
Emerging Therapies for Glioblastoma

 ,
,  ,
, 
Simple Summary
Abstract
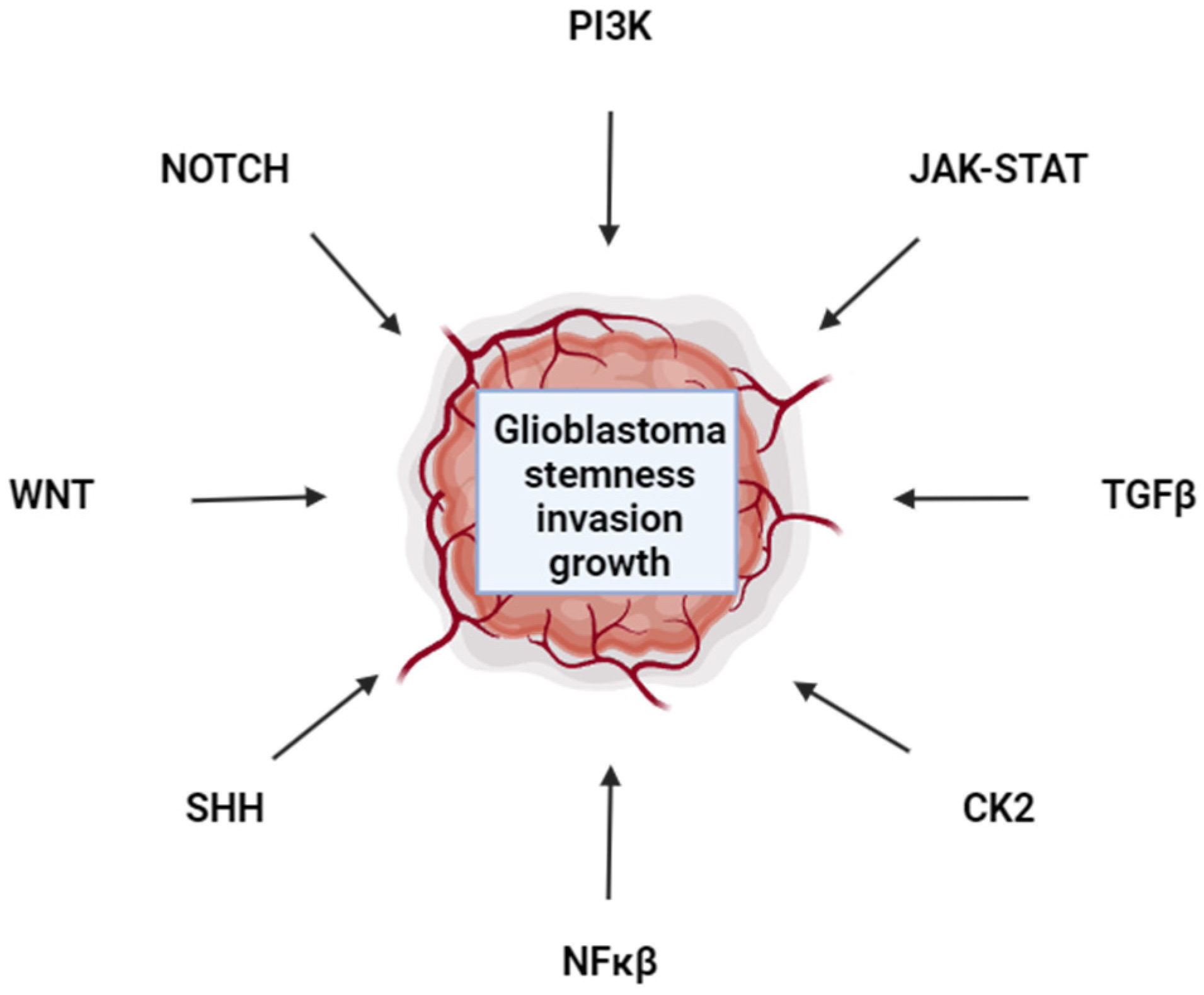
1. Introduction
2. Anti-Neoplastic Drugs and Surgical Interventions for Glioblastoma
2.1. Temozolomide (TMZ)
2.2. Carmustine
2.3. Surgical Resection
2.4. Fluorescent-Guided Surgical Resection
3. Tumor Treating Fields (TTFields)
4. Blocking Angiogenesis in Glioblastoma
5. Targeting EGFR in Glioblastoma
6. PI3K Is a Therapeutic Target for Glioblastoma
7. Targeting FOXO1 in Glioblastoma
8. The NF-κB Pathway as a Glioblastoma Therapeutic Target
9. Targeting CK2 in Glioblastoma
9.1. CK2 Biology
9.2. Biological Functions of CK2 in Glioblastoma Cancer
9.3. Inhibition of CK2 via CX-4945 as a Glioblastoma Therapy

{kind=link}
| Drug Name | Context | Impact |
|---|---|---|
| CX-4945 treatment alone and with TMZ | Mouse xenograft (GL261) and in vitro methods | Apoptosis induction, inhibition of cell migration, adhesion, and colony formation, inhibition of STAT3, NF-kB p56, and Akt; CX-4945 in combination TMZ had more robust impact [101] |
| CX-4945 treatment w/combined inhibitors: gefitinib and TMZ | In vivo mouse xenografts (GL261, SF767, U373, LN229) and in vitro methods | Inhibition of cell proliferation; CX-4945 sensitized TMZ-resistant cell lines (SF767) [101,103] |
| CX-4945 treatment w/combined inhibitor gefitinib | In vivo mouse xenograft (Xenograft X1046) and in vitro methods (X1016, X1046, X1066, U251-MG, U87MG) | Inhibition of tumor growth, cell proliferation, migration, adhesion; decreased activation of STAT3, NF-kB p56, and Akt; reduced stemness [104,105] |
9.4. Limitations of CX-4945 Therapies
10. Targeting JAK/STAT in Glioblastoma
| Drug Name | Context | Impact |
|---|---|---|
| Ruxolitinib | Human glioblastoma cells, U87MG | Ruxolitinib inhibited IFNs (IFN-α and IFN-γ)-dependent JAK/STAT signaling, decreased invasiveness and tumorigenesis [107] |
| Ruxolitinib | Human glioblastoma cells, U87MG | Inhibits IL-6 receptor-dependent JAK/STAT signaling (IL-6/JAK2/STAT3 axis) significantly inhibited tumor invasion (95.2%) [110] |
| JAK2 inhibitor SAR317461 | Human U87MG, U251 and A172 Glioblastoma | Potently inhibited STAT3 phosphorylation (10, 2, and 0.1 µM [108] |
11. Targeting LIF in Glioblastoma
12. WNT Pathway in Glioblastoma
13. Targeting the NOTCH Pathway in Glioblastoma
14. Hedgehog Signaling Pathway as Putative Glioblastoma Target
15. Targeting the TGFβ Pathway in Glioblastoma
16. Oncolytic Virotherapy’s Potential Role in Glioblastoma Multiform Prognosis
16.1. Oncolytic Virotherapy
16.2. Herpes Simplex Virus Type 1
16.3. DNX-2401 Clinical Trials
16.4. Limitations of Oncolytic Viral Therapy for Glioblastoma
17. Nanomaterials to Treat Glioblastoma
18. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, H.; Song, K.; Zhang, Y.; Zhang, J.; Wang, Y.; Sheng, X.; Chen, L.; Qin, Z. Tumor Treating Fields Combine with Temozolomide for Newly Diagnosed Glioblastoma: A Retrospective Analysis of Chinese Patients in a Single Center. J. Clin. Med. 2022, 11, 5855. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/beta-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, L.; Xu, H.; Long, S.; Jiang, J.; Wei, W.; Niu, X.; Li, X. Application of nanomaterials in diagnosis and treatment of glioblastoma. Front. Chem. 2022, 10, 1063152. [Google Scholar] [CrossRef]
- Chan, M.H.; Huang, W.T.; Satpathy, A.; Su, T.Y.; Hsiao, M.; Liu, R.S. Progress and Viewpoints of Multifunctional Composite Nanomaterials for Glioblastoma Theranostics. Pharmaceutics 2022, 14, 456. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef]
- Lau, Y.K.; Du, X.; Rayannavar, V.; Hopkins, B.; Shaw, J.; Bessler, E.; Thomas, T.; Pires, M.M.; Keniry, M.; Parsons, R.E.; et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5, 10503–10517. [Google Scholar] [CrossRef]
- Nichol, D.; Mellinghoff, I.K. PI3K pathway inhibition in GBM-is there a signal? Neuro-Oncol. 2015, 17, 1183–1184. [Google Scholar] [CrossRef] [PubMed]
