2. Materials and Methods
2.1. Cell Culture
SET-2, MARIMO, and K562 cells were cultured in 80% RPMI 1640 (Gibco™ RPMI-1640, Invitrogen-Life Technologies, Paisley, UK) and 20% fetal bovine serum (FBS, HyClone Fetal Bovine Serum, Thermo Fisher Scientific, Essex, UK) at 37 °C with 5% CO2. Additionally, the medium was supplemented with 1% penicillin/streptomycin (Gibco™ Penicillin-Streptomycin 10,000 U/mL, Invitrogen-Life Technologies, Paisley, UK) to prevent bacterial contamination.
2.2. Patient Samples
Patient samples used in this study were obtained with written informed consent from the patient and were completely anonymized. Mononuclear cells were extracted with Ficoll from the bone marrow (BM) or peripheral blood (PB) of 38 patients with ET and
CALR mutations (26 with type 1 mutations and 12 with type 2 mutations), 21 patients with ET and the p.V617F JAK2 mutation, and 10 healthy donors as controls. Most of the samples from the patients were taken at diagnosis (
Table S1).
2.3. qPCR
Total RNA was extracted using TRIzol
® (Thermo Fisher Scientific Inc., Paisley, UK) following the manufacturer’s specifications. The extracted RNA was purified with the commercial DNA-free
TM DNA Removal kit (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania) and reverse-transcribed with Invitrogen™ M-MLV-RT and random hexamers. The expression of the genes of interest was analyzed by qPCR using primers and PrimeTime™ probes from IDT (Integrated DNA Technologies Inc., Coralville, IA, USA) (
Table S2). All qPCR reactions were performed in 384-well plates using the CFX384™ Touch™ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) with an amplification profile of 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 95 °C for 15 s, followed by 1 min at 60 °C. The amplification reactions were performed in a final volume of 10 µL per well with 5 µL of iTaq™ Universal Probes Supermix (Bio-Rad Laboratories, Hercules, CA, USA), 0.5 µL of PrimeTime™ primers and probes (20×) (IDT, Integrated DNA Technologies Inc., Coralville, IA, USA), 1 µL of cDNA (12.5 ng/µL), and 3.5 µL of nuclease-free water. Each sample was analyzed in triplicate, and appropriate negative controls were included. C
T values were collected using the CFX Manager™ software v3.1 (Bio-Rad Laboratories, Hercules, CA, USA). The expression level of each gene was normalized to the expression of
TBP (#Hs.PT.58v.39858774 PrimeTime™ predesigned qPCR Assays; Integrated DNA Technologies Inc., Coralville, IA, USA).
2.4. Western Blot
Protein extraction was performed on 500,000 cells per cell line in triplicate. After washing with PBS (Gibco™, Thermo Fisher Scientific Inc., Paisley, UK) and discarding the supernatant, cells were lysed in 80 μL of RIPA Buffer (25 mM Tris-HCl at pH 7.6, 150 mM NaCl, 0.1% (w/v) sodium dodecyl sulfate (SDS), 1% (w/v) sodium deoxycholate, 1% (v/v) IGEPAL® CA-630 (Sigma-Aldrich Co., St. Louis, MO, USA)), and protease inhibitors (cOmplete™, Roche® Diagnostics GmbH, Mannheim, Germany). Samples were sonicated, and the protein concentration was quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) following the specifications from the manufacturer.
