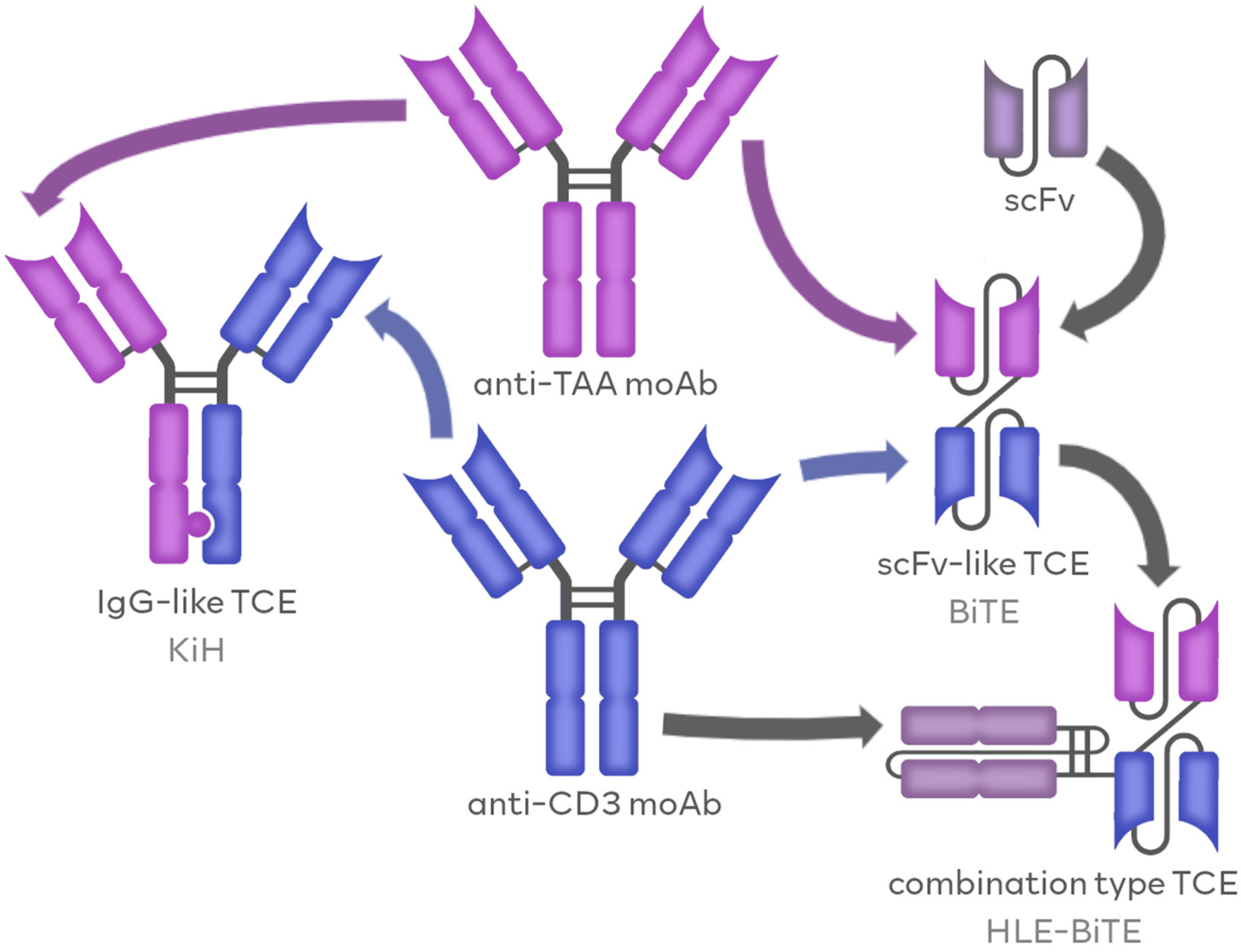
1.1. TCEs as Members of BsAbs Family
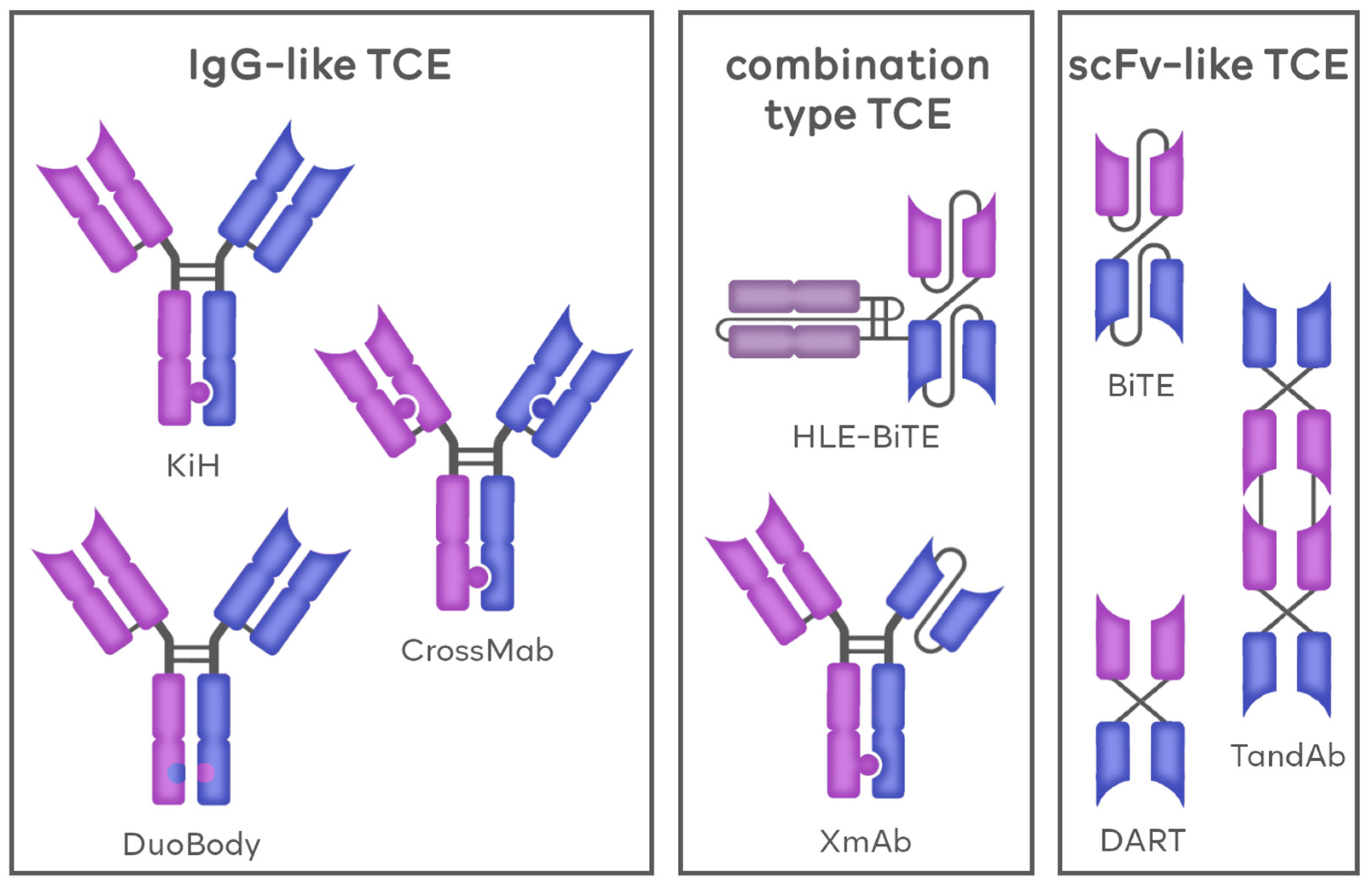
1.1.1. IgG-like TCEs
Knobs-into-Holes (KiH)
CrossMab
DuoBody
1.1.2. Fv-Based TCEs
Bi-Specific T-Cell Engagers (BiTE)
Dual-Affinity Retargeting Antibody (DART)
TandAb
1.1.3. Combination-Based TCEs
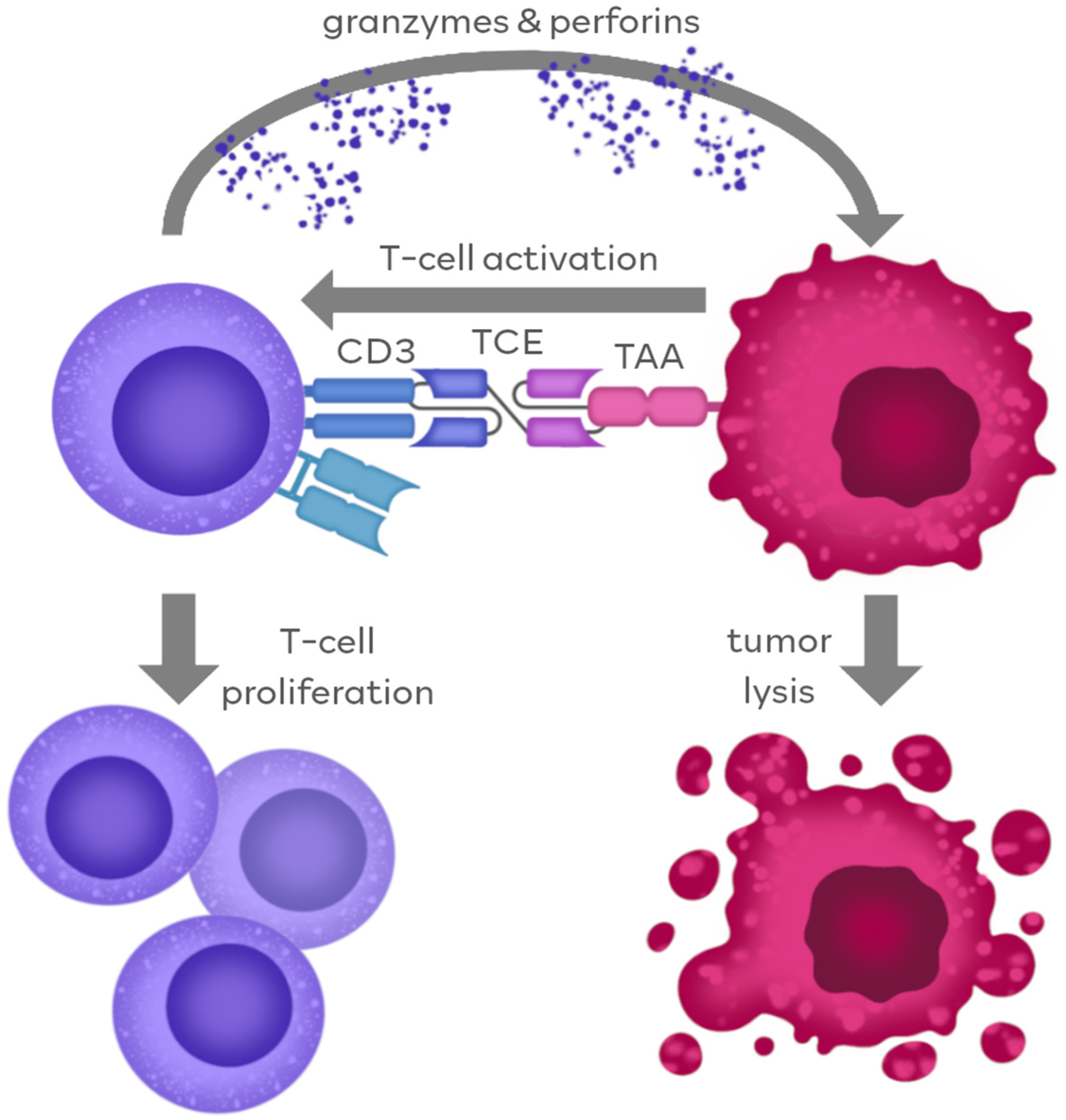
2. Mechanism of TCEs’ Anti-Tumor Action
2.1. TCE Action through Antigen Binding
2.2. Fc-Dependent TCE Action
3. TCEs in the Treatment of Hematological Malignancies
3.1. Antigens Frequently Targeted in Hematology
3.1.1. CD19
3.1.2. CD20
3.1.3. B-Cell Maturation Antigen (BCMA)
3.1.4. CD33
3.1.5. FMS-like Tyrosine Kinase 3 (FLT3)
4. Currently Approved TCE Therapies in Hematology (November 2023)
4.1. Blinatumomab
4.2. Mosunetuzumab
4.3. Teclistamab
4.4. Glofitamab
4.5. Epcoritamab
4.6. Talquetamab
4.7. Elranatamab
5. Challenges and Perspectives
5.1. On Target/Off Tumor Killing
5.2. Choice of Optimal Affinities
5.3. Tumor Immune Escape
5.4. Immunotoxicity
5.5. New Agents with Improved Properties
5.6. Secreted TCE
5.7. Combination Approaches
5.7.1. TCE in Combination with Chemotherapy
5.7.2. TCE in Combination with Immune Checkpoint Inhibitors
5.7.3. TCE in Combination with CAR-T Therapy
5.7.4. TCE in Combination with Oncolytic Viruses (OVs)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (MAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of Action of Therapeutic Antibodies for Cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Foster, L.H.; Lum, L.G. Treatment of Hematological Malignancies with T Cell Redirected Bispecific Antibodies: Current Status and Future Needs. Expert Opin. Biol. Ther. 2019, 19, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Nisonoff, A.; Wissler, F.C.; Lipman, L.N. Properties of the Major Component of a Peptic Digest of Rabbit Antibody. Science 1960, 132, 1770–1771. [Google Scholar] [CrossRef]
- Nisonoff, A.; Rivers, M.M. Recombination of a Mixture of Univalent Antibody Fragments of Different Specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Milstein, C.; Cuello, A.C. Hybrid Hybridomas and Their Use in Immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein Engineering of Antibody Binding Sites: Recovery of Specific Activity in an Anti-Digoxin Single-Chain Fv Analogue Produced in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An Efficient Route to Human Bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Jimeno, A. Bispecific Antibodies for Cancer Therapy: A Review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T Cells to Hematological Malignancies with Bispecific Antibodies. Blood 2018, 131, 30. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.L.; Profitós-Pelejà, N.; Santos, J.C.; Blecua, P.; Reyes-Garau, D.; Armengol, M.; Fernández-Serrano, M.; Miskin, H.P.; Bosch, F.; Esteller, M.; et al. G Protein-Coupled Receptor 183 Mediates the Sensitization of Burkitt Lymphoma Tumors to CD47 Immune Checkpoint Blockade by Anti-CD20/PI3Kδi Dual Therapy. Front. Immunol. 2023, 14, 1130052. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific Pd-1/Ctla4 Antibody That Enhances Ctla4 Blockade on Pd-1+ Activated t Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Zhou, Y.; Liu, Y.; Zhang, R.; Jiang, X.; Ren, C.; Gao, X.; Luo, L. PD-1/LAG-3 Bispecific Antibody Potentiates T Cell Activation and Increases Antitumor Efficacy. Front. Immunol. 2022, 13, 1047610. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific Antibody Based Therapeutics: Strengths and Challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of Bispecific Antibodies by Chemical Recombination of Monoclonal Immunoglobulin G1 Fragments. Science 1985, 229, 81–83. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering Therapeutic Bispecific Antibodies Using CrossMab Technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lee, J.; Tran, C.; Heibeck, T.H.; Wang, W.D.; Yang, J.; Stafford, R.L.; Steiner, A.R.; Sato, A.K.; Hallam, T.J.; et al. Production of Bispecific Antibodies in “Knobs-into-Holes” Using a Cell-Free Expression System. MAbs 2015, 7, 231–242. [Google Scholar] [CrossRef]