- Netland, I.A.; Forde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S.; Miletic, H.; Enger, P.O.; Goplen, D. Treatment with the PI3K inhibitor buparlisib (NVP-BKM120) suppresses the growth of established patient-derived GBM xenografts and prolongs survival in nude rats. J. Neuro-Oncol. 2016, 129, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.M.; Vandenberg, S.; Cheng, S.Y.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.W.; Cavenee, W.K.; Furnari, F.B. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Lu, Z. The role of protein kinase CK2 in glioblastoma development. Clin. Cancer Res. 2013, 19, 6335–6337. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Krishnan, S.; Lee, S.; Amoozgar, Z.; Subudhi, S.; Kumar, A.; Posada, J.; Lindeman, N.; Lei, P.; Duquette, M.; et al. Wnt inhibition alleviates resistance to immune checkpoint blockade in glioblastoma. Res. Sq. 2023. preprint. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Braun, S.; Oppermann, H.; Mueller, A.; Renner, C.; Hovhannisyan, A.; Baran-Schmidt, R.; Gebhardt, R.; Hipkiss, A.; Thiery, J.; Meixensberger, J.; et al. Hedgehog signaling in glioblastoma multiforme. Cancer Biol. Ther. 2012, 13, 487–495. [Google Scholar] [CrossRef]
- Wen, P.Y.; Packer, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical implications. Neuro-Oncol. 2021, 23, 1215–1217. [Google Scholar] [CrossRef]
- Sun, X.; Turcan, S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells 2021, 10, 1225. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Gurda, D.; Glodowicz, P.; Nowak, S.; Naskret-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yamashita, D.; Inoue, A.; Suehiro, S.; Ohue, S.; Kunieda, T. Is Interstitial Chemotherapy with Carmustine (BCNU) Wafers Effective against Local Recurrence of Glioblastoma? A Pharmacokinetic Study by Measurement of BCNU in the Tumor Resection Cavity. Brain Sci. 2022, 12, 567. [Google Scholar] [CrossRef]
- Roux, A.; Peeters, S.; Zanello, M.; Bou Nassif, R.; Abi Lahoud, G.; Dezamis, E.; Parraga, E.; Lechapt-Zalcmann, E.; Dhermain, F.; Dumont, S.; et al. Extent of resection and Carmustine wafer implantation safely improve survival in patients with a newly diagnosed glioblastoma: A single center experience of the current practice. J. Neuro-Oncol. 2017, 135, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rozental, J.M.; Cohen, J.D.; Mehta, M.P.; Levine, R.L.; Hanson, J.M.; Nickles, R.J. Acute changes in glucose uptake after treatment: The effects of carmustine (BCNU) on human glioblastoma multiforme. J. Neuro-Oncol. 1993, 15, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.Z.; Wang, Z.F.; Lan, T.; Huang, W.H.; Zhao, Y.H.; Ma, C.; Li, Z.Q. Carmustine as a Supplementary Therapeutic Option for Glioblastoma: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 1036. [Google Scholar] [CrossRef]
- Kumabe, T.; Shibahara, I.; Saito, R. Results for Treatment of Newly-Diagnosed Glioblastoma Using Carmustine Wafers (Gliadel((R))). No Shinkei Geka 2018, 46, 367–376. [Google Scholar] [CrossRef]
- Cruz, N.; Herculano-Carvalho, M.; Roque, D.; Faria, C.C.; Cascao, R.; Ferreira, H.A.; Reis, C.P.; Matela, N. Highlighted Advances in Therapies for Difficult-To-Treat Brain Tumours Such as Glioblastoma. Pharmaceutics 2023, 15, 928. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.F.; Rojas, L.; Wills, B.; Ruiz-Patino, A.; Abril, L.; Hakim, F.; Jimenez, E.; Useche, N.; Bermudez, S.; Mejia, J.A.; et al. A comprehensive analysis of factors related to carmustine/bevacizumab response in recurrent glioblastoma. Clin. Transl. Oncol. 2019, 21, 1364–1373. [Google Scholar] [CrossRef]
- Akiyama, Y.; Kimura, Y.; Enatsu, R.; Mikami, T.; Wanibuchi, M.; Mikuni, N. Advantages and Disadvantages of Combined Chemotherapy with Carmustine Wafer and Bevacizumab in Patients with Newly Diagnosed Glioblastoma: A Single-Institutional Experience. World Neurosurg. 2018, 113, e508–e514. [Google Scholar] [CrossRef]
