

DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care


Abstract
Simple Summary
Abstract
1. Introduction
2. Endocrine Disruptor Synthetic Estrogens Increase the Risk for Certain Cancers and Cardiovascular Complications
2.1. Controversial Correlations between Menopausal Hormone Therapy (MHT) and Women’s Health
2.2. Oral Contraceptives Are Endocrine Disruptors Inducing either Increased or Decreased Cancer Risk in Different Organs
3. In BRCA Gene Mutation Carriers, the Defect of Liganded ER Activation Is the Initiator of DNA Damage and Cancer Development
Both Healthy Cells and Tumor Cells with BRCA Mutation Show Compensatory Molecular Changes, Improving Genomic Stability
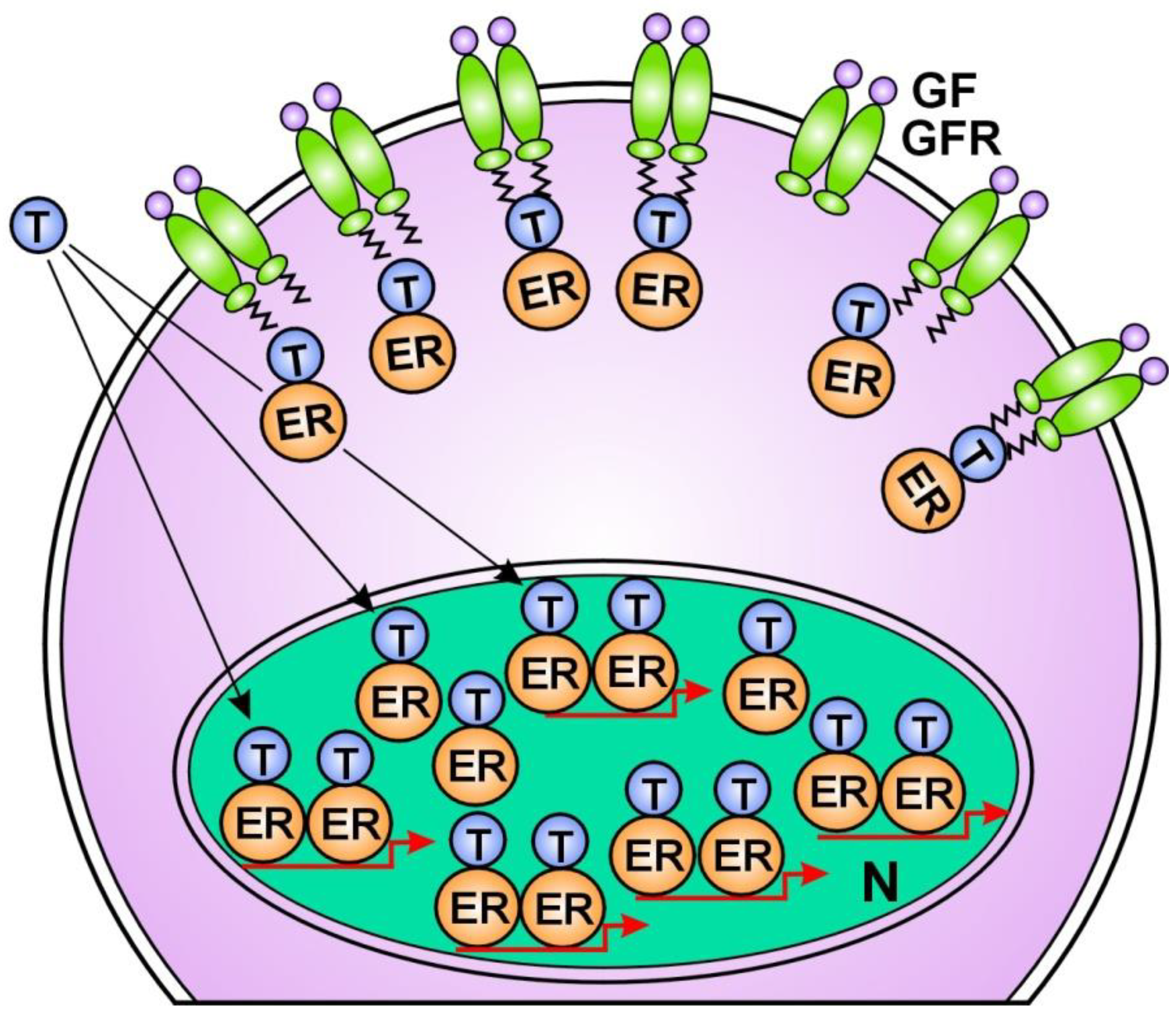
4. Estrogens Are the Principal Regulators of Genomic Machinery in Mammalian Cells
5. Estrogens Are Master Regulators of Metabolism and Energy Homeostasis via Orchestrating Adipose Tissue Functions
Secretory Activities of Visceral Adipose Tissue in Healthy Lean and Obese Cases
6. The Tumor Cell Itself Is the Frontline of Anticancer Combat
7. Peritumoral Microenvironment: The Second Line of the Antitumor Battle
8. Molecular Changes in Tumors Responsive and Non-Responsive to Endocrine Therapy
8.1. Successful Fight of Anti-estrogen Responsive Tumors against the Endocrine Disruptor Treatment
8.2. Unsuccessful Fight of Tumors Non Responsive to Endocrine Disruptor Treatment
9. Estrogen Induced Apoptosis Is Promising in Both the Prevention and Therapy of Cancer
10. Conclusions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Klapp, V.; Álvarez-Abril, B.; Leuzzi, G.; Kroemer, G.; Ciccia, A.; Galluzzi, L. The DNA Damage Response and Inflammation in Cancer. Cancer Discov. 2023, 13, 1521–1545. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, M. Mutational landscape of cancer-driver genes across human cancers. Sci. Rep. 2023, 13, 12742. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Valencia, A.; Godzik, A. Understanding oncogenicity of cancer driver genes and mutations in the cancer genomics era. FEBS Lett. 2020, 594, 4233–4246. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- García-Nieto, P.E.; Morrison, A.J.; Fraser, H.B. The somatic mutation landscape of the human body. Genome Biol. 2019, 20, 298. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, T.; Jia, Y.; Luo, X.; Gopal, P.; Li, L.; Odewole, M.; Renteria, V.; Singal, A.G.; Jang, Y.; et al. Somatic mutations increase hepatic clonal fitness and regeneration in chronic liver disease. Cell 2019, 177, 608–621.e12. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit? Int. J. Mol. Sci. 2022, 23, 13217. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Compensatory estrogen signal is capable of DNA repair in antiestrogen-responsive cancer cells via activating mutations. J. Oncol. 2020, 2020, 5418365. [Google Scholar] [CrossRef] [PubMed]
- Coelingh-Bennink, H.J.; Verhoeven, C.; Dutman, A.E.; Thijssen, J. The use of high-dose estrogens for the treatment of breast cancer. Maturitas 2017, 95, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Rogan, E.; Cavalieri, E. Mechanism of metabolic activation and DNA adduct formation by the human carcinogen diethylstilbestrol: The defining link to natural estrogens. Int. J. Cancer 2009, 124, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; de Assis, S.; Warri, A. Exposures to Synthetic Estrogens at Different Times during the Life, and Their Effect on Breast Cancer Risk. J. Mammary Gland. Biol. Neoplasia 2013, 18, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.M.; Rasanayagam, S.; Engel, C.; Rizzo, J. State of the evidence 2017: An update on the connection between breast cancer and the environment. Environ. Health 2017, 16, 94. [Google Scholar] [PubMed]
- Suba, Z. Amplified crosstalk between estrogen binding and GFR signaling mediated pathways of ER activation drives responses in tumors treated with endocrine disruptors. Recent Pat. Anticancer Drug Discov. 2018, 13, 428–444. [Google Scholar] [CrossRef]
- Stefanick, M.L. Estrogens and progestins: Background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am. J. Med. 2005, 118, 64–73. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar]
- Anderson, G.L.; Limacher, M.; Assaf, A.R. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004, 291, 1701–1712. [Google Scholar]
