Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect

 , , , ,
, , , ,  , ,
, ,  ,
,  and
and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
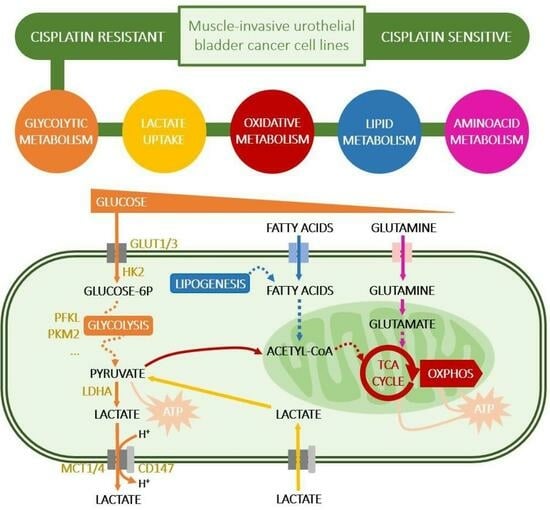
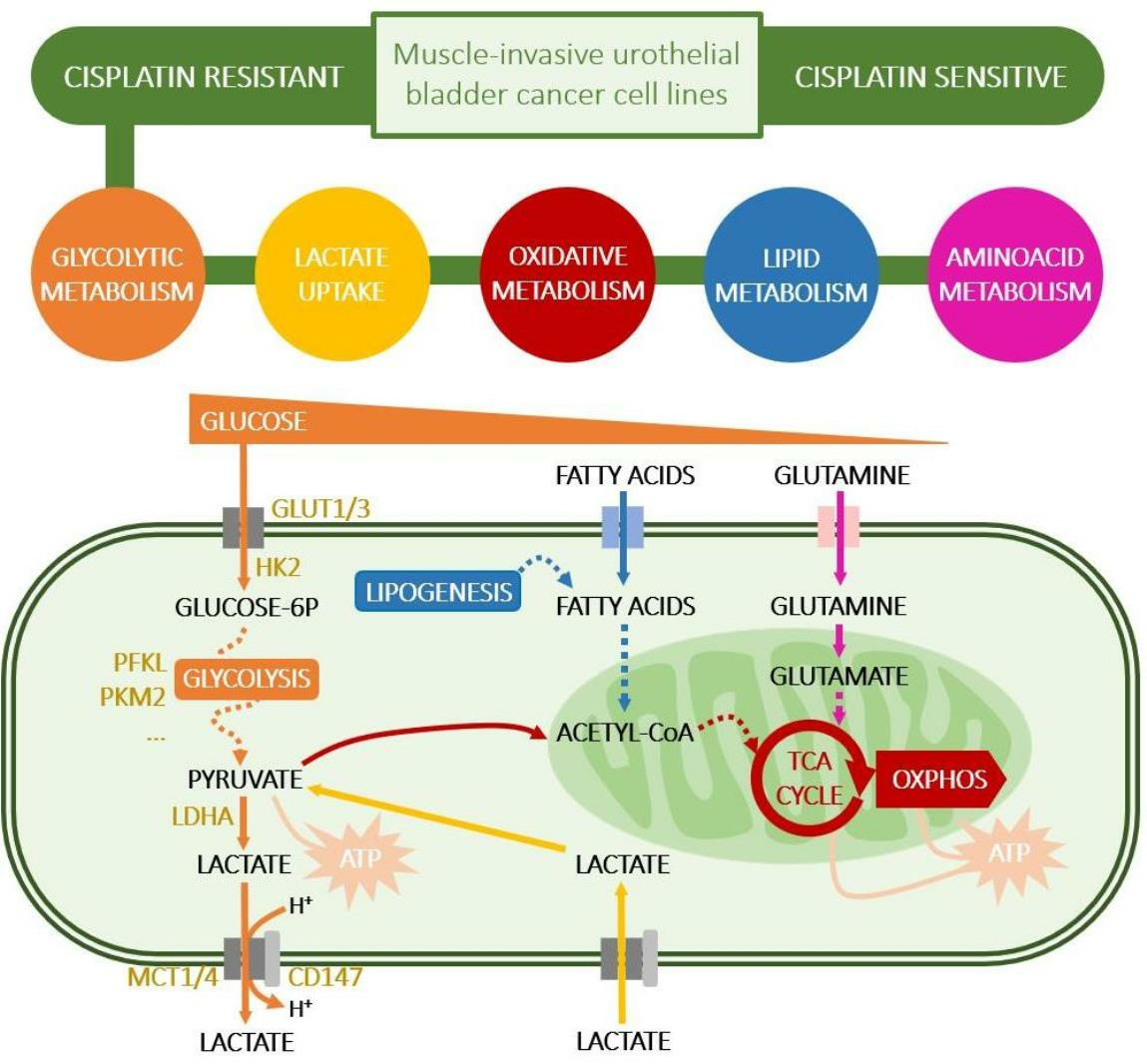
1. Introduction
2. Materials and Methods
2.1. Cell Lines and General Cell Culture Procedures
2.2. Cell Viability Assay
2.3. Protein Extraction and Western Blotting
2.4. Immunofluorescence
2.5. Glucose Uptake Analysis by Flow Cytometry
2.6. Colorimetric Analysis of Extracellular Glucose
2.7. ATP Quantification by Bioluminescence
2.8. Analysis of Mitochondrial Activity by Flow Cytometry
2.9. Fluorometric Analysis of Reactive Oxygen Species (ROS) Production
2.10. Colorimetric Analysis of Glutathione Content
2.11. Analysis of Neutral Lipid Content and Palmitate Uptake by Flow Cytometry
2.12. Seahorse Analysis of Mitochondrial Respiration