- Zeppa, P.; De Marco, R.; Monticelli, M.; Massara, A.; Bianconi, A.; Di Perna, G.; Greco Crasto, S.; Cofano, F.; Melcarne, A.; Lanotte, M.M.; et al. Fluorescence-Guided Surgery in Glioblastoma: 5-ALA, SF or Both? Differences between Fluorescent Dyes in 99 Consecutive Cases. Brain Sci. 2022, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Specchia, F.M.C.; Monticelli, M.; Zeppa, P.; Bianconi, A.; Zenga, F.; Altieri, R.; Pugliese, B.; Di Perna, G.; Cofano, F.; Tartara, F.; et al. Let Me See: Correlation between 5-ALA Fluorescence and Molecular Pathways in Glioblastoma: A Single Center Experience. Brain Sci. 2021, 11, 795. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, L.C.; Krabbenhoft, M.G.; Hansen, R.W.; Mikic, N.; Pedersen, C.B.; Poulsen, F.R.; Korshoej, A.R. Effect of 5-Aminolevulinic Acid and Sodium Fluorescein on the Extent of Resection in High-Grade Gliomas and Brain Metastasis. Cancers 2022, 14, 617. [Google Scholar] [CrossRef] [PubMed]
- Hadjipanayis, C.G.; Stummer, W. 5-ALA and FDA approval for glioma surgery. J. Neuro-Oncol. 2019, 141, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, H.; Ge, P.; Zhao, J.; Li, W.; Gu, H.; Wang, G.; Luo, Y.; Chen, D. Gross total resection of glioma with the intraoperative fluorescence-guidance of fluorescein sodium. Int. J. Med. Sci. 2012, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Ballo, M.T.; Conlon, P.; Lavy-Shahaf, G.; Kinzel, A.; Vymazal, J.; Rulseh, A.M. Association of Tumor Treating Fields (TTFields) therapy with survival in newly diagnosed glioblastoma: A systematic review and meta-analysis. J. Neuro-Oncol. 2023, 164, 1–9. [Google Scholar] [CrossRef]
- Deuschl, C.; Moenninghoff, C.; Goericke, S.; Kirchner, J.; Koppen, S.; Binse, I.; Poeppel, T.D.; Quick, H.H.; Forsting, M.; Umutlu, L.; et al. Response assessment of bevacizumab therapy in GBM with integrated 11C-MET-PET/MRI: A feasibility study. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1285–1295. [Google Scholar] [CrossRef]
- Nelson, S.J.; Li, Y.; Lupo, J.M.; Olson, M.; Crane, J.C.; Molinaro, A.; Roy, R.; Clarke, J.; Butowski, N.; Prados, M.; et al. Serial analysis of 3D H-1 MRSI for patients with newly diagnosed GBM treated with combination therapy that includes bevacizumab. J. Neuro-Oncol. 2016, 130, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neuro-Oncol. 2016, 126, 185–192. [Google Scholar] [CrossRef]
- Carlson, J.A.; Reddy, K.; Gaspar, L.E.; Ney, D.; Kavanagh, B.D.; Damek, D.; Lillehei, K.; Chen, C. Hypofractionated-intensity modulated radiotherapy (hypo-IMRT) and temozolomide (TMZ) with or without bevacizumab (BEV) for newly diagnosed glioblastoma multiforme (GBM): A comparison of two prospective phase II trials. J. Neuro-Oncol. 2015, 123, 251–257. [Google Scholar] [CrossRef]
- Farid, N.; Almeida-Freitas, D.B.; White, N.S.; McDonald, C.R.; Muller, K.A.; Vandenberg, S.R.; Kesari, S.; Dale, A.M. Restriction-Spectrum Imaging of Bevacizumab-Related Necrosis in a Patient with GBM. Front. Oncol. 2013, 3, 258. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.B.; Qiao, X.J.; Kim, H.J.; Lai, A.; Nghiemphu, P.; Xue, X.; Ellingson, B.M.; Schiff, D.; Aregawi, D.; Cha, S.; et al. Apparent diffusion coefficient histogram analysis stratifies progression-free and overall survival in patients with recurrent GBM treated with bevacizumab: A multi-center study. J. Neuro-Oncol. 2012, 108, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Ananthnarayan, S.; Bahng, J.; Roring, J.; Nghiemphu, P.; Lai, A.; Cloughesy, T.; Pope, W.B. Time course of imaging changes of GBM during extended bevacizumab treatment. J. Neuro-Oncol. 2008, 88, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhou, Z.; Huang, X.; Chen, Z.; Zhang, L.; Zhang, J.; Hua, W.; Mao, Y. Use of Bevacizumab in recurrent glioblastoma: A scoping review and evidence map. BMC Cancer 2023, 23, 544. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib alone or in combination with bevacizumab in patients with recurrent glioblastoma: Phase I/II clinical trial data. Neuro-Oncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef]