- Yu, J.; Song, Y.; Tian, W. How to Select IgG Subclasses in Developing Anti-Tumor Therapeutic Antibodies. J. Hematol. Oncol. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Ridgway, J.B.B.; Presta, L.G.; Carter, P. “Knobs-into-Holes” Engineering of Antibody CH3 Domains for Heavy Chain Heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; De Goeij, B.E.C.G.; Van Den Bremer, E.T.J.; Neijssen, J.; Van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient Generation of Stable Bispecific IgG1 by Controlled Fab-Arm Exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin Domain Crossover as a Generic Approach for the Production of Bispecific IgG Antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef]
- Regula, J.T.; Imhof-Jung, S.; Mølhøj, M.; Benz, J.; Ehler, A.; Bujotzek, A.; Schaefer, W.; Klein, C. Variable Heavy–Variable Light Domain and Fab-Arm CrossMabs with Charged Residue Exchanges to Enforce Correct Light Chain Assembly. Protein Eng. Des. Sel. 2018, 31, 289. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The Use of CrossMAb Technology for the Generation of Bi- and Multispecific Antibodies. MAbs 2016, 8, 1010. [Google Scholar] [CrossRef]
- Vu, M.D.; Moser, S.; Delon, C.; Latzko, M.; Gianotti, R.; Lüoend, R.; Friang, C.; Murr, R.; Duerner, L.J.; Weinzierl, T.; et al. A New Class of T-Cell Bispecific Antibodies for the Treatment of Multiple Myeloma, Binding to B Cell Maturation Antigen and CD3 and Showing Potent, Specific Antitumor Activity in Myeloma Cells and Long Duration of Action in Cynomolgus Monkeys. Blood 2015, 126, 2998. [Google Scholar] [CrossRef]
- Gramer, M.J.; Van Den Bremer, E.T.J.; Van Kampen, M.D.; Kundu, A.; Kopfmann, P.; Etter, E.; Stinehelfer, D.; Long, J.; Lannom, T.; Noordergraaf, E.H.; et al. Production of Stable Bispecific IgG1 by Controlled Fab-Arm Exchange: Scalability from Bench to Large-Scale Manufacturing by Application of Standard Approaches. MAbs 2013, 5, 962. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific Antibodies and Their Applications. J. Hematol. Oncol. 2015, 8, 1–14. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. MAbs 2017, 9, 182. [Google Scholar] [CrossRef]
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F.; Rahimi-Jamnani, F. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol. Ther. Oncolytics 2019, 14, 38. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.J.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Le Gall, F.; et al. A Tetravalent Bispecific TandAb (CD19/CD3), AFM11, Efficiently Recruits T Cells for the Potent Lysis of CD19+ Tumor Cells. MAbs 2015, 7, 584. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.J.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Circosta, P.; Elia, A.R.; Landra, I.; Machiorlatti, R.; Todaro, M.; Aliberti, S.; Brusa, D.; Deaglio, S.; Chiaretti, S.; Bruna, R.; et al. Tailoring CD19xCD3-DART Exposure Enhances T-Cells to Eradication of B-Cell Neoplasms. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Sherman, M.; McElroy, P.L.; Lofgren, J.A.; Moody, G.; Baeuerle, P.A.; Coxon, A.; Arvedson, T. Bispecific T Cell Engager (BiTE®) Antibody Constructs Can Mediate Bystander Tumor Cell Killing. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific Antibody Constructs with Unique Anti-Tumor Activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, C.P.M.; Frerichs, K.A.; Broekmans, M.; Absalah, S.; Maas-Bosman, P.W.C.; Kruyswijk, S.; Nijhof, I.S.; Mutis, T.; Zweegman, S.; van de Donk, N.W.C.J. T-Cell Redirecting Bispecific Antibodies Targeting BCMA for the Treatment of Multiple Myeloma. Oncotarget 2020, 11, 4076. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kroenke, J.; Facon, T.; Salnikov, A.; Lesley, R.; et al. Evaluation of AMG 420, an Anti-BCMA Bispecific T-Cell Engager (BiTE) Immunotherapy, in R/R Multiple Myeloma (MM) Patients: Updated Results of a First-in-Human (FIH) Phase I Dose Escalation Study. HemaSphere 2019, 37, 8007. [Google Scholar] [CrossRef]
- Aigner, M.; Feulner, J.; Schaffer, S.; Kischel, R.; Kufer, P.; Schneider, K.; Henn, A.; Rattel, B.; Friedrich, M.; Baeuerle, P.A.; et al. T Lymphocytes Can Be Effectively Recruited for Ex Vivo and in Vivo Lysis of AML Blasts by a Novel CD33/CD3-Bispecific BiTE Antibody Construct. Leukemia 2013, 27, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Stein, A.S.; Kantarjian, H.M.; Walter, R.B.; Paschka, P.; Jongen-Lavrencic, M.; Ossenkoppele, G.J.; Yang, Z.; Mehta, B.; Subklewe, M. A Phase 1 First-in-Human Study of AMG 330, an Anti-CD33 Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). Blood 2018, 132, 25. [Google Scholar] [CrossRef]