- Suba, Z. Synthetic Estrogens Deregulate Estrogen Receptors Inducing Thromboembolic Complications and Cancer. In Topics in Anti-Cancer Research; Rahman, A., Zaman, K., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2019; Volume 8, Chapter 2; pp. 44–73. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L.; Aragaki, A.K.; Manson, J.E.; Stefanick, M.L.; Pan, K.; Barrington, W.; Kuller, L.H.; Simon, M.S.; Lane, D.; et al. Association of Menopausal Hormone Therapy with Breast Cancer Incidence and Mortality during Long Term Follow-up of Women’s Health Initiative Randomized Clinical Trials. JAMA 2020, 324, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Aragaki, A.K.; Pan, K. Breast Cancer Prevention: Time for Change. JCO Oncol. Pract. 2021, 17, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Cortés, M.E.; Alfaro, A.A. The effects of hormonal contraceptives on glycemic regulation. Linacre Q. 2014, 81, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Mørch, L.S.; Skovlund, C.W.; Hannaford, P.C.; Iversen, L.; Fielding, S.; Lidegaard, Ø. Contemporary Hormonal Contraception and the Risk of Breast Cancer. N. Engl. J. Med. 2017, 377, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.; Boggs, D.A.; Wise, L.A.; Adams-Campbell, L.L.; Palmer, J.R. Oral contraceptive use and estrogen/progesterone receptor-negative breast cancer among African American women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, Y.; Sullivan-Halley, J.; Weiss, L.; Marchbanks, P.A.; Spirtas, R.; Ursin, G.; Burkman, R.T.; Simon, M.S.; Malone, K.E.; et al. Use of four biomarkers to evaluate the risk of breast cancer subtypes in the women’s contraceptive and reproductive experiences study. Cancer Res. 2010, 70, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Epidemiological Studies on Endometrial Cancer. Endometrial cancer and oral contraceptives: An individual participant meta-analysis of 27,276 women with endometrial cancer from 36 epidemiological studies. Lancet Oncol. 2015, 16, 1061–1070. [Google Scholar] [CrossRef]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian cancer and oral contraceptives: Collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008, 371, 303–314. [Google Scholar] [CrossRef]
- Bosetti, C.; Bravi, F.; Negri, E.; La Vecchia, C. Oral contraceptives and colorectal cancer risk: A systematic review and meta-analysis. Hum. Reprod. Update 2009, 15, 489–498. [Google Scholar] [CrossRef]
- Peng, H.; Qi, X.; Wang, Q. Long term use of oral contraceptives comprising synthetic estrogens induces an excessive breast cancer risk in BRCA mutation carrier women: A meta-analysis. Clin. Exp. Obstet. Gynecol. 2022, 49, 1. [Google Scholar] [CrossRef]
- Huber, D.; Seitz, S.; Kast, K.; Emons, G.; Ortmann, O. Use of oral contraceptives in BRCA mutation carriers and risk for ovarian and breast cancer: A systematic review. Arch. Gyn. Obst. 2020, 301, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.J.; Kennedy, R.D.; Hosey, A.M.; Harkin, D.P. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin. Cancer Res. 2009, 15, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Di, L.J. BRCA1 and estrogen/estrogen receptor in breast cancer: Where they interact? Int. J. Biol. Sci. 2014, 10, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Bégin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Spillman, M.A.; Bowcock, A.M. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene 1996, 13, 1639–1645. [Google Scholar]
- Suba, Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: Preventive and therapeutic implications. OncoTargets Ther. 2014, 7, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wang, J.; Yuan, R.; Ma, Y.; Meng, Q.; Erdos, M.R.; Pestell, R.G.; Yuan, F.; Auborn, K.J.; Goldberg, I.D.; et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999, 284, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, S.; Rosen, E.M. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology 2005, 146, 2031–2047. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fan, S.; Hu, C.; Meng, Q.; Fuqua, S.A.; Pestell, R.G.; Tomita, Y.A.; Rosen, E.M. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol. Endocrinol. 2010, 24, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.-Q.; Meng, Q.; Wang, J.-A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002, 62, 141–151. [Google Scholar] [PubMed]
- Wang, C.; Fan, S.; Li, Z.; Fu, M.; Rao, M.; Ma, Y.; Lisanti, M.P.; Albanese, C.; Katzenellenbogen, B.S.; Kushner, P.J.; et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer Res. 2005, 65, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. DNA stabilization by the upregulation of estrogen signaling in BRCA gene mutation carriers. Drug Des. Dev. Ther. 2015, 9, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef]
- Oktay, K.; Kim, J.Y.; Barad, D.; Babayev, S.N. Association of BRCA1 mutations with occult primary ovarian insufficiency: A possible explanation for the link between infertility and breast/ovarian cancer risks. J. Clin. Oncol. 2010, 28, 240–244. [Google Scholar] [CrossRef]