2.13. Chromatographic Analysis of Extracellular Glucose, Pyruvate and Lactate
2.14. Proton Nuclear Magnetic Resonance (1H NMR)-Based Metabolomics for Intracellular Metabolome Analysis
2.15. Statistical Analysis
3. Results
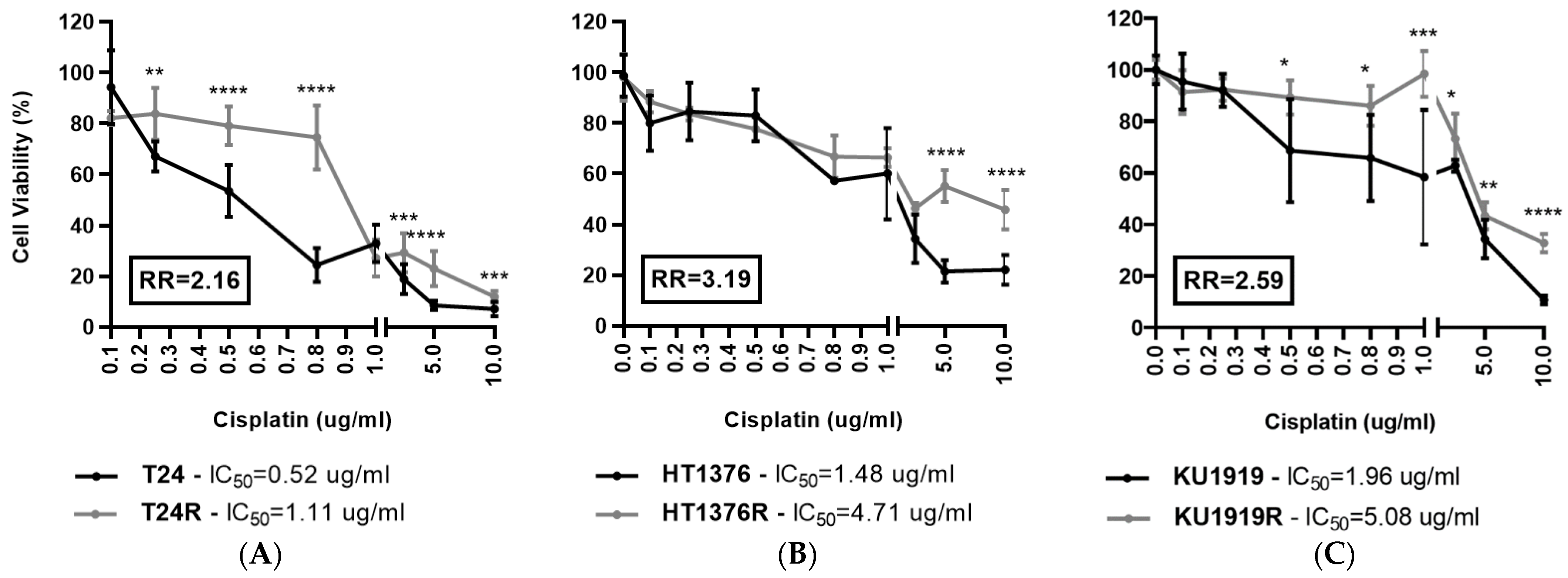
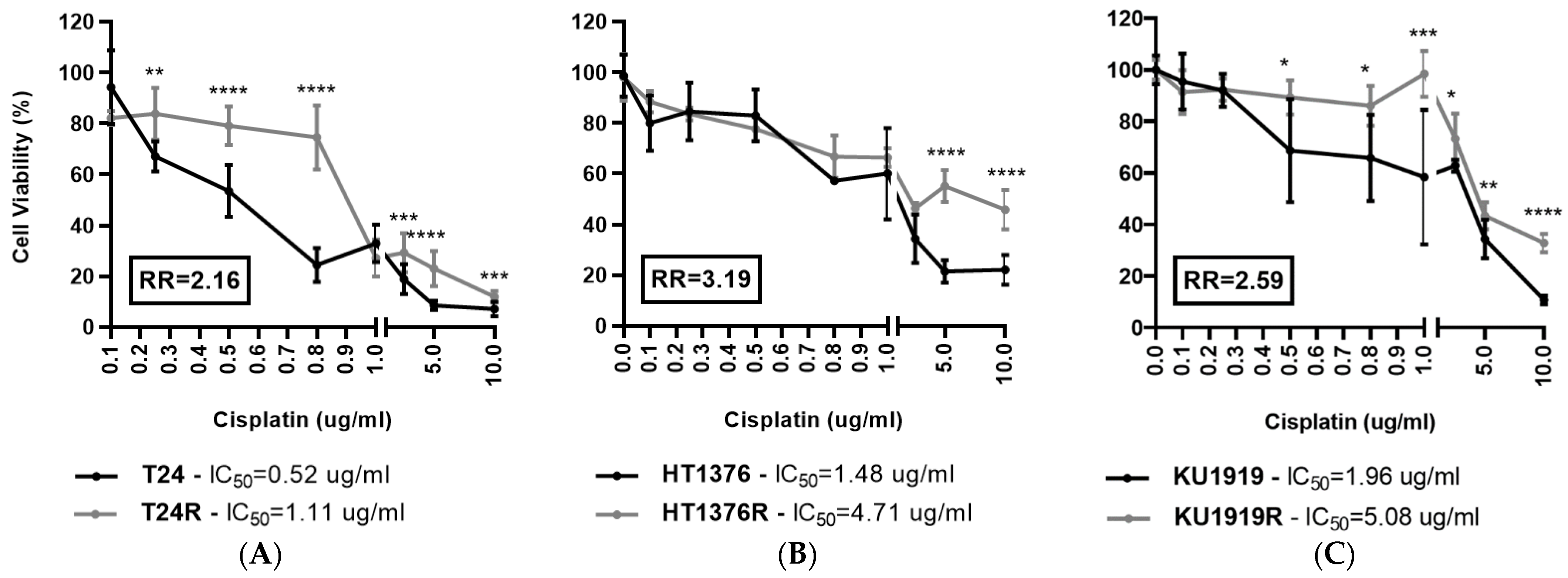
3.1. Effect of Cisplatin on Cell Viability
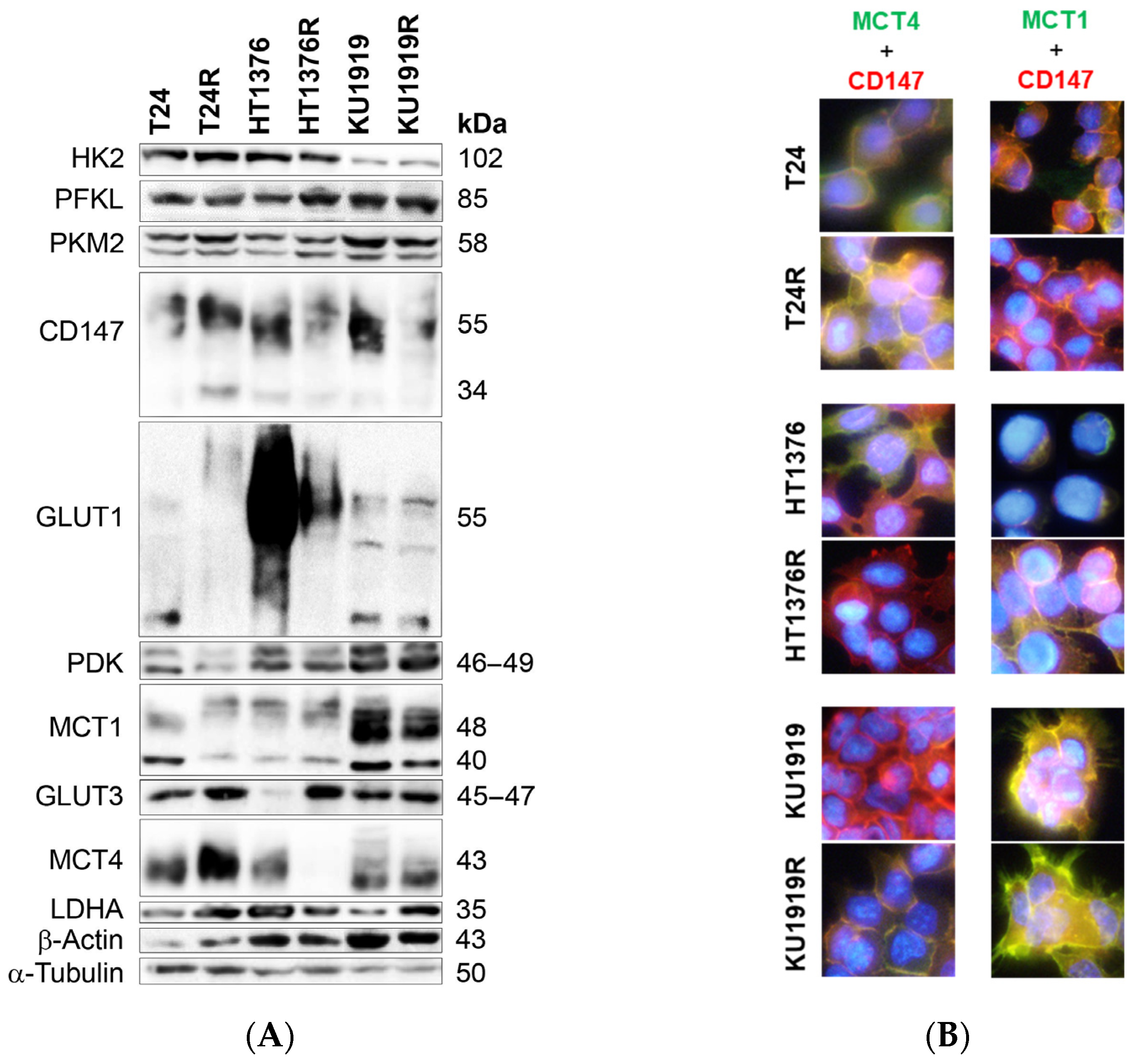
3.2. Immunoexpression of Glycometabolism and Cell Death-Related Biomarkers
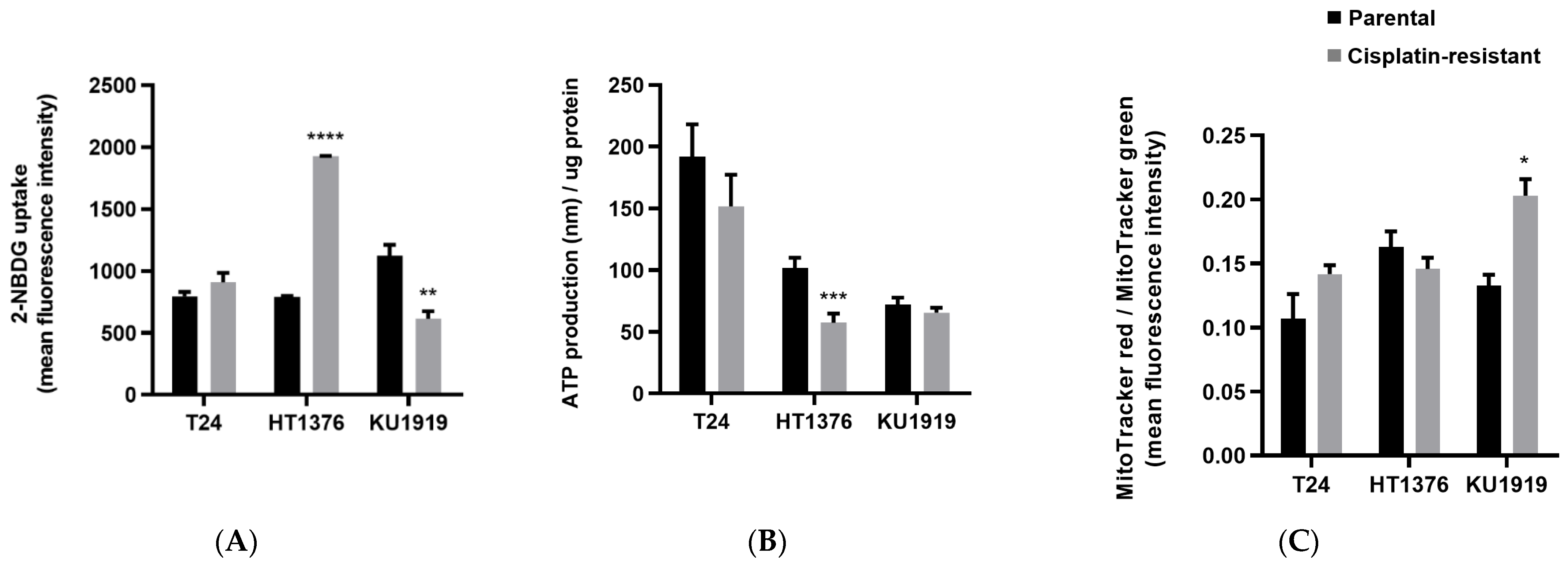
3.3. Glycolytic Metabolism and Mitochondrial Function
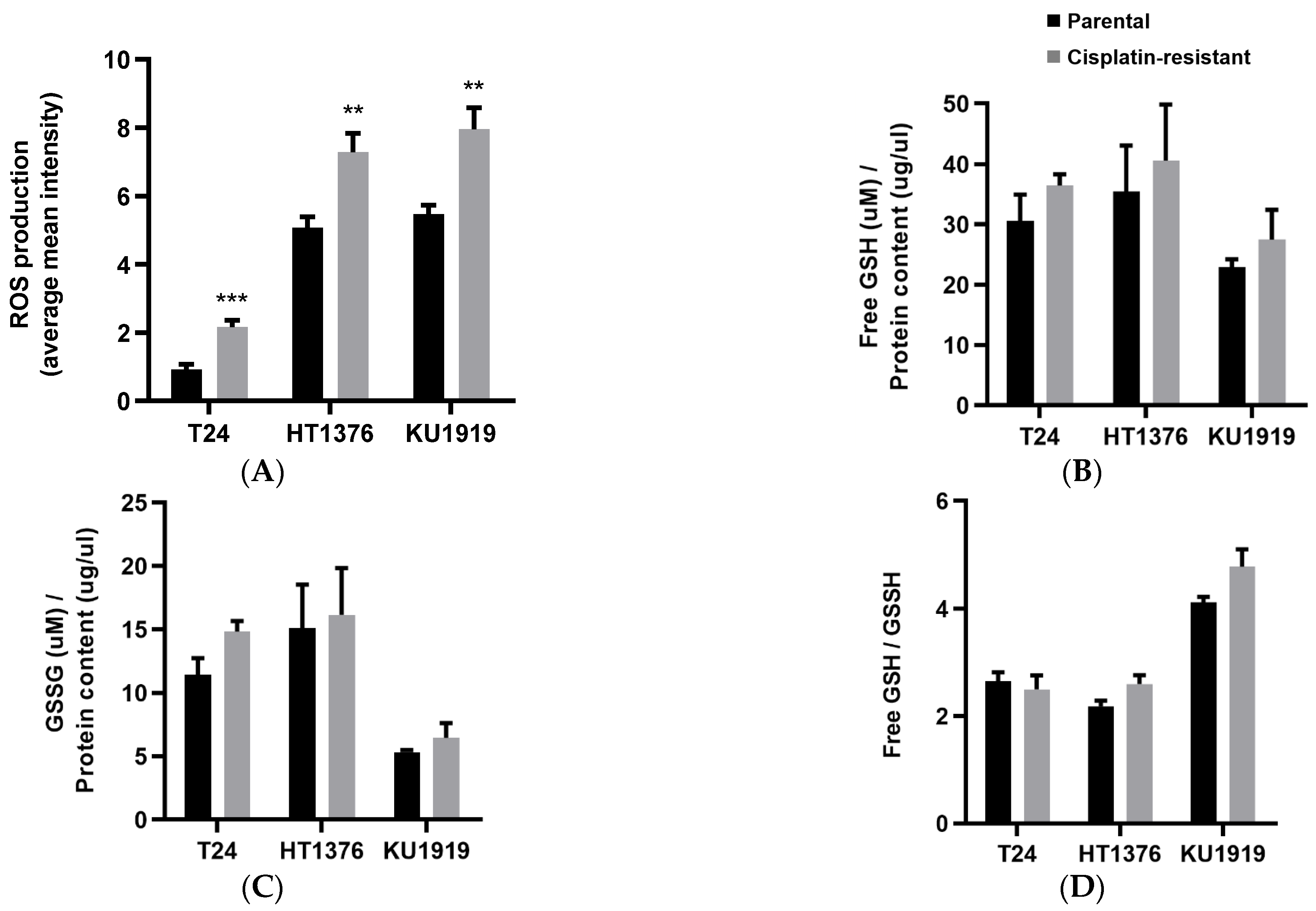
3.4. Antioxidant Activity
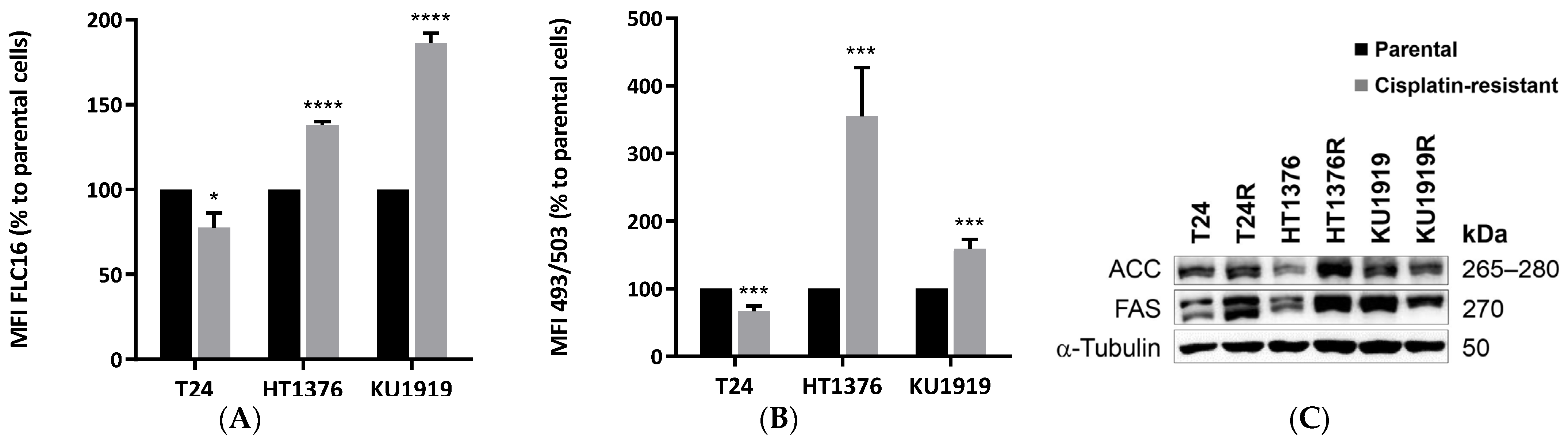
3.5. Lipid Metabolism
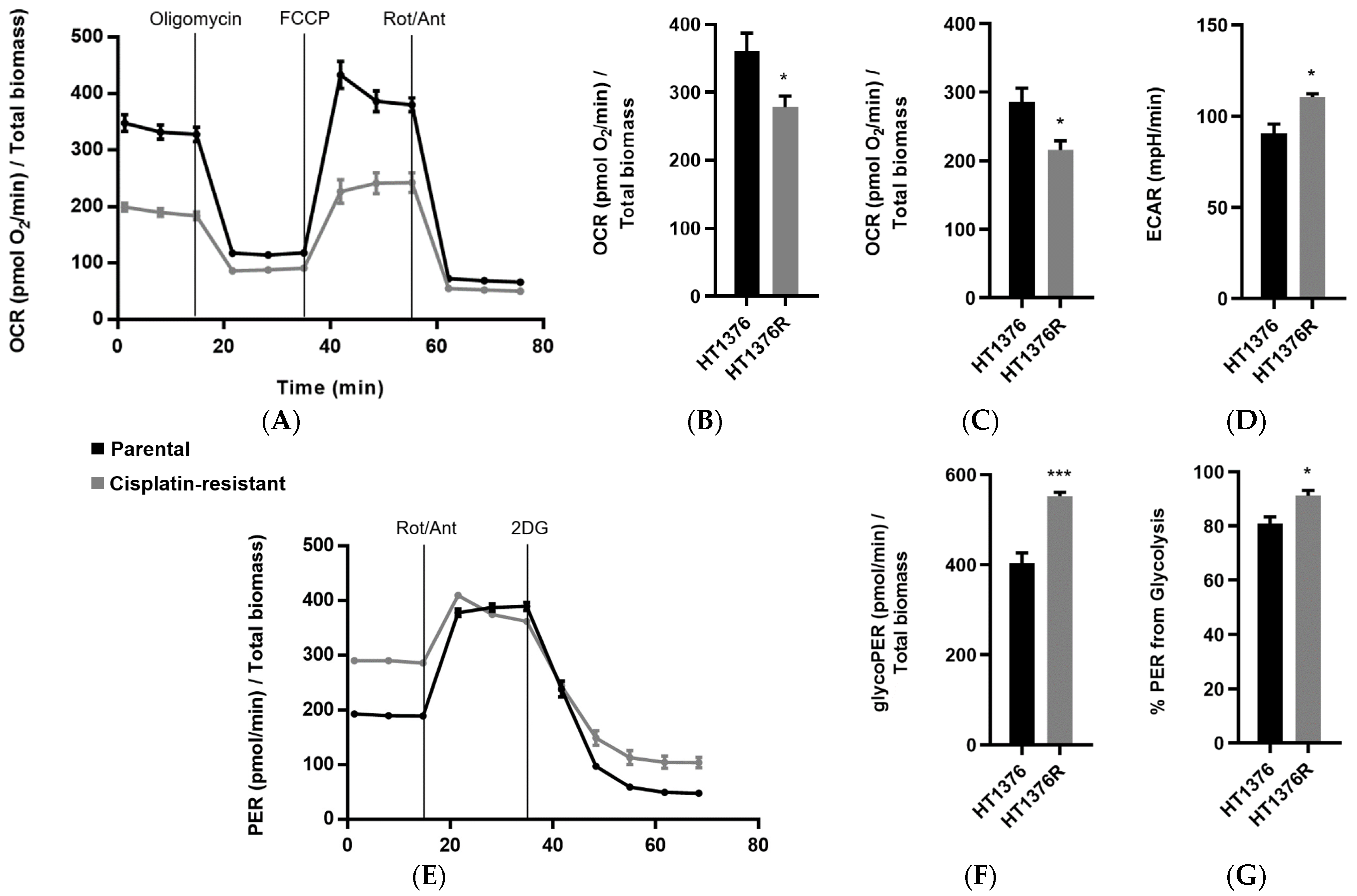
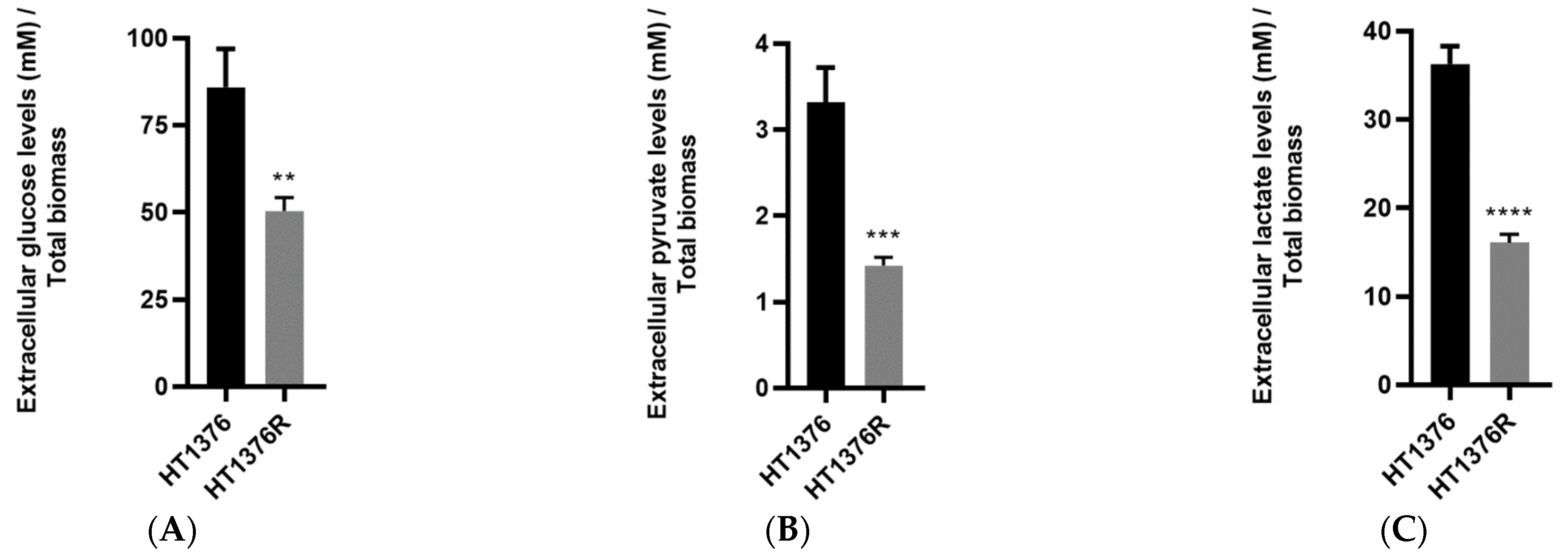
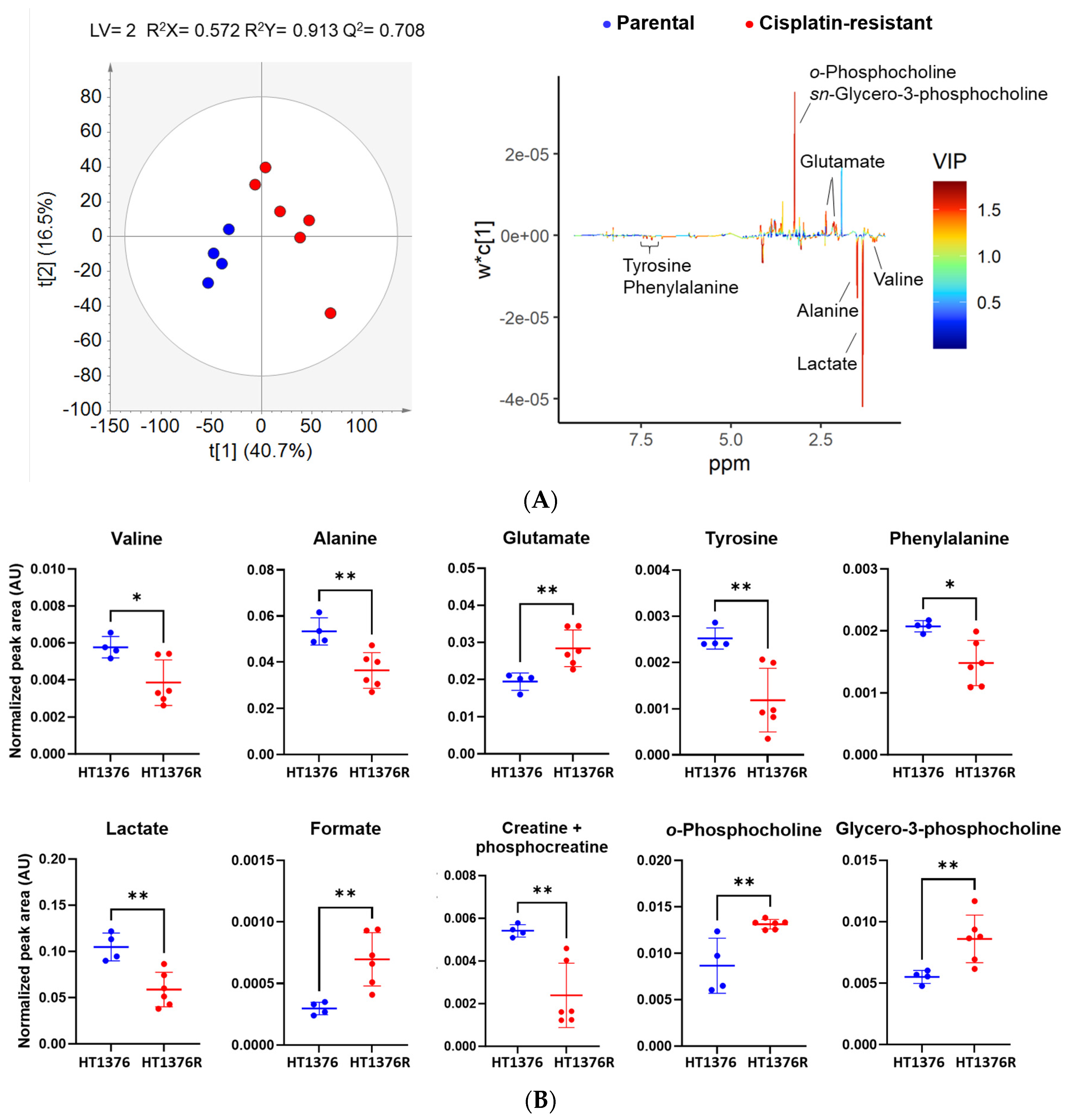
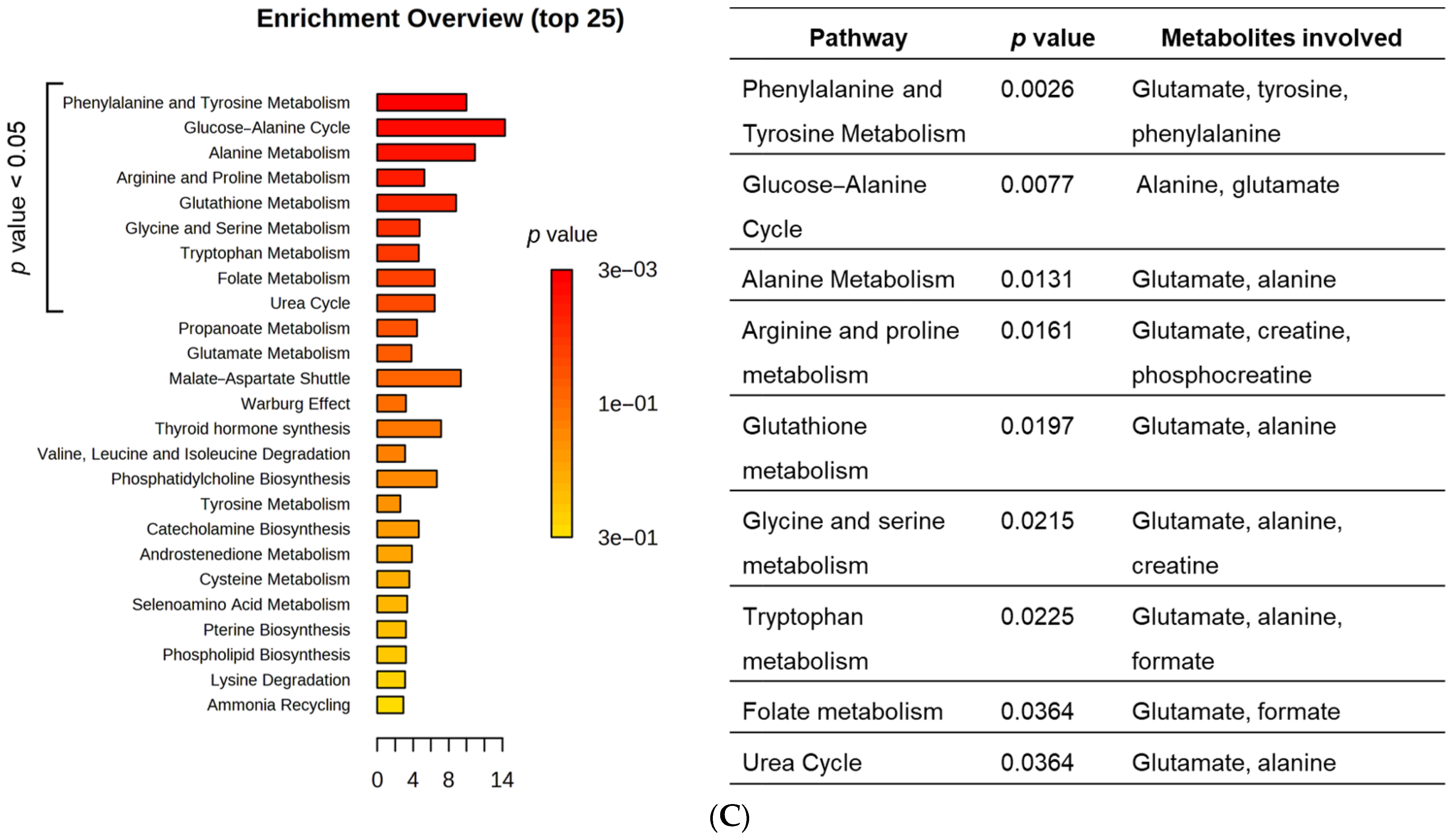