- Arvedson, T.L.; Balazs, M.; Bogner, P.; Black, K.; Graham, K.; Henn, A.; Friedrich, M.; Hoffmann, P.; Kischel, R.; Kufer, P.; et al. Abstract 55: Generation of Half-Life Extended Anti-CD33 BiTE® Antibody Constructs Compatible with Once-Weekly Dosing. Cancer Res. 2017, 77, 55. [Google Scholar] [CrossRef]
- Einsele, H.; Borghaei, H.; Orlowski, R.Z.; Subklewe, M.; Roboz, G.J.; Zugmaier, G.; Kufer, P.; Iskander, K.; Kantarjian, H.M. The BiTE (Bispecific T-Cell Engager) Platform: Development and Future Potential of a Targeted Immuno-Oncology Therapy across Tumor Types. Cancer 2020, 126, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.A.; Chendamarai, E.; Ritchey, J.K.; Rettig, M.P.; Gehrs, L.; Moore, P.; Alderson, R.F.; Bonvini, E.; Christopher, M.; DiPersio, J.F. Modeling Flotetuzumab-Associated CRS in AML Using InVitro and InVivo Preclinical Models. Blood 2022, 140, 8795–8796. [Google Scholar] [CrossRef]
- Winer, E.S.; Maris, M.; Sharma, M.R.; Kaminker, P.; Zhao, E.; Ward, A.; Sochacki, A.L. A Phase 1, First-in-Human, Dose-Escalation Study of MGD024, a CD123 x CD3 Bispecific Dart® Molecule, in Patients with Relapsed or Refractory CD123-Positive (+) Hematologic Malignancies. Blood 2022, 140, 11753–11754. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of Dual Affinity Retargeting Molecules to Achieve Optimal Redirected T-Cell Killing of B-Cell Lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von Der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific Tandem Diabody for Tumor Therapy with Improved Antigen Binding and Pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef]
- Topp, M.; Dlugosz-Danecka, M.; Skotnicki, A.B.; Salogub, G.; Viardot, A.; Klein, A.K.; Hess, G.; Michel, C.S.; Grosicki, S.; Gural, A.; et al. Safety of AFM11 in the Treatment of Patients with B-Cell Malignancies: Findings from Two Phase 1 Studies. Trials 2023, 24, 4. [Google Scholar] [CrossRef] [PubMed]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE® Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130, 2815. [Google Scholar] [CrossRef]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (Bispecific T-Cell Engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Patel, K.; Riedell, P.A.; Tilly, H.; Ahmed, S.; Michot, J.-M.; Ghesquieres, H.; Schiano de Collela, J.M.; Chanan-Khan, A.; Bouabdallah, K.; Tessoulin, B.; et al. A Phase 1 Study of Plamotamab, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma: Recommended Dose Safety/Efficacy Update and Escalation Exposure-Response Analysis. Blood 2022, 140, 9470–9472. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Godar, M.; de Haard, H.; Blanchetot, C.; Rasser, J. Therapeutic Bispecific Antibody Formats: A Patent Applications Review (1994-2017). Expert Opin. Ther. Pat. 2018, 28, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cheung, N.V. T Cell Engaging Bispecific Antibody (T-BsAb): From Technology to Therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.E.; Bargou, R. Blinatumomab: A CD19/CD3 Bispecific T Cell Engager (BiTE) with Unique Anti-Tumor Efficacy. Leuk. Lymphoma 2016, 57, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.H.; Paulos, C.M. Novel Immunotherapies for Hematological Malignancies. Immunol. Rev. 2015, 263, 90. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive Immunity Maintains Occult Cancer in an Equilibrium State. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, N.; Li, Y.; Wang, Y.; Liu, S.; Hwu, P.; Liu, Y.J.; Dong, C.; Radvanyi, L. Melanoma Cells Express ICOS Ligand to Promote the Activation and Expansion of T-Regulatory Cells. Cancer Res. 2010, 70, 9581. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A Recombinant Bispecific Single-Chain Antibody, CD19 x CD3, Induces Rapid and High Lymphoma-Directed Cytotoxicity by Unstimulated T Lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Infiltrating Lymphocytes as Antitumor Effector Cells. Biotherapy 1992, 5, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical Development and Future Directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Jäger, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Stroöhlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring Results of a Phase II/III Study of Malignant Ascites Patients Treated with the Trifunctional Antibody Catumaxomab (Anti-EpCAM x Anti-CD3). Cancer Res. 2012, 72, 24–32. [Google Scholar] [CrossRef]