- Lin, W.T.; Beattie, M.; Chen, L.M.; Oktay, K.; Crawford, S.L.; Gold, E.B.; Cedars, M.; Rosen, M. Comparison of age at natural menopause in BRCA1/2 mutation carriers with a non-clinic-based sample of women in northern California. Cancer 2013, 119, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Chand, A.L.; Simpson, E.R.; Clyne, C.D. Aromataseexpression is increased in BRCA1mutationcarriers. BMC Cancer 2009, 9, 148. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. Toward a Unified Concept of Mammary Carcinogenesis; Aldaz, M.C., Gould, M.N., McLachlan, J., Slaga, T.J., Eds.; Progress in Clinical and Biological Research; Wiley-Liss: New York, NY, USA, 1997; pp. 1–16. [Google Scholar]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef]
- Suba, Z. Activating mutations of ESR1, BRCA1 and CYP19 aromatase genes confer tumor response in breast cancers treated with antiestrogens. Recent Pat. Anticancer Drug Discov. 2017, 12, 136–147. [Google Scholar] [CrossRef]
- Burga, L.N.; Hu, H.; Juvekar, A.; Tung, N.M.; Troyan, S.L.; Hofstatter, E.W.; Wulf, G.M. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Res. 2011, 13, R30. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lu, Y.; Katz, A.; Hu, Y.; Li, R. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am. J. Physiol. Endocrinol. Metab. 2007, 292, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hu, C.; Riegel, A.T.; Fan, S.; Rosen, E.M. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol. Endocrinol. 2007, 21, 1905–1923. [Google Scholar] [CrossRef]
- Zheng, L.; Annab, L.A.; Afshari, C.A.; Lee, W.H.; Boyer, T.G. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar] [CrossRef]
- Arnold, A.; Papanikolaou, A. Cyclin D1 in Breast Cancer Pathogenesis. J. Clin. Oncol. 2005, 23, 4215–4224. [Google Scholar] [CrossRef]
- Sau, A.; Lau, R.; Cabrita, M.A.; Nolan, E.; Crooks, P.A.; Visvader, J.E.; Pratt, M.C. Persistent Activation of NF-κB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA damage. Cell Stem Cell 2016, 19, 52–65. [Google Scholar] [CrossRef]
- Kim, J.; Jeong, K.; Jun, H.; Kim, K.; Bae, J.M.; Song, M.G.; Yi, H.; Park, S.; Woo, G.U.; Lee, D.W.; et al. Mutations of TP53 and genes related to homologous recombination repair in breast cancer with germline BRCA1/2 mutations. Hum. Genom. 2023, 17, 2. [Google Scholar] [CrossRef]
- Maggi, A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim. Biophys Acta 2011, 1812, 1054–1060. [Google Scholar] [CrossRef]
- Charlier, T.D.; Cornil, C.A.; Patte-Mensah, C.; Meyer, L.; Mensah-Nyagan, A.G.; Balthazart, J. Local modulation of steroid action: Rapid control of enzymatic activity. Front. Neurosci. 2015, 9, 83. [Google Scholar] [CrossRef]
- Levin, E.R. Bidirectional Signaling between the Estrogen Receptor and the Epidermal Growth Factor Receptor. Mol. Endocrinol. 2003, 17, 309–317. [Google Scholar] [CrossRef]
- Ohlsson, C.; Mohan, S.; Sjögren, K.; Tivesten, A.; Isgaard, J.; Isaksson, O.; Jansson, J.-O.; Svensson, J. The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 2009, 30, 494–535. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.B.; Franke, T.F.; Stoica, G.E.; Chambon, F.; Katzenellenbogen, B.S.; Stoica, B.A.; McLemore, M.S.; Olivo, S.E.; Stoica, A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology 2000, 141, 4503–4511. [Google Scholar] [CrossRef]
- Chan, T.W.; Pollak, M.; Huynh, H. Inhibition of insulin-like growth factor signaling pathways in mammary gland by pure antiestrogen ICI 182,780. Clin. Cancer Res. 2001, 7, 2545–2554. [Google Scholar] [PubMed]
- Sato, T.; Wang, G.; Hardy, M.P.; Kurita, T.; Cuncha, G.R.; Cooke, P.S. Role of systemic and local IGF-I in the effects of estrogen on growth and epithelial proliferation of mouse uterus. Endocrinology 2002, 143, 2673–2679. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klotz, D.M.; Hewitt, S.C.; Ciana, P.; Raviscioni, M.; Lindzey, J.K.; Foley, J.; Maggi, A.; DiAugustine, R.P.; Korach, K.S. Requirement of estrogen receptor-α in insulin-like growth factor-1 (IGF-1)-induced uterine responses and in vivo evidence for IGF-1/estrogen receptor cross talk. J. Biol. Chem. 2002, 277, 8531–8537. [Google Scholar] [CrossRef]
- DiAugustine, R.P.; Petrusz, P.; Bell, G.I.; Brown, C.F.; Korach, K.S.; McLachlan, J.A.; Teng, C.T. Influence of estrogens on mouse uterine epidermal growth factor precursor protein and messenger ribonucleic acid. Endocrinology 1988, 122, 2355–2363. [Google Scholar] [CrossRef]