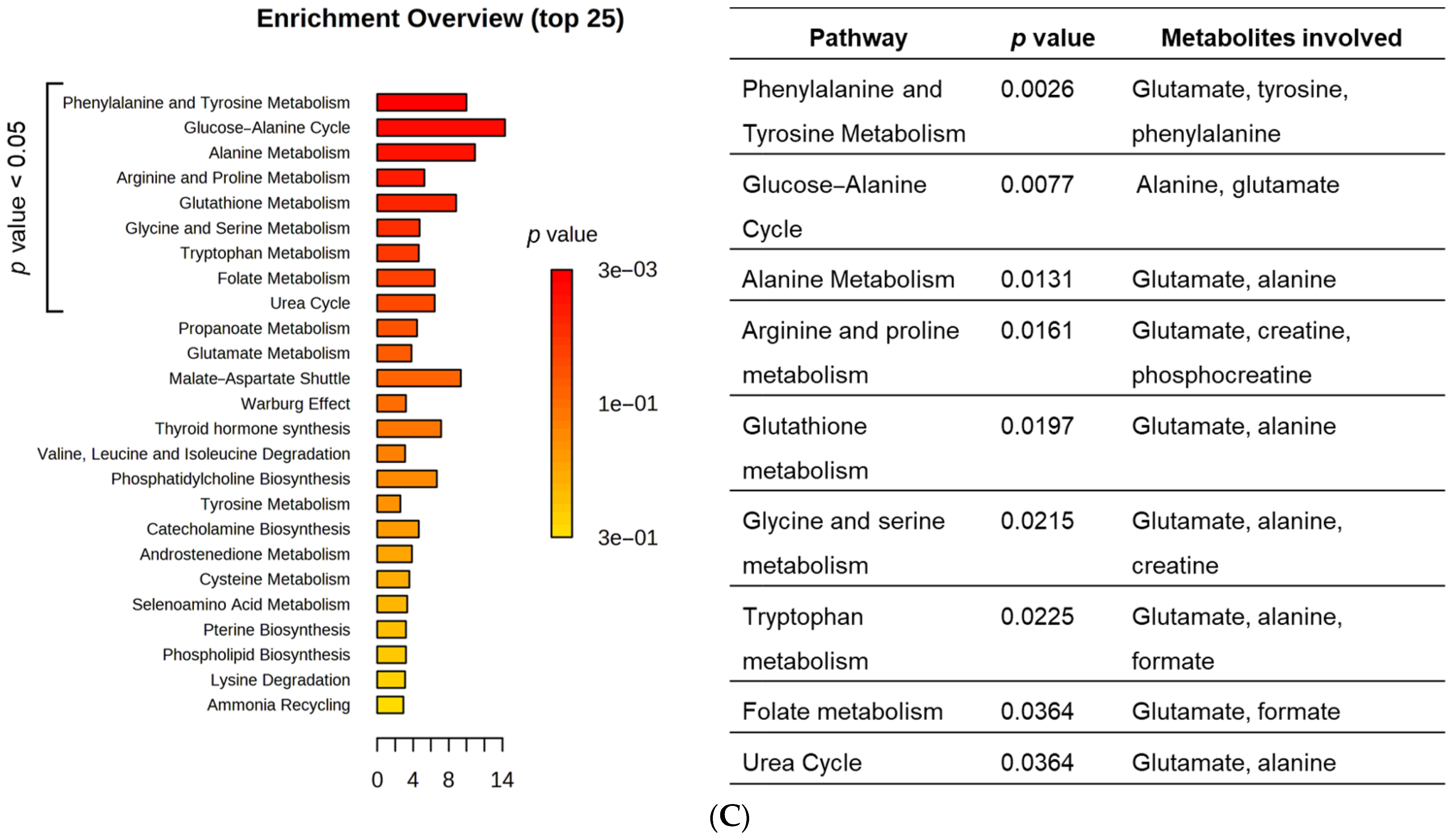
3.6. HT1376 versus HT1376R Cell Lines—Detailed Analysis of Metabolic Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Lobo, N.; Afferi, L.; Moschini, M.; Mostafid, H.; Porten, S.; Psutka, S.P.; Gupta, S.; Smith, A.B.; Williams, S.B.; Lotan, Y. Epidemiology, Screening, and Prevention of Bladder Cancer. Eur. Urol. Oncol. 2022, 5, 628–639. [Google Scholar] [CrossRef]
- Joyce, D.D.; Sharma, V.; Williams, S.B. Cost-Effectiveness and Economic Impact of Bladder Cancer Management: An Updated Review of the Literature. Pharmacoeconomics 2023, 41, 751–769. [Google Scholar] [CrossRef] [PubMed]
- Facchini, G.; Cavaliere, C.; Romis, L.; Mordente, S.; Facchini, S.; Iovane, G.; Capasso, M.; D’Errico, D.; Liguori, C.; Formato, R.; et al. Advanced/metastatic bladder cancer: Current status and future directions. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11536–11552. [Google Scholar] [CrossRef] [PubMed]