- Hristodorov, D.; Fischer, R.; Linden, L. With or without Sugar? (A)Glycosylation of Therapeutic Antibodies. Mol. Biotechnol. 2013, 54, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lam, C.Y.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-Specific Molecule Incorporating Extended Circulating Half-Life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Ellerman, D. Bispecific T-Cell Engagers: Towards Understanding Variables Influencing the in Vitro Potency and Tumor Selectivity and Their Modulation to Enhance Their Efficacy and Safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; Van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, R.H.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z.; Herishanu, Y. Therapeutic Targeting of CD19 in Hematological Malignancies: Past, Present, Future and Beyond. Leuk. Lymphoma 2014, 55, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fairfax, K.; Light, A.; Huntington, N.D.; Tarlinton, D.M. CD19 Differentially Regulates BCR Signalling through the Recruitment of PI3K. Autoimmunity 2014, 47, 430–437. [Google Scholar] [CrossRef]
- Gaballa, M.R.; Banerjee, P.; Milton, D.R.; Jiang, X.; Ganesh, C.; Khazal, S.; Nandivada, V.; Islam, S.; Kaplan, M.; Daher, M.; et al. Blinatumomab Maintenance after Allogeneic Hematopoietic Cell Transplantation for B-Lineage Acute Lymphoblastic Leukemia. Blood 2022, 139, 1908–1919. [Google Scholar] [CrossRef]
- Coyle, L.; Morley, N.J.; Rambaldi, A.; Mason, K.D.; Verhoef, G.; Furness, C.L.; Zhang, A.; Jung, A.S.; Cohan, D.; Franklin, J.L. Open-Label, Phase 2 Study of Blinatumomab as Second Salvage Therapy in Adults with Relapsed/Refractory Aggressive B-Cell Non-Hodgkin Lymphoma. Leuk. Lymphoma 2020, 61, 2103–2112. [Google Scholar] [CrossRef]
- Dufner, V.; Sayehli, C.M.; Chatterjee, M.; Hummel, H.D.; Gelbrich, G.; Bargou, R.C.; Goebeler, M.E. Long-Term Outcome of Patients with Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Treated with Blinatumomab. Blood Adv. 2019, 3, 2491. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Prince, H.M.; Tam, C.S.; Ku, M.; Thieblemont, C.; Popplewell, L.L.; Wermke, M.; Haioun, C.; Wong Doo, N.; Viardot, A.; et al. A Phase 1b Study of Blinatumomab Including Subcutaneous Administration in Relapsed / Refractory (R/R) Indolent Non Hodgkin’s Lymphoma (NHL). Blood 2021, 138, 2436. [Google Scholar] [CrossRef]
- Popplewell, L.; Verhoef, G.; Kuruvilla, J.; Tuglus, C.; Kischel, R.; Stieglmaier, J.; Ghobadi, A. A First-In-Human Study Of A Half-Life Extended Cd19-Targeting Bite in Relapsed/Refractory Diffuse Large B Cell Lymphoma, Mantle Cell Lymphoma or Follicular Lymphoma. Hematol. Oncol. 2019, 37, 566–567. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Janas, E.; Priest, R.; Wilde, J.I.; White, J.H.; Malhotra, R. Rituxan (Anti-CD20 Antibody)-Induced Translocation of CD20 into Lipid Rafts Is Crucial for Calcium Influx and Apoptosis. Clin. Exp. Immunol. 2005, 139, 439. [Google Scholar] [CrossRef] [PubMed]
- Bannerji, R.; Arnason, J.E.; Advani, R.H.; Brown, J.R.; Allan, J.N.; Ansell, S.M.; Barnes, J.A.; O’Brien, S.M.; Chávez, J.C.; Duell, J.; et al. Odronextamab, a Human CD20×CD3 Bispecific Antibody in Patients with CD20-Positive B-Cell Malignancies (ELM-1): Results from the Relapsed or Refractory Non-Hodgkin Lymphoma Cohort in a Single-Arm, Multicentre, Phase 1 Trial. Lancet. Haematol. 2022, 9, e327–e339. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Taszner, M.; Cho, S.-G.; Novelli, S.; Le Gouill, S.; Poon, M.L.; Villasboas, J.C.; Champion, R.; Bachy, E.; Guidez, S.; et al. Odronextamab in Patients with Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Grade 1-3a: Results from a Prespecified Analysis of the Pivotal Phase II Study ELM-2. Blood 2022, 140, 2280–2282. [Google Scholar] [CrossRef]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL System: Emerging Functions beyond B Cell Biology and Autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Bounds, D.; Paterson, J.; Herledan, G.; Sully, K.; Seestaller-Wehr, L.M.; Fieles, W.E.; Tunstead, J.; McCahon, L.; Germaschewski, F.M.; et al. Evaluation of B Cell Maturation Antigen as a Target for Antibody Drug Conjugate Mediated Cytotoxicity in Multiple Myeloma. Br. J. Haematol. 2016, 174, 911–922. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Andrews, R.; Torok-Storb, B.; Bernstein, I. Myeloid-Associated Differentiation Antigens on Stem Cells and Their Progeny Identified by Monoclonal Antibodies. Blood 1983, 62, 124–132. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An Overview on the Role of FLT3-Tyrosine Kinase Receptor in Acute Myeloid Leukemia: Biology and Treatment. Oncol. Rev. 2012, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 Mutations in Acute Myeloid Leukemia: Therapeutic Paradigm beyond Inhibitor Development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef]