- Vignon, F.; Bouton, M.M.; Rochefort, H. Antiestrogens inhibit the mitogenic effect of growth factors on breast cancer cells in the total absence of estrogens. Biochem. Biophys Res. Commun. 1987, 146, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Tsukamura, H.; Paria, B.C.; Andrews, G.K.; Dey, S.K. Differential expression of epidermal growth factor receptor (EGF-R) gene and regulation of EGF-R bioactivity by progesterone and estrogen in the adult mouse uterus. Endocrinology 1994, 134, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996, 15, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Zwijsen, R.M.; Buckle, R.S.; Hijmans, E.M.; Loomans, C.J.; Bernards, R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. 1998, 12, 3488–3498. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.; Suthiphongchai, T.; DiRenzo, J.; Ewen, M.E. P/CAF associates with cyclin D1 and potentiates its activation of the estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 5382–5387. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Parks, S.; Levin, E.R. Proximal events in signaling by plasma membrane estrogen receptors. J. Biol. Chem. 2003, 278, 2701–2712. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.W.; Levin, E.R. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef]
- Wilson, M.A.; Chrysogelos, S.A. Identification and characterization of a negative regulatory element within the epidermal growth factor receptor gene first intron in hormone-dependent breast cancer cells. J. Cell Biochem. 2002, 85, 601–614. [Google Scholar] [CrossRef]
- de Fazio, A.; Chiew, Y.E.; Sini, R.L.; Janes, P.W.; Sutherland, R.L. Expression of c-erbB receptors, heregulin and oestrogen receptor in human breast cell lines. Int. J. Cancer 2000, 87, 487–498. [Google Scholar] [CrossRef]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Interplay between insulin resistance and estrogen deficiency as co-activators in carcinogenesis. Pathol. Oncol. Res. 2012, 18, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.B.; Jang, J.S.; Park, S. Estrogen and exercise may enhance beta-cell function and mass via insulin receptor substrate-2 induction in ovariectomized diabetic rats. Endocrinology 2005, 146, 4786–4794. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M.; Hedo, J.A.; Yamada, K.M.; Kahn, C.R. The structure of insulin receptor and its subunits. Evidence for multiple non reduced forms and a 210,000 possible proreceptor. J. Biol. Chem. 1982, 257, 10392–10399. [Google Scholar] [CrossRef]
- Campello, R.S.; Fátima, L.A.; Barreto-Andrade, J.N.; Lucas, T.F.; Mori, R.C.; Porto, C.S.; Machado, U.F. Estradiol-induced regulation of GLUT4 in 3T3-L1 cells: Involvement of ESR1 and AKT activation. J. Mol. Endocrinol. 2017, 59, 257–268. [Google Scholar] [CrossRef]
- Richards, R.G.; DiAugustine, R.P.; Petrusz, P.; Clark, G.C.; Sebastian, J. Estradiol stimulates tyrosine phosphorylation of the insulin-like growth factor-1 receptor and insulin receptor substrate-1 in the uterus. Proc. Natl. Acad. Sci. USA 1996, 93, 12002–12007. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.; Salerno, M.; Panno, M.L.; Bellizzi, D.; Sisci, D.; Miglietta, A.; Surmacz, E.; Andò, S. Estradiol increases IRS-1 gene expression and insulin signaling in breast cancer cells. Biochem. Biophys. Res. Commun. 2001, 288, 685–689. [Google Scholar] [CrossRef]
- Medina, R.A.; Meneses, A.M.; Vera, J.C.; Guzman, C.; Nualart, F.; Astuya, A.; García, M.d.L.A.; Kato, S.; Carvajal, A.; Pinto, M.; et al. Estrogen and progesterone up-regulate glucose transporter expression in ZR-75-1 human breast cancer cells. Endocrinology 2003, 144, 4527–4535. [Google Scholar] [CrossRef]
- Garrido, P.; Morán, J.; Alonso, A.; González, S.; González, C. 17β-estradiol activates glucose uptake via GLUT4 translocation and PI3K/Akt signaling pathway in MCF-7 cells. Endocrinology 2013, 154, 1979–1989. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signaling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Mahboobifard, F.; Pourgholami, M.H.; Jorjani, M.; Dargahi, L.; Amiri, M.; Sadeghi, S.; Tehrani, F.R. Estrogen as a key regulator of energy homeostasis and metabolic health. Biomed. Pharmacother. 2022, 156, 113808. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Low estrogen exposure and/or defective estrogen signaling induces disturbances in glucose uptake and energy expenditure. J. Diabet. Metab. 2013, 4, 272–281. [Google Scholar] [CrossRef]
- Donohoe, C.L.; Doyle, S.L.; Reynolds, J.V. Visceral adiposity, insulin resistance and cancer risk. Diabetol. Metab. Syndr. 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Rajsheker, S.; Manka, D.; Blomkalns, A.L.; Chatterjee, T.K.; Stoll, L.L.; Weintraub, N. Crosstalk between perivascular adipose tissue and blood vessels. Curr. Opin. Pharmacol. 2010, 10, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Roca-Rivada, A.; Alonso, J.; Al-Massadi, O.; Castelao, C.; Peinado, J.R.; Seoane, L.M.; Casanueva, F.F.; Pardo, M. Secretome analysis of rat adipose tissues shows location-specific roles for each depot type. J. Proteom. 2011, 74, 1068–1079. [Google Scholar] [CrossRef]