For Western Blot, proteins were first separated by SDS-PAGE on 12% denaturing acrylamide gels. For this, 30 μg of protein was diluted in 8 μL of 4× Laemmli loading buffer (Bio-Rad Laboratories, Hercules, CA, USA), and 10% of β-mercaptoethanol and distilled water were immediately added to the final volume of 35 μL. Samples were then heated at 100 °C for 5 min and loaded onto the gel. Electrophoresis was performed for 1.5 h at 120 V and room temperature. After that, proteins were transferred to 0.45 μm pore nitrocellulose membranes (Whatman® Protran®, Merck-Millipore, Billerica, MA, USA) for 1 h at 350 mA and 4 °C in a Mini Trans-Blot® Cell system (Bio-Rad Laboratories, Hercules, CA, USA) with a transfer buffer composed of 25 mM Tris, 192 mM glycine, and 20% methanol. Next, non-specific binding sites were blocked by incubating the membrane in 10–20 mL of 5% (w/v) skimmed milk or BSA dissolved in TBS (20 mM Tris and 150 mM NaCl) with 0.1% Tween at RT for at least 1 h with agitation. After that, the membranes were incubated at 4 °C overnight with shaking and 5 mL of a solution of primary antibodies, which were specific for the detection of each protein, diluted in the same solution prepared for blocking: anti-RXRα (#3085, Cell Signaling Technology®, Danvers, MA, USA), diluted 1:1000 in 5% milk; anti-PPARγ (#2435, Cell Signaling Technology®, Danvers, MA, USA), diluted 1:1000 in 5% BSA; and anti-β-actin (#8457, Cell Signaling Technology®, Danvers, MA, USA), diluted 1:10,000 in 5% BSA. The next day, the membranes were washed three times with TBS with 0.1% Tween for 10 min to remove excess primary antibody. Subsequently, they were incubated with shaking for 1 h at room temperature in 5 mL of secondary anti-rabbit IgG antibody linked to HRP (#7074, Cell Signaling Technology®, Danvers, MA, USA), diluted 1:2000 (for anti-RXRα and anti-PPARγ) or 1:10,000 (for anti-β-actin) in 5% BSA. The membranes were then additionally washed three more times with TBS with 0.1% Tween for 10 min to remove excess secondary antibody.
For detection, the commercial system Lumi-Light
PLUS Western Blotting Substrate (Roche Diagnostics GmbH, Mannheim, Germany) was used according to the instructions from the manufacturer. The chemiluminescent signal intensity was detected using a ChemiDoc XRS+ system coupled with the Bio-Rad Universal Hood II system (Bio-Rad Laboratories, Hercules, CA, USA) and quantified using ImageJ v1.53 software [
3]. The expression levels of the target proteins were normalized by dividing the signal obtained for the protein in each sample by that obtained for β-actin in the same sample from the same gel. Membrane images were cropped to show results that were specific to the proteins of interest. All uncropped Western Blot images can be found in
Figure S1.
2.5. Cell Line Treatments and Viability Assessment
Cell line treatments were carried out in 96-well ELISA plates, with a final volume of 100 μL per well (300 cells/μL). All compounds were dissolved in DMSO at concentrations that allowed for the exposure of the cells to the desired doses of compound, while adding less than 0.5% DMSO to avoid its cytotoxic effects. Controls consisted of wells containing cells without DMSO and with the maximum amount of DMSO added in each treatment. Each treatment and control were tested in triplicate on each of the cell lines. Plates were incubated for 72 h in a Galaxy B incubator (RS Biotech Laboratory Equipment Ltd., Irvine, UK) at 37 °C with 5% CO2.
To determine the number of viable cells after exposure to the different treatments, a colorimetric assay was performed using the commercial system CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI, USA). To do this, 20 μL of the reagent was added to each well that already contained a volume of 100 μL. The plates were incubated for 2.5 h at 37 °C with 5% CO2. Subsequently, absorbance at 492 nm was measured using a Multiskan Ex Microplate Photometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). It was verified that the added DMSO had no cytotoxic effects on the cells by comparing the viability obtained in the control of cells without compound with that of cells with the highest percentage of DMSO used. Finally, the percentage of live cells after exposure to each treatment was calculated, considering 100% viability for the wells to which DMSO without compound was added.
2.6. Statistics
Statistical calculations were performed using StataSE v12 software (StataCorp LP, College Station, TX, USA). The significance level (α) was set at 0.05. Differences were considered non-significant (ns) when p > 0.05, significant (*) when p < 0.05, very significant (**) when p < 0.01, and highly significant (***) when p < 0.001. All data obtained in this study were analyzed using an ANOVA test followed by multiple comparisons, except for the gene expression data of patients. In this case, the median test was used, followed by multiple comparisons, as the residuals did not fit a normal distribution or follow a similar distribution between groups for any of the genes analyzed. For the statistical analysis of qPCR, when no expression was detected in any of the samples, it was considered that the CT value was 40, since it is the detection limit of the system (above this cycle, no signal is detected). In these cases, it is important to note that the differences are at least as significant as indicated in the graphs, but it cannot be ruled out that they may be higher than indicated (if the CT value is greater than 40).