- Mehta, N.K.; Pfluegler, M.; Meetze, K.; Li, B.; Sindel, I.; Vogt, F.; Marklin, M.; Heitmann, J.S.; Kauer, J.; Osburg, L.; et al. A Novel IgG-Based FLT3xCD3 Bispecific Antibody for the Treatment of AML and B-ALL. J. Immunother. cancer 2022, 10, e003882. [Google Scholar] [CrossRef]
- Demichelis-Gómez, R.; Pérez-Sámano, D.; Bourlon, C. Bispecific Antibodies in Hematologic Malignancies: When, to Whom, and How Should Be Best Used? Curr. Oncol. Rep. 2019, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and Activity of Blinatumomab for Adult Patients with Relapsed or Refractory B-Precursor Acute Lymphoblastic Leukaemia: A Multicentre, Single-Arm, Phase 2 Study. Lancet. Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase II, Single-Arm, Multicenter Study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 Study of the Bispecific T-Cell Engager (BiTE) Antibody Blinatumomab in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Löffler, A.; Gruen, M.; Wuchter, C.; Schriever, F.; Kufer, P.; Dreier, T.; Hanakam, F.; Baeuerle, P.A.; Bommert, K.; Karawajew, L.; et al. Efficient Elimination of Chronic Lymphocytic Leukaemia B Cells by Autologous T Cells with a Bispecific Anti-CD19/Anti-CD3 Single-Chain Antibody Construct. Leukemia 2003, 17, 900. [Google Scholar] [CrossRef]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab Induces Autologous T-Cell Killing of Chronic Lymphocytic Leukemia Cells. Haematologica 2013, 98, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T Cell-Dependent Bispecific Antibody for the Treatment of B Cell Malignancies. Sci. Transl. Med. 2015, 7, 287ra70. [Google Scholar] [CrossRef] [PubMed]
- Eltantawy, A.; Vallejos, X.; Sebea, E.; Evans, K. Copanlisib: An Intravenous Phosphatidylinositol 3-Kinase (PI3K) Inhibitor for the Treatment of Relapsed Follicular Lymphoma. Ann. Pharmacother. 2019, 53, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.E.; Assouline, S.; Sehn, L.H.; Schuster, S.J.; Yoon, S.S.; Yoon, D.H.; Matasar, M.J.; Bosch, F.; Kim, W.S.; Nastoupil, L.J.; et al. Single-Agent Mosunetuzumab Shows Durable Complete Responses in Patients with Relapsed or Refractory B-Cell Lymphomas: Phase I Dose-Escalation Study. J. Clin. Oncol. 2022, 40, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab Is an Active T Cell–Redirecting Bispecific Antibody against B-Cell Maturation Antigen for Multiple Myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Teclistamab: First Approval. Drugs 2022, 82, 1613–1619. [Google Scholar] [CrossRef]
- Bacac, M.; Colombetti, S.; Herter, S.; Sam, J.; Perro, M.; Chen, S.; Bianchi, R.; Richard, M.; Schoenle, A.; Nicolini, V.; et al. CD20-TCB with Obinutuzumab Pretreatment as next-Generation Treatment of Hematologic Malignancies. Clin. Cancer Res. 2018, 24, 4785–4797. [Google Scholar] [CrossRef]
- Dickinson, M.J.; Carlo-Stella, C.; Morschhauser, F.; Bachy, E.; Corradini, P.; Iacoboni, G.; Khan, C.; Wróbel, T.; Offner, F.; Trněný, M.; et al. Glofitamab for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 387, 2220–2231. [Google Scholar] [CrossRef]
- Shirley, M. Glofitamab: First Approval. Drugs 2023, 83, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 Induces Potent T-Cell-Mediated Killing of Malignant B Cells in Preclinical Models and Provides Opportunities for Subcutaneous Dosing. EBioMedicine 2020, 52. [Google Scholar] [CrossRef] [PubMed]
- Thieblemont, C.; Phillips, T.; Ghesquieres, H.; Cheah, C.Y.; Clausen, M.R.; Cunningham, D.; Do, Y.R.; Feldman, T.; Gasiorowski, R.; Jurczak, W.; et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell–Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J. Clin. Oncol. 2023, 41, 2238. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Epcoritamab: First Approval. Drugs 2023, 83, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Nambu, T.; Shimomura, T. The RAIG Family Member, GPRC5D, Is Associated with Hard-Keratinized Structures. J. Invest. Dermatol. 