- Pelekanou, V.; Leclercq, G. Recent insights into the effect of natural and environmental estrogens on mammary development and carcinogenesis. Int. J. Dev. Biol. 2011, 55, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Leng, Y.; Gong, Y. Bone Marrow Fat and Hematopoiesis. Front. Endocrinol. 2018, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Is There a Brain Microbiome? Neurosci. Insights 2021, 16, 26331055211018709. [Google Scholar] [CrossRef] [PubMed]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadalsites of estrogenbiosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Bélanger, A.; Luu-The, V.; Labrie, C.; Simard, J.; Cusan, L.; Gomez, J.-L.; Candas, B. DHEA and the intracrine formation of androgens and estrogens in peripheral target tissues: Its role during aging. Steroids 1998, 63, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Bjune, J.-I.; Strømland, P.P.; Jersin, R.Å.; Mellgren, G.; Dankel, S.N. Metabolic and Epigenetic Regulation by Estrogen in Adipocytes. Front. Endocrinol. 2022, 13, 828780. [Google Scholar] [CrossRef] [PubMed]
- Dieudonné, M.N.; Leneveu, M.C.; Giudicelli, Y.; Pecquery, R. Evidence for functional estrogen receptors α and β in human adipose cells: Regional specificities and regulation by estrogens. Am. J. Physiol. Cell Physiol. 2004, 286, C655–C661. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mariman, E.; Renes, J.; Keijer, J. The Secretory Function of Adipocytes in the Physiology of White Adipose Tissue. J. Cell Physiol. 2008, 216, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Brown, L.M.; Woods, S.C.; Benoit, S.C. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes 2006, 55, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.R.; Misso, M.; Hewitt, K.N.; Hill, R.A.; Boon, W.C.; Jones, M.E.; Kovacic, A.; Zhou, J.; Clyne, C.D. Estrogen—The good, the bad, and the unexpected. Endocr. Rev. 2005, 26, 322–330. [Google Scholar] [CrossRef]
- Combs, T.P.; Pajvani, U.B.; Berg, A.H.; Lin, Y.; Jelicks, L.A.; Laplante, M.; Nawrocki, A.R.; Rajala, M.W.; Parlow, A.F.; Cheeseboro, L.; et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 2004, 145, 367–383. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Suba, Z. Crossroad between obesity and cancer: A defective signaling function of heavily lipid laden adipocytes. In Crosstalk in Biological Processes; El-Esawi, M.A., Ed.; InTechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Armani, A.; Berry, A.; Cirulli, F.; Caprio, M. Molecular mechanisms underlying metabolic syndrome: The expanding role of the adipocyte. FASEB J. 2017, 31, 4240–4255. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between Adipocytes and Immune Cells in Adipose Tissue Inflammation and Metabolic Dysregulation in Obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar] [CrossRef]
- Purohit, A.; Newman, S.P.; Reed, M.J. The role of cytokines in regulating estrogen synthesis: Implications for the etiology of breast cancer. Breast Cancer Res. 2002, 4, 65. [Google Scholar] [CrossRef]
- Ye, J.; McGuinness, O.P. Inflammation during obesity is not all bad: Evidence from animal and human studies. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E466–E477. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Tseng, J.-H.; Kahn, C.R. Insulin and insulin-like growth factor receptors act as ligand-specific amplitude modulators of a common pathway regulating gene. J. Biol. Chem. 2010, 285, 17235–17245. [Google Scholar] [CrossRef]
- Smith, C.L. Cross-talk between peptide growth factor and estrogen receptor signaling pathways. Biol. Reprod. 1998, 58, 627–632. [Google Scholar] [CrossRef]
- Xu, J.; Xiang, Q.; Lin, G.; Fu, X.; Zhou, K.; Jiang, P.; Zheng, S.; Wang, T. Estrogen improved metabolic syndrome through down-regulation of VEGF and HIF-1α to inhibit hypoxia of periaortic and intra-abdominal fat in ovariectomized female rats. Mol. Biol. Rep. 2012, 39, 8177–8185. [Google Scholar] [CrossRef]
- Caizzi, L.; Ferrero, G.; Cutrupi, S.; Cordero, F.; Ballaré, C.; Miano, V.; Reineri, S.; Ricci, L.; Friard, O.; Testori, A.; et al. Genome-wide activity of unliganded estrogen receptor-α in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4892–4897. [Google Scholar] [CrossRef]
- Stubbins, R.E.; Najjar, K.; Holcomb, V.B.; Hong, J.; Núñez, N.P. Oestrogen alters adipocyte biology and protects female mice from adipocyte inflammation and insulin resistance. Diabetes Obes. Metab. 2012, 14, 58–66. [Google Scholar] [CrossRef]
- Suba, Z. Rosetta Stone for Cancer Cure: Comparison of the Anticancer Capacity of Endogenous Estrogens, Synthetic Estrogens and Antiestrogens. Oncol. Rev. 2023, 17, 10708. [Google Scholar] [CrossRef]
- Thomas, E.T.; Mar, C.D.; Glasziou, P.; Wright, G.; Barratt, A.; Bell, K.J.L. Prevalence of incidental breast cancer and precursor lesions in autopsy studies: A systematic review and meta-analysis. BMC Cancer 2017, 17, 808. [Google Scholar] [CrossRef]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef]