3. Results and Discussion
When performing a BLASTP search of the NHR-2 protein sequence provided by UniProt (Q10902) using UniProtKB/Swiss-Prot reference proteomes for
Homo sapiens [NCBI:txid9606] as the target database, many homologous sequences were found (
Figure S2). From all of them, we selected the genes with a percentage of identity in the encoded protein sequence above 30% with NHR-2 that could be of interest for ET patients (previously related to hematological disorders) (
Table S3) and analyzed their expression in cell lines derived from
CALR- or
JAK2-mutated patients with leukemic transformation of ET by qPCR. We focused on the cell lines SET-2 (with a heterozygous p.V617F mutation in
JAK2 [
4] and established from an ET patient at megakaryoblastic leukemic transformation [
5]) and MARIMO (with a heterozygous type 1
CALR mutation [
6] and established from an ET patient with a therapy-related acute myeloid leukemia [
7]). We also included in the analysis the cell line K562 (with the fusion
BCR::ABL1 established from a pleural effusion of a patient with chronic myeloid leukemia in terminal blastic crisis [
8]) as a control of a myeloproliferative neoplasm other than from ET. As we hypothesized that the overexpression of
nhr-2 counterparts could be due to a JAK2/STAT-independent mechanism derived from mutant calreticulin, we expected that these orthologs would only be overexpressed in the MARIMO cell line (
CALR-mutated) and not in the SET-2 cell line (