- Patel, N.V.; Wong, T.; Fralin, S.R.; Li, M.; McKeown, A.; Gruber, D.; D’Amico, R.S.; Patsalides, A.; Tsiouris, A.; Stefanov, D.G.; et al. Repeated superselective intraarterial bevacizumab after blood brain barrier disruption for newly diagnosed glioblastoma: A phase I/II clinical trial. J. Neuro-Oncol. 2021, 155, 117–124. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Ren, X.; Ai, D.; Li, T.; Xia, L.; Sun, L. Effectiveness of Lomustine Combined with Bevacizumab in Glioblastoma: A Meta-Analysis. Front. Neurol. 2020, 11, 603947. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Weppler, S.A.; Li, Y.; Dubois, L.; Lieuwes, N.; Jutten, B.; Lambin, P.; Wouters, B.G.; Lammering, G. Expression of EGFR variant vIII promotes both radiation resistance and hypoxia tolerance. Radiother. Oncol. 2007, 83, 333–339. [Google Scholar] [CrossRef]
- Rutkowska, A.; Stoczynska-Fidelus, E.; Janik, K.; Wlodarczyk, A.; Rieske, P. EGFR(vIII): An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: A phase III randomized clinical trial. Neuro-Oncol. 2023, 25, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Abousaud, M.; Faroqui, N.M.; Lesser, G.; Strowd, R.E.; Ramkissoon, S.H.; Kwatra, M.; Houston, K.S.; Hsu, F.C.; Carter, A.; Petro, R.; et al. Clinical Experience using Osimertinib in Patients with Recurrent Malignant Gliomas Containing EGFR Alterations. J. Cancer Sci. Clin. Ther. 2021, 5, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncol. 2015, 17, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncol. 2010, 12, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, B.; Eriksen, J.G.; Broholm, H.; Christensen, I.J.; Grunnet, K.; Horsman, M.R.; Poulsen, H.S.; Stockhausen, M.T.; Lassen, U. Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS 2010, 118, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, E.; Chapusot, C.; Tournier, B.; Sentis, J.; Marion, E.; Remond, A.; Aubry, M.; Pioche, C.; Bergeron, A.; Primois, C.; et al. Idylla EGFR assay on extracted DNA: Advantages, limits and place in molecular screening according to the latest guidelines for non-small-cell lung cancer (NSCLC) patients. J. Clin. Pathol. 2023, 76, 698–704. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.R.; Picone, A.; Ferraro, G.; Zanghi, M.; Toscano, G.; Giordano, A.; Adamo, V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget 2015, 6, 26814–26825. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.W.; Pedersen, N.; Ottesen, L.H.; Poulsen, H.S. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br. J. Cancer 2005, 93, 915–923. [Google Scholar] [CrossRef]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef] [PubMed]
- Gallia, G.L.; Rand, V.; Siu, I.M.; Eberhart, C.G.; James, C.D.; Marie, S.K.; Oba-Shinjo, S.M.; Carlotti, C.G.; Caballero, O.L.; Simpson, A.J.; et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol. Cancer Res. 2006, 4, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Tohma, Y.; Gratas, C.; Biernat, W.; Peraud, A.; Fukuda, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 684–689. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Wen, P.Y.; Yung, W.K.A.; Mellinghoff, I.K.; Ramkissoon, S.; Alexander, B.; Rinne, M.; Colman, H.; Omuro, A.M.; DeAngelis, L.M.; et al. Phase ii trial of the phosphatidyinositol-3 kinase (pi3k) inhibitor buparlisib (bkm120) in recurrent glioblastoma conducted by the ivy foundation early phase clinical trials consortium. Neuro-Oncol. 2014, 16, iii47. [Google Scholar] [CrossRef]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neuro-Oncol. 2019, 144, 303–311. [Google Scholar] [CrossRef]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.; Parsons, R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef]