- Tan, P.H.; Ellis, I.; Allison, K.; Brogi, E.; Fox, S.B.; Lakhani, S.; Lazar, A.J.; Morris, E.A.; Sahin, A.; Salgado, R.; et al. The 2019 World Health Organization Classification of Tumours of the Breast. Histopathology 2020, 77, 181–185. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Matta, J.; Morales, L.; Ortiz, C.; Adams, D.; Vargas, W.; Casbas, P.; Dutil, J.; Echenique, M.; Suárez, E. Estrogen Receptor Expression Is Associated with DNA Repair Capacity in Breast Cancer. PLoS ONE 2016, 11, e0152422. [Google Scholar] [CrossRef]
- Hayes, D.F. Tamoxifen: Dr. Jekyll and Mr. Hyde? J. Natl. Cancer Inst. 2004, 96, 895–897. [Google Scholar] [CrossRef][Green Version]
- Munzone, E.; Colleoni, M. Optimal management of luminal breast cancer: How much endocrine therapy is long enough? Ther. Adv. Med. Oncol. 2018, 10, 1758835918777437. [Google Scholar] [CrossRef]
- Arpino, G.; Weiss, H.; Lee, A.V.; Schiff, R.; De Placido, S.; Osborne, C.K.; Elledge, R.M. Estrogen receptor-positive, progesterone receptor-negative breast cancer: Association with growth factor receptor expression and tamoxifen resistance. J. Natl. Cancer Inst. 2005, 97, 1254–1261. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Rouanet, P.; Roger, P.; Rousseau, E.; Thibault, S.; Romieu, G.; Mathieu, A.; Cretin, J.; Barneon, G.; Granier, M.; Maran-Gonzalez, A.; et al. HER2 overexpression a major risk factor for recurrence in pT1a-bN0M0 breast cancer: Results from a French regional cohort. Cancer Med. 2014, 3, 134–142. [Google Scholar] [CrossRef]
- Suba, Z. Light deficiency confers breast cancer risk by endocrine disorders. Recent Pat. Anticancer Discov. 2012, 7, 337–344. [Google Scholar] [CrossRef]
- Chew, V.; Toh, H.; Abastado, J. Immune Microenvironment in Tumor Progression: Characteristics and Challenges for Therapy. J. Oncol. 2012, 2012, 2312956. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef]
- Linares, J.; Marín-Jiménez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome during Cancer progression and Therapy. Front. Cell Dev. Biol. 2021, 8, 621070. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Pejerrey, S.M.; Dustin, D.; Kim, J.A.; Gu, G.; Rechoum, Y.; Fuqua, S.A.W. The impact of ESR1 mutations on the treatment of metastatic breast cancer. Horm. Cancer 2018, 9, 215–228. [Google Scholar] [CrossRef]
- Miano, V.; Ferrero, G.; Reineri, S.; Caizzi, L.; Annaratone, L.; Ricci, L.; Cutrupi, S.; Castellano, I.; Cordero, F.; De Bortoli, M. Luminal long non-coding RNAs regulated by estrogen receptor alpha in a ligand-independent manner show functional roles in breast cancer. Oncotarget 2016, 7, 3201–3216. [Google Scholar] [CrossRef]
- Lei, J.T.; Gou, X.; Seker, S.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef]
- Stellato, C.; Porreca, I.; Cuomo, D.; Tarallo, R.; Nassa, G.; Ambrosino, C. The “busy life” of unliganded estrogen receptors. Proteomics 2016, 16, 288–300. [Google Scholar] [CrossRef]
- Fan, P.; Jordan, V.C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019, 2, 198–209. [Google Scholar] [CrossRef]
- Yu, B.; Wu, K.; Wang, X.; Zhang, J.; Wang, L.; Jiang, Y.; Zhu, X.; Chen, W.; Yan, M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Giusti, I.; Francesco, M.; Di D’Ascenzo, S.; Palmerini, M.G.; Macchiarelli, G.; Carta, G.; Dolo, V. Ovarian cancer-derived extracellular vesicles affect normal human fibroblast behavior. Cancer Biol. Ther. 2018, 19, 722. [Google Scholar] [CrossRef]
- Dai, G.; Yao, X.; Zhang, Y.; Gu, J.; Geng, Y.; Xue, F.; Zhang, J. Colorectal cancer cell–derived exosomes containing miR-10b regulate fibroblast cells via the PI3K/Akt pathway. Bull. Cancer 2018, 105, 336–349. [Google Scholar] [CrossRef]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef]
- Rothenberger, N.J.; Somasundaram, A.; Stabile, L.P. The Role of the Estrogen Pathway in the Tumor Microenvironment. Int. J. Mol. Sci. 2018, 19, 611. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Reed, M.J.; Purohit, A. Breast Cancer and the Role of Cytokines in Regulating Estrogen Synthesis: An Emerging Hypothesis. Endocr. Rev. 1997, 18, 701–715. [Google Scholar] [CrossRef]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Dörsam, B.; Bösl, T.; Reiners, K.S.; Barnert, S.; Schubert, R.; Shatnyeva, O.; Zigrino, P.; Engert, A.; Hansen, H.P.; Von Strandmann, E.P. Hodgkin lymphoma-derived extracellular vesicles change the secretome of fibroblasts toward a CAF phenotype. Front. Immunol. 2018, 9, 1358. [Google Scholar] [CrossRef]