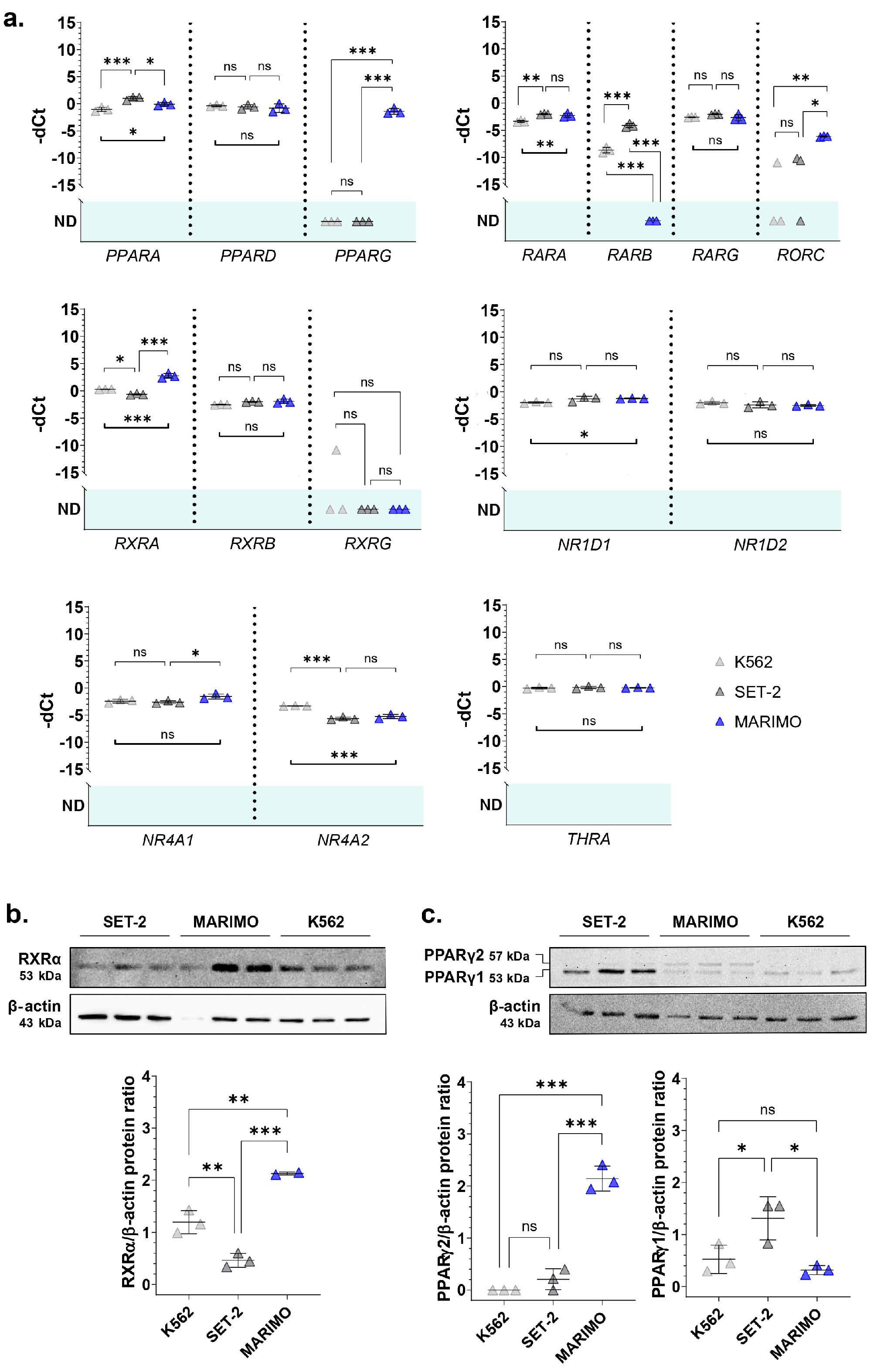
JAK2-mutated) when compared to the control cell line K562. The results of the qPCR analyses showed that this pattern of expression was true for
RXRA,
PPARG, and
RORC (
Figure 1a). These three genes encode nuclear receptors that regulate the transcription of multiple target genes, yet their regulatory mechanisms and functions differ. Thus, RXRα (retinoid X receptor alpha) and PPARγ (peroxisome proliferator-activated receptor gamma) are activated by retinoids and fatty acids, respectively. In addition, RXRα can bind to its target genes by forming homodimers or heterodimers, while PPARγ generally binds DNA as heterodimers with RXRXα [
9,
10]. Interestingly, both genes have previously been found to be altered in myeloid leukemia cells and proposed as treatments for myeloid cancers [
11,
12,
13]. Although the functions of RORC (nuclear receptor ROR-gamma) are not well understood, it is well known that this nuclear receptor has key functions in lymphoid organogenesis and thymopoiesis, but no function has been described in the myeloid tissue [
14]. Therefore, we decided to focus our attention on a deeper analysis of
RXRA and
PPARG.
Then, we verified that the
RXRA and
PPARG expression data were correlated at the protein level using Western Blot. The results show that RXRα was also overexpressed in the MARIMO cell line (
Figure 1b). Regarding PPARγ, differences were found in the isoform PPARγ2, which is also only overexpressed in the MARIMO cell line (
Figure 1c).