- Martinez, E.; Vazquez, N.; Lopez, A.; Fanniel, V.; Sanchez, L.; Marks, R.; Hinojosa, L.; Cuello, V.; Cuevas, M.; Rodriguez, A.; et al. The PI3K pathway impacts stem gene expression in a set of glioblastoma cell lines. J. Cancer Res. Clin. Oncol. 2020, 146, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yalcin, S.; Lee, D.F.; Yeh, T.Y.; Lee, S.M.; Su, J.; Mungamuri, S.K.; Rimmele, P.; Kennedy, M.; Sellers, R.; et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 1092–1099. [Google Scholar] [CrossRef]
- Flores, D.; Lopez, A.; Udawant, S.; Gunn, B.; Keniry, M. The FOXO1 inhibitor AS1842856 triggers apoptosis in glioblastoma multiforme and basal-like breast cancer cells. FEBS Open Bio 2023, 13, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Shabason, J.E.; Camphausen, K. Cancer stem cells as a prognostic indicator for glioblastoma multiforme. Biomark. Med. 2010, 4, 127–128. [Google Scholar] [CrossRef]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Xu, C.; Shen, G.; Chen, C.; Gelinas, C.; Kong, A.N. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.U.; Ravi, S.; Lee, D.W.; McFadden, K.; Kamradt, M.L.; Toussaint, L.G.; Sitcheran, R. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr. Biol. 2016, 26, 3288–3302. [Google Scholar] [CrossRef] [PubMed]
- Avci, N.G.; Ebrahimzadeh-Pustchi, S.; Akay, Y.M.; Esquenazi, Y.; Tandon, N.; Zhu, J.J.; Akay, M. NF-kappaB inhibitor with Temozolomide results in significant apoptosis in glioblastoma via the NF-kappaB(p65) and actin cytoskeleton regulatory pathways. Sci. Rep. 2020, 10, 13352. [Google Scholar] [CrossRef]
- Kamradt, M.L.; Jung, J.U.; Pflug, K.M.; Lee, D.W.; Fanniel, V.; Sitcheran, R. NIK promotes metabolic adaptation of glioblastoma cells to bioenergetic stress. Cell Death Dis. 2021, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ho, W.S.; Lu, R. Targeting Mitochondrial Oxidative Phosphorylation in Glioblastoma Therapy. Neuromolecular Med. 2022, 24, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Firnau, M.-B.; Brieger, A. CK2 and the Hallmarks of Cancer. Biomedicines 2022, 10, 1987. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, B.M.; Boewe, A.S.; Gotz, C.; Philipp, S.E.; Urbschat, S.; Oertel, J.; Menger, M.D.; Laschke, M.W.; Ampofo, E. CK2 Activity Mediates the Aggressive Molecular Signature of Glioblastoma Multiforme by Inducing Nerve/Glial Antigen (NG)2 Expression. Cancers 2021, 13, 1678. [Google Scholar] [CrossRef]
- Yde, C.W.; Olsen, B.B.; Meek, D.; Watanabe, N.; Guerra, B. The regulatory beta-subunit of protein kinase CK2 regulates cell-cycle progression at the onset of mitosis. Oncogene 2008, 27, 4986–4997. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Roffey, S.E.; Litchfield, D.W. CK2 Regulation: Perspectives in 2021. Biomedicines 2021, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front. Pharmacol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Borgo, C.; Bosello-Travain, V.; Vilardell, J.; Salizzato, V.; Pinna, L.A.; Venerando, A.; Salvi, M. Deciphering the role of protein kinase CK2 in the maturation/stability of F508del-CFTR. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165611. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—potential clinical relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Lettieri, A.; Borgo, C.; Zanieri, L.; D’Amore, C.; Oleari, R.; Paganoni, A.; Pinna, L.A.; Cariboni, A.; Salvi, M. Protein Kinase CK2 Subunits Differentially Perturb the Adhesion and Migration of GN11 Cells: A Model of Immature Migrating Neurons. Int. J. Mol. Sci. 2019, 20, 5951. [Google Scholar] [CrossRef]
- Pucko, E.B.; Ostrowski, R.P. Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms. Pharmaceutics 2022, 14, 331. [Google Scholar] [CrossRef]