- Dannenfelser, R.; Nome, M.; Tahiri, A.; Ursini-Siegel, J.; Vollan, H.K.M.; Haakensen, V.D.; Helland, A.; Naume, B.; Caldas, C.; Borresen-Dale, A.L.; et al. Data-driven analysis of immune infiltrate in a large cohort of breast cancer and its association with disease progression, er activity, and genomic complexity. Oncotarget 2017, 8, 57121–57133. [Google Scholar] [CrossRef]
- Suzuki, T.; Miki, Y.; Akahira, J.I.; Moriya, T.; Ohuchi, N.; Sasano, H. Review: Aromatase in human breast carcinoma as a key regulator of intratumoral sex steroid concentrations. Endocr. J. 2008, 55, 455–463. [Google Scholar] [CrossRef]
- Bollet, M.A.; Savignoni, A.; De Koning, L.; Tran-Perennou, C.; Barbaroux, C.; Degeorges, A.; Sigal-Zafrani, B.; Almouzni, G.; Cottu, P.; Salmon, R.; et al. Tumor aromatase expression as a prognostic factor for local control in young breast cancer patients after breast-conserving treatment. Breast Cancer Res. 2009, 11, R54. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Ghosh, S.; Procopio, M.G.; Mazzeo, L.; Bordignon, P.; Ostano, P.; Goruppi, S.; Bottoni, G.; Katarkar, A.; Levesque, M.; et al. Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation. J. Clin. Investig. 2018, 128, 5465–5478. [Google Scholar] [CrossRef]
- Vivacqua, A.; Muoio, M.G.; Miglietta, A.M.; Maggiolini, M. Differential microRNA landscape triggered by estrogens in cancer associated fibroblasts (CAFs) of primary and metastatic breast tumors. Cancers 2019, 11, 412. [Google Scholar] [CrossRef]
- Zhang, Y.; Cong, X.; Li, Z.; Xue, Y. Estrogen facilitates gastric cancer cell proliferation and invasion through promoting the secretion of interleukin-6 by cancer-associated fibroblasts. Int. Immunopharmacol. 2020, 78, 105937. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFbetasignalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.; Xu, L.; Dai, Y.; Teng, Y.; Ma, S.; Ho, S.-H.; Kim, J.-M.; Yu, S.S.; Kim, S.; et al. Multicenter, randomized study of genetically modified recombinant human interleukin-11 to prevent chemotherapy-induced thrombocytopenia in cancer patients receiving chemotherapy. Support. Care Cancer 2012, 20, 1875–1884. [Google Scholar] [CrossRef]
- da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Facilitators of cancer and obesity-induced cancer. Annu. Rev. Cancer Biol. 2021, 5, 17–38. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Lorvik, K.B.; Hammarstrom, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific th1 cells protects against b-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. Cd4(+) t cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Fish, E.N. The x-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008, 8, 737–744. [Google Scholar] [CrossRef]
- Khan, D.; Ansar Ahmed, S. The immune system is a natural target for estrogen action: Opposing effects of estrogen in two prototypical autoimmune diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef]
- Ali, H.R.; Provenzano, E.; Dawson, S.J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between cd8+ t-cell infiltration and breast cancer survival in 12,439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef]
- Bentrem, D.J.; Jordan, V.C. Role of antiestrogens and aromatase inhibitors in breast cancer treatment. Curr. Opin. Obstet. Gynecol. 2002, 14, 5–12. [Google Scholar] [CrossRef]
- Jordan, V.C.; Lewis-Wambi, J.S.; Patel, R.R.; Kim, H.; Ariazi, E.A. New hypotheses and opportunities in endocrine therapy: Amplification of oestrogen-induced apoptosis. Breast. 2009, 18 (Suppl. S3), S10–S17. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef]
- Fan, P.; Agboke, F.A.; Cunliffe, H.E.; Ramos, P.; Jordan, V.C. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur. J. Cancer 2014, 50, 2866–2876. [Google Scholar] [CrossRef]
- Tilghman, S.L.; Sabnis, G.; Brodie, A.M.H. Upregulation of AIB1, aromatase and ERα provides long-term estrogen-deprived human breast cancer cells with a mechanistic growth advantage for survival. Horm. Mol. Biol. Clin. Investig. 2011, 3, 357–366. [Google Scholar] [CrossRef]
- Ishii, Y.; Waxman, S.; Germain, D. Tamoxifen Stimulates the Growth of Cyclin D1–Overexpressing Breast Cancer Cells by Promoting the Activation of Signal Transducer and Activator of Transcription 3. Cancer Res. 2008, 68, 852–860. [Google Scholar] [CrossRef]
- Zhou, Y.; Yau, C.; Joe, W.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Eppenberger, U.; Eppenberger-Castori, S.; Benz, C.C. Enhanced NFκB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007, 7, 59. [Google Scholar] [CrossRef]
- Frasor, J.; El-Shennawy, L.; Stender, J.D.; Kastrati, I. NFκB Affects Estrogen Receptor Expression and Activity in Breast Cancer through Multiple Mechanisms. Mol. Cell Endocrinol. 2015, 418, 235–239. [Google Scholar] [CrossRef]