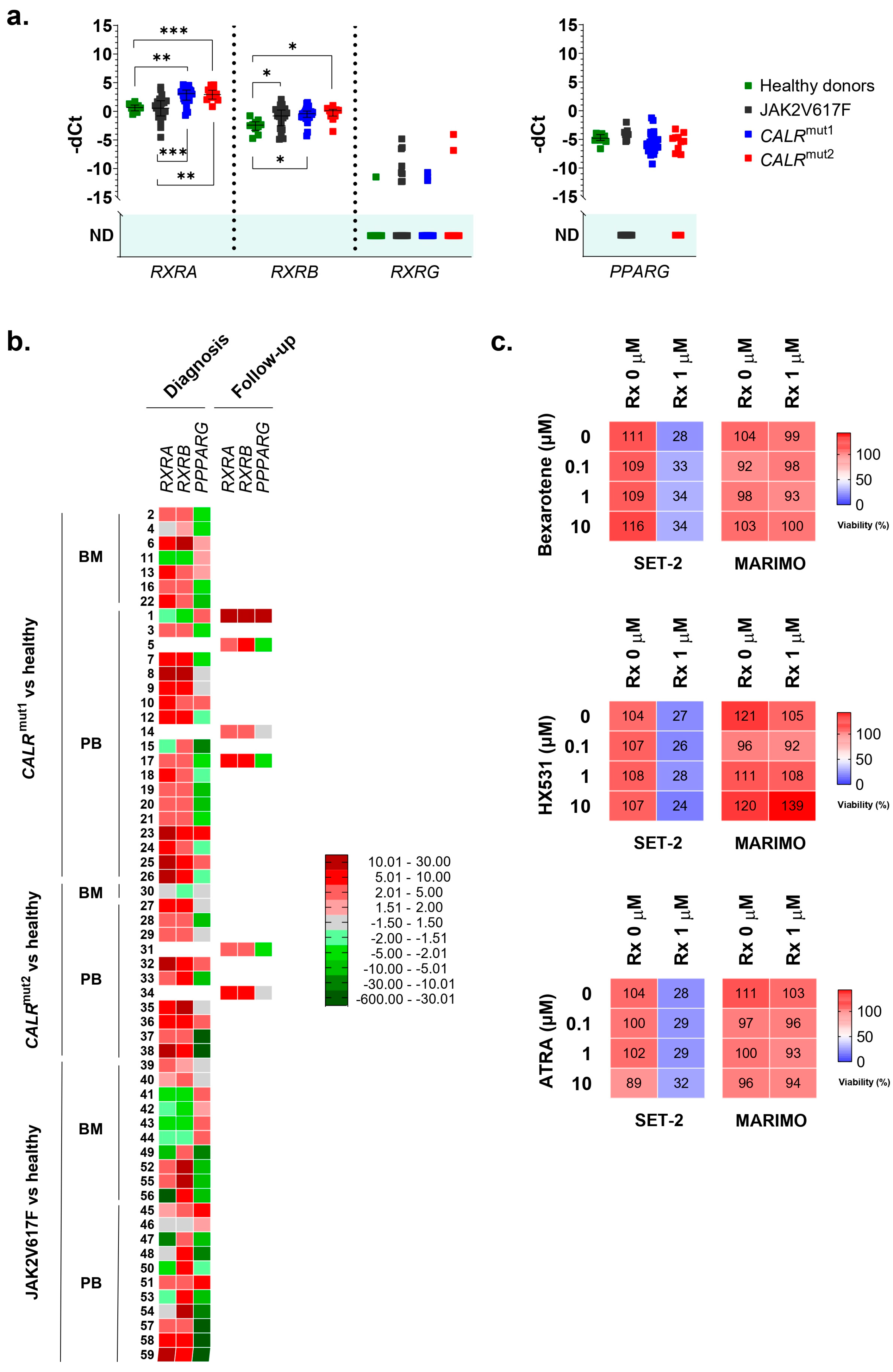
Given these results, using qPCR, we analyzed the expression of RXRA and PPARG in mononuclear cells extracted with Ficoll from the BM or PB of 38 patients with ET and CALR mutations (26 with type 1 mutations and 12 with type 2 mutations), 21 patients with ET and the p.V617F JAK2 mutation, and 10 healthy donors as controls.
In the series of analyzed samples, no differences were found for
PPARG. On the contrary,
RXRA was highly overexpressed in
CALR-mutated patients, but not in
JAK2-mutated patients, compared to healthy controls (
Figure 2a,b). This overexpression was observed in 23 of 26 patients with type 1
CALR mutations (88.5%) and in 11 of 12 patients with type 2
CALR mutations (91.7%). In contrast, only 9 of the 21 patients with the
JAK2 mutation (42.8%) had
RXRA overexpressed compared to healthy donors. No significant differences were found in the
RXRA expression between samples taken from BM and those from PB. Additionally, all samples taken during the disease follow-up from patients with
CALR mutations (6/6) were overexpressing
RXRA, regardless of the type of mutation. This result is consistent with the results obtained in cell lines and corroborates that the overexpression of
RXRA could be triggered by mutant type 1 and type 2 calreticulins in a
JAK2-independent manner.