- Ferrer-Font, L.; Villamanan, L.; Arias-Ramos, N.; Vilardell, J.; Plana, M.; Ruzzene, M.; Pinna, L.A.; Itarte, E.; Arus, C.; Candiota, A.P. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals 2017, 10, 24. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Li, W.; Hang, C.; Dai, Y. Inhibition of Casein Kinase II by CX-4945, But Not Yes-associated protein (YAP) by Verteporfin, Enhances the Antitumor Efficacy of Temozolomide in Glioblastoma. Transl. Oncol. 2020, 13, 70–78. [Google Scholar] [CrossRef]
- Rowse, A.L.; Gibson, S.A.; Meares, G.P.; Rajbhandari, R.; Nozell, S.E.; Dees, K.J.; Hjelmeland, A.B.; McFarland, B.C.; Benveniste, E.N. Protein kinase CK2 is important for the function of glioblastoma brain tumor initiating cells. J. Neuro-Oncol. 2017, 132, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.S.; Zeng, J.; Mei, J.; Wang, P.Y. JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Delen, E.; Doganlar, O. The Dose Dependent Effects of Ruxolitinib on the Invasion and Tumorigenesis in Gliomas Cells via Inhibition of Interferon Gamma-Depended JAK/STAT Signaling Pathway. J. Korean Neurosurg. Soc. 2020, 63, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Mukthavaram, R.; Ouyang, X.; Saklecha, R.; Jiang, P.; Nomura, N.; Pingle, S.C.; Guo, F.; Makale, M.; Kesari, S. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J. Transl. Med. 2015, 13, 269. [Google Scholar] [CrossRef] [PubMed]
- Smedley, W.; Patra, A. JAK3 Inhibition Regulates Stemness and Thereby Controls Glioblastoma Pathogenesis. Cells 2023, 12, 2547. [Google Scholar] [CrossRef]
- Delen, E.; Doganlar, O.; Doganlar, Z.B.; Delen, O. Inhibition of the Invasion of Human Glioblastoma U87 Cell Line by Ruxolitinib: A Molecular Player of miR-17 and miR-20a Regulating JAK/STAT Pathway. Turk. Neurosurg. 2020, 30, 182–189. [Google Scholar] [CrossRef]
- Spencer, N.; Rodriguez Sanchez, A.L.; Gopalam, R.; Subbarayalu, P.; Medina, D.M.; Yang, X.; Ramirez, P.; Randolph, L.; Aller, E.J.; Santhamma, B.; et al. The LIFR Inhibitor EC359 Effectively Targets Type II Endometrial Cancer by Blocking LIF/LIFR Oncogenic Signaling. Int. J. Mol. Sci. 2023, 24, 7426. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.Z.; Li, N.Z.; Hu, X.B.; Xie, Y.Y.; Huang, Q.H.; Zhang, J.; Chen, Z.; Chen, S.J.; Wang, F.; Sun, X.J. The LIFR-targeting small molecules EC330/EC359 are potent ferroptosis inducers. Genes Dis. 2023, 10, 735–738. [Google Scholar] [CrossRef]
- Viswanadhapalli, S.; Luo, Y.; Sareddy, G.R.; Santhamma, B.; Zhou, M.; Li, M.; Ma, S.; Sonavane, R.; Pratap, U.P.; Altwegg, K.A.; et al. EC359: A First-in-Class Small-Molecule Inhibitor for Targeting Oncogenic LIFR Signaling in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2019, 18, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.R.; Cannon, A.; Thompson, C.; Santhamma, B.; Chavez-Riveros, A.; Bhatia, R.; Nair, H.B.; Nickisch, K.; Batra, S.K.; Kumar, S. Utilizing cell line-derived organoids to evaluate the efficacy of a novel LIFR-inhibitor, EC359 in targeting pancreatic tumor stroma. Genes Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Parte, S.; Kshirsagar, P.; Muniyan, S.; Nair, H.B.; Batra, S.K.; Seshacharyulu, P. The Pleiotropic role, functions and targeted therapies of LIF/LIFR axis in cancer: Old spectacles with new insights. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188737. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Wu, F.; Wang, J.; Kim, K.; Santhamma, B.; Dileep, K.V.; Zhang, K.Y.J.; Viswanadhapalli, S.; Vadlamudi, R.K.; Ahmed, G.; et al. EC330, a small-molecule compound, is a potential novel inhibitor of LIF signaling. J. Mol. Cell Biol. 2020, 12, 477–480. [Google Scholar] [CrossRef]
- Liu, S.C.; Tsang, N.M.; Chiang, W.C.; Chang, K.P.; Hsueh, C.; Liang, Y.; Juang, J.L.; Chow, K.P.; Chang, Y.S. Leukemia inhibitory factor promotes nasopharyngeal carcinoma progression and radioresistance. J. Clin. Investig. 2013, 123, 5269–5283. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Fer, N.; Galeas, J.; Collisson, E.A.; Kim, S.E.; Sharib, J.; McCormick, F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat. Commun. 2019, 10, 3055. [Google Scholar] [CrossRef]