- Giles, M.; Crabb, S.J. Systemic Treatment-Decision Algorithms in Muscle-Invasive Bladder Cancer: Clinical Complexities and Navigating for Improved Outcomes. Res. Rep. Urol. 2023, 15, 321–331. [Google Scholar] [CrossRef]
- Ghatalia, P.; Kaur, J.; Sonpavde, G. Muscle invasive bladder cancer: Where is the field headed? Expert Opin. Biol. Ther. 2023, 23, 913–927. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Drayton, R.M.; Catto, J.W. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev. Anticancer Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef]
- Yue, P.; Han, B.; Zhao, Y. Focus on the molecular mechanisms of cisplatin resistance based on multi-omics approaches. Mol. Omics 2023, 19, 297–307. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xia, L.; Liang, J.; Han, Y.; Wang, H.; Oyang, L.; Tan, S.; Tian, Y.; Rao, S.; Chen, X.; et al. The roles of glucose metabolic reprogramming in chemo- and radio-resistance. J. Exp. Clin. Cancer Res. 2019, 38, 218. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Links between cancer metabolism and cisplatin resistance. Int. Rev. Cell Mol. Biol. 2020, 354, 107–164. [Google Scholar] [CrossRef] [PubMed]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, M.; Szczudlo, J.; Pietrzyk, A.; Shah, J.; Trojan, S.E.; Ostrowska, B.; Kocemba-Pilarczyk, K.A. The Warburg effect: A score for many instruments in the concert of cancer and cancer niche cells. Pharmacol. Rep. PR 2023, 75, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Ciccarese, C.; Santoni, M.; Iacovelli, R.; Mazzucchelli, R.; Piva, F.; Scarpelli, M.; Berardi, R.; Tortora, G.; Lopez-Beltran, A.; et