2004, 122, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Atamaniuk, J.; Gleiss, A.; Porpaczy, E.; Kainz, B.; Grunt, T.W.; Raderer, M.; Hilgarth, B.; Drach, J.; Ludwig, H.; Gisslinger, H.; et al. Overexpression of G Protein-Coupled Receptor 5D in the Bone Marrow Is Associated with Poor Prognosis in Patients with Multiple Myeloma. Eur. J. Clin. Invest. 2012, 42, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.-V.; Costa, L.J.; Caers, J.; et al. Talquetamab, a T-Cell–Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl. J. Med. 2022, 387, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Talquetamab: First Approval. Drugs 2023, 83. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Tomasson, M.H.; Arnulf, B.; Bahlis, N.J.; Miles Prince, H.; Niesvizky, R.; Rodrίguez-Otero, P.; Martinez-Lopez, J.; Koehne, G.; Touzeau, C.; et al. Elranatamab in Relapsed or Refractory Multiple Myeloma: Phase 2 MagnetisMM-3 Trial Results. Nat. Med. 2023, 29, 2259–2267. [Google Scholar] [CrossRef]
- Grosicki, S.; Mellqvist, U.-H.; Pruchniewski, Ł.; Crafoord, J.; Trudel, S.; Min, C.-K.; White, D.; Alegre, A.; Hansson, M.; Ikeda, T.; et al. Elranatamab in Combination with Daratumumab for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Results from the Phase 3 Magnetismm-5 Study Safety Lead-in Cohort. Blood 2022, 140, 4407–4408. [Google Scholar] [CrossRef]
- Grosicki, S.; Yeh, S.-P.; Huang, J.S.Y.; Byun, J.M.; DiRienzo, C.; Viqueira, A. MagnetisMM-6: An Open-Label, Multicenter, Randomized Phase 3 Study of Elranatamab + Daratumumab + Lenalidomide (EDR) versus Daratumumab + Lenalidomide + Dexamethasone (DRd) in Transplant Ineligible (TI) Patients with Newly Diagnosed Multiple Myeloma (NDMM). J. Clin. Oncol. 2023, 41, TPS8065. [Google Scholar] [CrossRef]
- Mateos Manteca, M.V.; Grosicki, S.; Kim, K.; Negre, E.; Vandendries, E. MagnetisMM-7: An Open-Label, Multicenter, Randomized Phase 3 Study of Elranatamab versus Lenalidomide in Post-Transplant Patients with Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS8066. [Google Scholar] [CrossRef]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of Regulatory T Cells Determines the Outcome of the T-Cell-Engaging Antibody Blinatumomab in Patients with B-Precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, U.; Boudreau, A.; Buelow, B.; Clarke, S.; Dang, K.; Davison, L.; Aldred, S.F.; Harris, K.; Iyer, S.; Jorgensen, B.; et al. A Novel T-Cell Bispecific Antibody Platform for Efficient T-Cell Mediated Killing of Tumor Cells with Minimal Cytokine Release. J. Clin. Oncol. 2018, 36 (Suppl. S5), 209. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 Axis Augments Lysis of AML Cells by the CD33/CD3 BiTE Antibody Construct AMG 330: Reversing a T-Cell-Induced Immune Escape Mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Walter, R.B. T-Cell Ligands Modulate the Cytolytic Activity of the CD33/CD3 BiTE Antibody Construct, AMG 330. Blood Cancer J. 2015 58 2015, 5, e340. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Krupka, C.; Tischer, J.; Knösel, T.; Subklewe, M. Increase of PD-L1 Expressing B-Precursor ALL Cells in a Patient Resistant to the CD19/CD3-Bispecific T Cell Engager Antibody Blinatumomab. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350. [Google Scholar] [CrossRef]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-Cell Responses against CD19+ Pediatric Acute Lymphoblastic Leukemia Mediated by Bispecific T-Cell Engager (BiTE) Are Regulated Contrarily by PD-L1 and CD80/CD86 on Leukemic Blasts. Oncotarget 2016, 7, 76902. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ Regulatory T Cells Suppress Tumor Immunity but Are Sensitive to Cyclophosphamide Which Allows Immunotherapy of Established Tumors to Be Curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.T.; Goicoechea, I.; Alameda, D.; José-Eneriz, E.S.; Merino, J.; Rodríguez-Otero, P.; et al. Immunogenomic Identification and Characterization of Granulocytic Myeloid-Derived Suppressor Cells in Multiple Myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted Therapy with the T-Cell—Engaging Antibody Blinatumomab of Chemotherapy-Refractory Minimal Residual Disease in B-Lineage Acute Lymphoblastic Leukemia Patients Results in High Response Rate and Prolonged Leukemia-Free Survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II Trial of the Anti-CD19 Bispecific T Cell-Engager Blinatumomab Shows Hematologic and Molecular Remissions in Patients with Relapsed or Refractory B-Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor-Intrinsic and -Extrinsic Determinants of Response to Blinatumomab in Adults with B-ALL. Blood 2021, 137, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, A.; Winkler, B.; zur Stadt, U.; Müller, I.; Escherich, G. Lineage Switch Under Blinatumomab of a Relapsed Common ALL Co-Expressing Myeloid Markers without MLL Rearrangement. Blood 2016, 128, 5196. [Google Scholar] [CrossRef]