As
RXRA was found to be overexpressed in these patients, we decided to study the expression of other members of the RXR family (
RXRB and
RXRG). Regarding
RXRB, we found it to be overexpressed in samples from patients with the
JAK2 mutation (16 out of 21 patients analyzed, 76.2%) and also in samples from patients with
CALR mutations (36 out of 38 patients analyzed, 94.7%) (
Figure 2a,b). Therefore, this expression cannot be considered an event that is independent of the activation of the JAK2 axis. In this case, no different expression patterns were observed depending on the origin of the sample (BM or PB) or the moment of their collection (at diagnosis or during disease follow-up) (
Figure 2b). Finally,
RXRG expression was undetectable in the majority of samples, and no significant differences were found between healthy donors and patients with ET, regardless of whether they had
CALR or
JAK2 mutations (
Figure 2a).
Thus, the results point to the fact that the overexpression of
RXRA seems to be a consequence of mutant calreticulin in humans. For this reason, we tried to test an RXR-targeted treatment in SET-2 and MARIMO cell lines, expecting to find some delay in proliferation in the MARIMO cell line due to
RXRA overexpression. Specifically, bexarotene (#SML0282, Sigma-Adrich Co., St. Louis, MO, USA), HX531 (#SML2170, Sigma-Adrich Co., St. Louis, MO, USA), and ATRA (#R2625, Sigma-Adrich Co., St. Louis, MO, USA) were tested as, respectively, a pan-RXR agonist, a pan-RXR antagonist, and a compound derived from vitamin A that acts as a ligand for retinoic acid receptors and X retinoid receptors. Each of these compounds were also tested in combination with ruxolitinib (#HY-50856, MedChemExpress MCE
®, Monmouth Junction, NJ, USA), a JAK1/JAK2 inhibitor that is approved for patients with MF post-ET, to find possible synergies and interactions. It is important to note that MARIMO cells are resistant to ruxolitinib [
6]. Our results show that bexarotene, HX531, and ATRA had no effects at the tested concentrations on the viability of the
CALR-mutated MARIMO cell line, either alone or in combination with ruxolitinib (
Figure 2c). As expected, a decrease in viability was observed in the SET-2 cell line after exposure to 1 μM ruxolitinib [
15], but the combination of this compound with increasing doses of bexarotene, HX531, and ATRA also did not change the effects of ruxolitinib (
Figure 2c).
These results led us to consider whether the
CALR-mutated MARIMO cell line was indeed a suitable model for evaluating the action of these compounds. It should be noted that this cell line shows other alterations in addition to a
CALR mutation [
16], which could dampen the effects produced by compounds targeting RXRs. In fact, although it shows an
MPL mutation (p.S505N), it does not show the canonical activation of the JAK2/STAT5 cascade that is present in patients with mutations in
CALR, probably due to these cells not expressing MPL. The replicative potential of MARIMO cells seems to be more related to the activation of the MAPK pathway by a p.Q61K mutation in
NRAS [
6,
16,
17].
Despite this, the magnitude of
RXRA overexpression that was observed in almost all the samples analyzed in patients with ET and
CALR mutations and recent studies that highlight the importance of RXRα and retinoids in myeloid malignancies [
12,
18,
19,
20] point to a possible relevant effect on the disease. Among the results of these studies, it is worth noting that an RXRα/RXRβ deficiency has been shown to induce the exhaustion of HSCs and the differentiation of myeloid cells, causing a myeloproliferative-type disease in mice [
18]. Therefore, the overexpression of
RXRA in
CALR-mutated ET patients could be a protective mechanism against the progression of the disease, which might be potentiated pharmacologically. For this reason, it does not seem appropriate to completely discard the RXR-targeted treatments for ET, either alone or in combination with ruxolitinib, although further analyses in a more suitable model are necessary.



 ,
, 
{kind=link}
{kind=link}