- McCord, M.; Mukouyama, Y.S.; Gilbert, M.R.; Jackson, S. Targeting WNT Signaling for Multifaceted Glioblastoma Therapy. Front. Cell Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef]
- Hussain, B.; Fang, C.; Huang, X.; Feng, Z.; Yao, Y.; Wang, Y.; Chang, J. Endothelial beta-Catenin Deficiency Causes Blood-Brain Barrier Breakdown via Enhancing the Paracellular and Transcellular Permeability. Front. Mol. Neurosci. 2022, 15, 895429. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Schiff, D.; Henson, J.W.; Muzikansky, A.; Gigas, D.C.; Doherty, L.; Batchelor, T.T.; Longtine, J.A.; Ligon, K.L.; Weaver, S.; et al. Phase II study of temozolomide, thalidomide, and celecoxib for newly diagnosed glioblastoma in adults. Neuro-Oncol. 2008, 10, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Penas-Prado, M.; Hess, K.R.; Fisch, M.J.; Lagrone, L.W.; Groves, M.D.; Levin, V.A.; De Groot, J.F.; Puduvalli, V.K.; Colman, H.; Volas-Redd, G.; et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro-Oncol. 2015, 17, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Gonzalez, J.; Hunter, K.; Hess, K.; Giglio, P.; Chang, E.; Puduvalli, V.; Groves, M.D.; Colman, H.; Conrad, C.; et al. A phase I factorial design study of dose-dense temozolomide alone and in combination with thalidomide, isotretinoin, and/or celecoxib as postchemoradiation adjuvant therapy for newly diagnosed glioblastoma. Neuro-Oncol. 2010, 12, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, K.H.; Lee, J.; Lee, Y.A.; Kim, M.; Lee, S.J.; Park, K.; Yang, H.; Jin, J.; Joo, K.M.; et al. Wnt activation is implicated in glioblastoma radioresistance. Lab. Investig. 2012, 92, 466–473. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, A.; Valensin, S.; Rossi, M.; Tunici, P.; Verani, M.; De Rosa, A.; Giordano, C.; Varrone, M.; Nencini, A.; Pratelli, C.; et al. Identification and characterization of a small-molecule inhibitor of Wnt signaling in glioblastoma cells. Mol. Cancer Ther. 2013, 12, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Suwala, A.K.; Koch, K.; Natsumeda, M.; Orr, B.A.; Hayashi, M.; Maciaczyk, J.; Eberhart, C.G. Pharmacologic Wnt Inhibition Reduces Proliferation, Survival, and Clonogenicity of Glioblastoma Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Gursel, D.B.; Berry, N.; Boockvar, J.A. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: The role of the endothelial network. Neurosurgery 2012, 70, N19–N21. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef]
- Raghu, H.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch-1 receptor. Mol. Cancer 2011, 10, 130. [Google Scholar] [CrossRef]
- Jeon, H.M.; Jin, X.; Lee, J.S.; Oh, S.Y.; Sohn, Y.W.; Park, H.J.; Joo, K.M.; Park, W.Y.; Nam, D.H.; DePinho, R.A.; et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 2008, 22, 2028–2033. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ye, X.; Mikkelsen, T.; Lesser, G.J.; Lieberman, F.S.; Robins, H.I.; Ahluwalia, M.S.; Sloan, A.E.; Grossman, S.A. A Phase II and Pharmacodynamic Trial of RO4929097 for Patients with Recurrent/Progressive Glioblastoma. Neurosurgery 2021, 88, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Sloan, A.E.; Nock, C.J.; Ye, X.; Buerki, R.; Chang, S.; Lesser, G.; Norden, A.; Cloughesy, T.; Olson, J.; Kerstetter-Fogle, A.; et al. ABTC-0904: Targeting glioma stem cells in GBM: A phase 0/II study of hedgehog pathway inhibitor GDC-0449. J. Neuro-Oncol. 2023, 161, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Drakulic, D.; Schwirtlich, M.; Petrovic, I.; Mojsin, M.; Milivojevic, M.; Kovacevic-Grujicic, N.; Stevanovic, M. Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma. Cells 2022, 11, 2530. [Google Scholar] [CrossRef] [PubMed]