- Frey, N.V.; Porter, D.L. Cytokine Release Syndrome with Novel Therapeutics for Acute Lymphoblastic Leukemia. Hematol. Am. Soc. Hematol. Educ. Progr. 2016, 2016, 567–572. [Google Scholar] [CrossRef]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic Adverse Events in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia Treated with Blinatumomab: Management and Mitigating Factors. Ann. Hematol. 2019, 98, 159. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Makino, K.; Nakata, J.; Kawachi, S.; Hayashi, T.; Nakajima, A.; Yokoyama, M. Treatment Strategy for Reducing the Risk of Rituximab-Induced Cytokine Release Syndrome in Patients with Intravascular Large B-Cell Lymphoma: A Case Report and Review of the Literature. J. Med. Case Rep. 2013, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.S.; Kasi, P.M. Rituximab and Cytokine Release Syndrome. Case Rep. Oncol. 2012, 5, 134. [Google Scholar] [CrossRef]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T Cell-Induced Cytokine Release Syndrome Is Mediated by Macrophages and Abated by IL-1 Blockade. Nat. Med. 2018, 24, 731. [Google Scholar] [CrossRef] [PubMed]
- Kebenko, M.; Goebeler, M.E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A Multicenter Phase 1 Study of Solitomab (MT110, AMG 110), a Bispecific EpCAM/CD3 T-Cell Engager (BiTE®) Antibody Construct, in Patients with Refractory Solid Tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Haas, C.; D’Argouges, S.; Fisch, T.; Kufer, P.; Brischwein, K.; Prang, N.; Bargou, R.; Suzich, J.A.; Baeuerle, P.A.; et al. The Effect of Dexamethasone on Polyclonal T Cell Activation and Redirected Target Cell Lysis as Induced by a CD19/CD3-Bispecific Single-Chain Antibody Construct. Cancer Immunol. Immunother. 2007, 56, 1551–1563. [Google Scholar] [CrossRef]
- Kauer, J.; Hörner, S.; Osburg, L.; Müller, S.; Märklin, M.; Heitmann, J.S.; Zekri, L.; Rammensee, H.G.; Salih, H.R.; Jung, G. Tocilizumab, but Not Dexamethasone, Prevents CRS without Affecting Antitumor Activity of Bispecific Antibodies. J. Immunother. Cancer 2020, 8, 621. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Zugmaier, G.; Nägele, V.; Goebeler, M.E.; Brandl, C.; Stelljes, M.; Lassmann, H.; von Stackelberg, A.; Bargou, R.C.; Kufer, P. Adhesion of T Cells to Endothelial Cells Facilitates Blinatumomab-Associated Neurologic Adverse Events. Cancer Res. 2020, 80, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, N.G.; Xie, H.; Bentzen, S.; Kesari, V.; Bukhari, A.; El Chaer, F.; Lutfi, F.; Siglin, J.; Hutnick, E.; Gahres, N.; et al. Immune Effector Cell–Associated Neurotoxicity Syndrome after Chimeric Antigen Receptor T-Cell Therapy for Lymphoma: Predictive Biomarkers and Clinical Outcomes. Neuro. Oncol. 2021, 23, 112. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific Antibodies Enhance the Therapeutic Efficacy of Tumor-Directed T Cells through T Cell Receptor Co-Stimulation. Nat. Cancer 2019 11 2019, 1, 86–98. [Google Scholar] [CrossRef]
- Del Giudice, I.; Anaclerico, B.; De Propris, M.S.; Mancini, F.; Pescarmona, E.; Bizzoni, L.; Levi, A.; Calabrese, E.; Pileri, S.; Tafuri, A.; et al. Diagnostic Approach to CD5+/CD23+ Leukemic Non-Hodgkin Lymphomas Lacking Lymphnode Histopathology. Blood 2005, 106, 4690. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape without Detectable Toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Richard-Carpentier, G.; Kantarjian, H.M.; Short, N.J.; Ravandi, F.; Ferrajoli, A.; Schroeder, H.M.; Garcia-Manero, G.; Montalban Bravo, G.; Cortes, J.E.; Kwari, M.; et al. Updated Results from the Phase II Study of Hyper-CVAD in Sequential Combination with Blinatumomab in Newly Diagnosed Adults with B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2019, 134, 3807. [Google Scholar] [CrossRef]