- Hayes, E.L.; Lewis-Wambi, J.S. Mechanisms of endocrine resistance in breast cancer: An overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015, 17, 40. [Google Scholar] [CrossRef]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Jänicke, F.; et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar] [CrossRef]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor alpha gene ESR1 amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef]
- Magnani, L.; Frige, G.; Gadaleta, R.M.; Corleone, G.; Fabris, S.; Kempe, H.; Verschure, P.J.; Barozzi, I.; Vircillo, V.; Hong, S.P.; et al. Acquired CYP19A1 amplification is an early specific mechanism of aromatase inhibitor resistance in ER alpha metastatic breast cancer. Nat. Genet. 2017, 49, 444–450. [Google Scholar] [CrossRef]
- Suba, Z. The pitfall of the transient, inconsistent anticancer capacity of antiestrogens and the mechanism of apparent antiestrogen resistance. Drug Des. Dev. Ther. 2015, 9, 4341–4353. [Google Scholar] [CrossRef]
- Jin, K.; Kong, X.; Shah, T.; Penet, M.-F.; Wildes, F.; Sgroi, D.C.; Ma, X.J.; Huang, Y.; Kallioniemi, A.; Landberg, G.; et al. The HOXB7 protein renders breast cancer cells resistant to tamoxifen through activation of the egfr pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 2736–2741. [Google Scholar] [CrossRef]
- Jin, K.; Park, S.; Teo, W.W.; Korangath, P.; Cho, S.S.; Yoshida, T.; Győrffy, B.; Goswami, C.P.; Nakshatri, H.; Cruz, L.A.; et al. HOXB7 is an ERA cofactor in the activation of HER2 and multiple er target genes leading to endocrine resistance. Cancer Discov. 2015, 5, 944–959. [Google Scholar] [CrossRef]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef]
- Chen, A.C.; Migliaccio, I.; Rimawi, M.; Lopez-Tarruella, S.; Creighton, C.J.; Massarweh, S.; Huang, C.; Wang, Y.-C.; Batra, S.K.; Gutierrez, M.C.; et al. Upregulation of MUCIN4 in ER-positive/HER2-overexpressing breast cancer xenografts with acquired resistance to endocrine and HER2-targeted therapies. Breast Cancer Res. Treat. 2012, 134, 583–593. [Google Scholar] [CrossRef]
- Fan, P.; Wang, J.; Santen, R.J.; Yue, W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res. 2007, 67, 1352–1360. [Google Scholar] [CrossRef]
- Song, R.X.; Barnes, C.J.; Zhang, Z.; Bao, Y.; Kumar, R.; Santen, R.J. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Moerkens, M.; Ramaiahgari, S.; de Bont, H.; Price, L.; Meerman, J.; van de Water, B. Elevated insulin-like growth factor-1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011, 13, R52. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, Q.; Li, S.; Shi, Q.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Farahmand, L.; Hosseinzade, A.; Eslami-S, Z.; Majidzadeh-A, K. Estrogen can restore Tamoxifen sensitivity in breast cancer cells amidst the complex network of resistance. Biomed. Pharmacother. 2017, 93, 1320–1325. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Linking estrogen induced apoptosis with decreases in mortality following long term adjuvant tamoxifen therapy. 2014, 106, dju296.
- Jordan, V.C. The new biology of estrogen induced apoptosis applied to treat and prevent breast cancer. Endocr. Relat. Cancer 2015, 22, R1–R31. [Google Scholar] [CrossRef]
- Jordan, V.C.; Fan, P.; Abderrahman, B.; Maximov, P.Y.; Hawsawi, Y.M.; Bhattacharya, P.; Pokharel, N. Sex steroid induced apoptosis as a rational strategy to treat anti-hormone resistant breast and prostate cancer. Discov. Med. 2016, 21, 411–427. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Subtype of Breast Cancer | Receptor Status | Signs of DNA Damage | Sigs of DNA Repair | Proliferative Activity Endocrine | Response to Therapy |
|---|---|---|---|---|---|
| Luminal A type | ER overexpression | no | ER overexpression | low | good in 50% |
| (50–60%) | PR positive | ||||
| Luminal B type | ER positive | PR negative | ER positive | increased | moderate/inverse |
| (10%) | PR pos/neg | PR positive | |||
| HER2 pos/neg | HER2 positive | ||||
| HER2 enriched | ER negative | ER negative | HER2 rich | high | no |
| (20%) | PR negative | PR negative | |||
| HER2 rich | |||||
| Triple negative | ER negative | ER negative | no | high | no |
| (10%) | PR negative | PR negative | |||
| HER2 negative | HER2 negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suba, Z. DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care. Cancers 2024, 16, 1573. https://doi.org/10.3390/cancers16081573
Suba Z. DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care. Cancers. 2024; 16(8):1573. https://doi.org/10.3390/cancers16081573
Chicago/Turabian StyleSuba, Zsuzsanna. 2024. "DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care" Cancers 16, no. 8: 1573. https://doi.org/10.3390/cancers16081573
APA StyleSuba, Z. (2024). DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care. Cancers, 16(8), 1573. https://doi.org/10.3390/cancers16081573