- Seystahl, K.; Papachristodoulou, A.; Burghardt, I.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P.; Weller, M. Biological Role and Therapeutic Targeting of TGF-beta(3) in Glioblastoma. Mol. Cancer Ther. 2017, 16, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.M.; Shin, Y.J.; Lee, J.; Chang, N.; Woo, D.H.; Lee, W.J.; Nguyen, D.; Kang, W.; Cho, H.J.; Yang, H.; et al. The semaphorin 3A/neuropilin-1 pathway promotes clonogenic growth of glioblastoma via activation of TGF-beta signaling. JCI Insight 2023, 8, 49. [Google Scholar] [CrossRef]
- Kim, B.G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-beta pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wei, M.; Wang, Y.; Nowicki, M.O.; Ha, Y.P.; Bergin, S.; Hwang, C.; Fernandez, S.A.; et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 2012, 86, 4566–4577. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Jung, B.K.; An, Y.H.; Jang, H. The phosphatase and tensin homolog gene inserted between NP and P gene of recombinant New castle disease virus oncolytic effect test to glioblastoma cell and xenograft mouse model. Virol. J. 2022, 19, 21. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Gesundheit, B.; Ben-David, E.; Posen, Y.; Ellis, R.; Wollmann, G.; Schneider, E.M.; Aigner, K.; Brauns, L.; Nesselhut, T.; Ackva, I.; et al. Effective Treatment of Glioblastoma Multiforme with Oncolytic Virotherapy: A Case-Series. Front. Oncol. 2020, 10, 702. [Google Scholar] [CrossRef]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- Roque, D.; Cruz, N.; Ferreira, H.A.; Reis, C.P.; Matela, N.; Herculano-Carvalho, M.; Cascao, R.; Faria, C.C. Nanoparticle-Based Treatment in Glioblastoma. J. Pers. Med. 2023, 13, 1328. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.C.; Chauhan, R.; Bates, P.J.; O’Toole, M.G. Optimization of Tumor Targeting Gold Nanoparticles for Glioblastoma Applications. Nanomaterials 2022, 12, 3869. [Google Scholar] [CrossRef] [PubMed]
- Koziara, J.M.; Lockman, P.R.; Allen, D.D.; Mumper, R.J. Paclitaxel nanoparticles for the potential treatment of brain tumors. J. Control Release 2004, 99, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Gazaille, C.; Sicot, M.; Saulnier, P.; Eyer, J.; Bastiat, G. Local Delivery and Glioblastoma: Why Not Combining Sustained Release and Targeting? Front. Med. Technol. 2021, 3, 791596. [Google Scholar] [CrossRef] [PubMed]
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncol. 2022, 24, 669–682. [Google Scholar] [CrossRef]

| Drug Name | Context | Impact |
|---|---|---|
| Gefitinib | Human glioblastoma clinical trial | Minimal to no activity [56] |
| Erlotinib | Human glioblastoma clinical trial | Minimal to no activity [58] |
| Afatinib | Human glioblastoma clinical trial | Minimal to no activity [55] |
| Lapatinib | Human glioblastoma clinical trial | Minimal to no activity [57] |
| Osimertinib | Human glioblastoma clinical trial | Minimal to no activity [53] |
| Cetuximab | Human glioblastoma clinical trial | Minimal to no activity [59] |
| Rindopepimut | Human glioblastoma clinical trial | Minimal to no activity [54] |
| Depatuxizumab mafodotin + TMZ | Human glioblastoma clinical trial | Minimal to no activity [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rios, S.A.; Oyervides, S.; Uribe, D.; Reyes, A.M.; Fanniel, V.; Vazquez, J.; Keniry, M. Emerging Therapies for Glioblastoma. Cancers 2024, 16, 1485. https://doi.org/10.3390/cancers16081485
Rios SA, Oyervides S, Uribe D, Reyes AM, Fanniel V, Vazquez J, Keniry M. Emerging Therapies for Glioblastoma. Cancers. 2024; 16(8):1485. https://doi.org/10.3390/cancers16081485
Chicago/Turabian StyleRios, Stella Aimé, Stephanie Oyervides, David Uribe, Angelica Maree Reyes, Victor Fanniel, Jonathan Vazquez, and Megan Keniry. 2024. "Emerging Therapies for Glioblastoma" Cancers 16, no. 8: 1485. https://doi.org/10.3390/cancers16081485
APA StyleRios, S. A., Oyervides, S., Uribe, D., Reyes, A. M., Fanniel, V., Vazquez, J., & Keniry, M. (2024). Emerging Therapies for Glioblastoma. Cancers, 16(8), 1485. https://doi.org/10.3390/cancers16081485