- Fleming, S.; Venn, N.; Reynolds, J.; Nguyen, U.; Kwan, J.; Moore, J.; Yeung, D.T.; Leahy, M.F.; Greenwood, M.; Verner, E.; et al. Preliminary Minimal Residual Disease Analysis of the Australasian Leukaemia & Lymphoma Group (ALLG) ALL8 Study of Front-Line Blinatumomab with Chemotherapy in Adults with Ph Negative B-Cell Acute Lymphoblastic Leukaemia. Blood 2019, 134, 1300. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Ravandi, F.; Huang, X.; Ferrajoli, A.; Kadia, T.M.; Thompson, P.A.; Alvarado, Y.; Jain, N.; Yilmaz, M.; et al. Hyper-CVAD and Sequential Blinatumomab in Adults with Newly Diagnosed Philadelphia Chromosome-Negative B-Cell Acute Lymphoblastic Leukemia: Results from a Phase II Study. Blood 2020, 136, 9–11. [Google Scholar] [CrossRef]
- Katz, D.A.; Chu, M.P.; David, K.A.; Thieblemont, C.; Morley, N.J.; Khan, S.S.; Chen, Y.; Kalabus, J.; Morris, J.; Anderson, A.; et al. Open-Label, Phase 2 Study of Blinatumomab after First-Line Rituximab-Chemotherapy in Adults with Newly Diagnosed, High-Risk Diffuse Large B-Cell Lymphoma. Blood 2019, 134, 4077. [Google Scholar] [CrossRef]
- Webster, J.; Luskin, M.R.; Prince, G.T.; DeZern, A.E.; DeAngelo, D.J.; Levis, M.J.; Blackford, A.; Sharon, E.; Streicher, H.; Luznik, L.; et al. Blinatumomab in Combination with Immune Checkpoint Inhibitors of PD-1 and CTLA-4 in Adult Patients with Relapsed/Refractory (R/R) CD19 Positive B-Cell Acute Lymphoblastic Leukemia (ALL): Preliminary Results of a Phase I Study. Blood 2018, 132, 557. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Macias, A.; Del Real, M.; Beltran, A.L.; Kim, Y.S.; Zhang, J.; Palmer, J.; Robbins, M.; Loomis, R.; Akhtari, M.; et al. Interim Results of a Phase 1/2 Study of Pembrolizumab Combined with Blinatumomab in Patients with Relapsed/Refractory (r/r) ALL. Blood 2022, 140, 8985–8986. [Google Scholar] [CrossRef]
- Shalabi, H.; Koegel, A.; Ponduri, A.; Qin, H.; Salem, D.; Stetler-Stevenson, M.; Yuan, C.; Yates, B.; Delbrook, C.; Loh, M.; et al. Case Report: Impact of BITE on CAR-T Cell Expansion. Adv. Cell Gene Ther. 2019, 2, e50. [Google Scholar] [CrossRef]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid Tumor Immunotherapy with T Cell Engager-Armed Oncolytic Viruses. Macromol. Biosci. 2018, 18, 1700187. [Google Scholar] [CrossRef]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.M.; Silk, A.W.; Zloza, A. Oncolytic Viruses-Natural and Genetically Engineered Cancer Immunotherapies. Front. Oncol. 2017, 7, 295393. [Google Scholar] [CrossRef] [PubMed]
- Speck, T.; Heidbuechel, J.P.W.; Veinalde, R.; Jaeger, D.; Von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted Bite Expression by an Oncolytic Vector Augments Therapeutic Efficacy against Solid Tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Drug Name | Targets | Indications | Platform | First Approved Date (Country) | Confirmatory Study | Primary Endpoint | The Most Common AEs |
|---|---|---|---|---|---|---|---|
| Blinatumomab | CD3/CD19 | r/r B-ALL | BiTE | December 2014 (USA) | NCT01207388 | CRR: 78% | Pyrexia: 89% Neurological events: 53% Headache: 38% |
| Mosunetuzumab | CD3/CD20 | r/r FL | KiH | June 2022 (EU) | NCT02500407 | CRR: 60% | Neutropenia: 28% CRS: 27% Hypophosphatemia: 23% |
| Teclistamab | CD3/BCMA | r/r MM | DuoBody | August 2022 (EU) | NCT04557098 | ORR: 63% | CRS: 72% Neutropenia: 71% Anemia: 51% |
| Glofitamab | CD3/CD20 | DLBCL | 2:1 Crossmab | March 2023 (Canada) | NCT03075696 | CRR: 39% | CRS: 66% Neutropenia: 38% Anemia: 31% |
| Epcoritamab | CD3/CD20 | DLBCL | DuoBody | May 2023 (USA) | NCT03625037 | ORR: 63% | CRS: 50% Pyrexia: 24% Fatigue: 23% |
| Talquetamab | CD3/GPRC5D | r/r MM | DuoBody | August 2023 (USA) | NCT03399799 | ORR: 70% *, 64% ** | CRS: 77% *, 80% ** Skin-related events: 67% *, 70% ** Dysgeusia: 63% *, 57% ** |
| Elranatamab | CD3/BCMA | r/r MM | DuoBody | August 2023 (USA) | NCT04649359 | ORR: 61% | CRS: 58% Anemia: 49% Neutropenia: 49% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cech, P.; Skórka, K.; Dziki, L.; Giannopoulos, K. T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies. Cancers 2024, 16, 1580. https://doi.org/10.3390/cancers16081580
Cech P, Skórka K, Dziki L, Giannopoulos K. T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies. Cancers. 2024; 16(8):1580. https://doi.org/10.3390/cancers16081580
Chicago/Turabian StyleCech, Paweł, Katarzyna Skórka, Laura Dziki, and Krzysztof Giannopoulos. 2024. "T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies" Cancers 16, no. 8: 1580. https://doi.org/10.3390/cancers16081580
APA StyleCech, P., Skórka, K., Dziki, L., & Giannopoulos, K. (2024). T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies. Cancers, 16(8), 1580. https://doi.org/10.3390/cancers16081580