al. Metabolic phenotype of bladder cancer. Cancer Treat. Rev. 2016, 45, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.E.; Hurst, C.D.; Knowles, M.A.; Phillips, R.M.; Allison, S.J. The Warburg effect as a therapeutic target for bladder cancers and intratumoral heterogeneity in associated molecular targets. Cancer Sci. 2021, 112, 3822–3834. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.J.; Thoms, J.; Sykes, J.; Ahmed, O.; Evans, A.; van Rhijn, B.W.; Mirtti, T.; Stakhovskyi, O.; Laato, M.; Margel, D.; et al. Hypoxia Marker GLUT-1 (Glucose Transporter 1) is an Independent Prognostic Factor for Survival in Bladder Cancer Patients Treated with Radical Cystectomy. Bladder Cancer 2016, 2, 101–109. [Google Scholar] [CrossRef]
- Sun, C.M.; Xiong, D.B.; Yan, Y.; Geng, J.; Liu, M.; Yao, X.D. Genetic alteration in phosphofructokinase family promotes growth of muscle-invasive bladder cancer. Int. J. Biol. Markers 2016, 31, 286–293. [Google Scholar] [CrossRef]
- Huang, C.; Huang, Z.; Bai, P.; Luo, G.; Zhao, X.; Wang, X. Expression of pyruvate kinase M2 in human bladder cancer and its correlation with clinical parameters and prognosis. Onco Targets Ther. 2018, 11, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ma, S.; Xue, Y.; Hou, J.; Zhang, Y. LDH-A promotes malignant progression via activation of epithelial-to-mesenchymal transition and conferring stemness in muscle-invasive bladder cancer. Biochem. Biophys. Res. Commun. 2016, 469, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A., 3rd. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Goncalves, C.; Costa, M.; Ferreira, D.; Santos, L.; Longatto-Filho, A.; Baltazar, F. Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy. Cancers 2023, 15, 982. [Google Scholar] [CrossRef]
- Xiang, A.P.; Chen, X.N.; Xu, P.F.; Shao, S.H.; Shen, Y.F. Expression and prognostic value of carbonic anhydrase IX (CA-IX) in bladder urothelial carcinoma. BMC Urol. 2022, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Santos, L.L.; Miranda-Goncalves, V.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, F. CD147 and MCT1-potential partners in bladder cancer aggressiveness and cisplatin resistance. Mol. Carcinog. 2015, 54, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Santos, L.L.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, F. Metabolic coupling in urothelial bladder cancer compartments and its correlation to tumor aggressiveness. Cell Cycle 2016, 15, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zeng, J.; Xie, R.; Schulz, M.J.; Tedesco, R.; Qu, J.; Erhard, K.F.; Mack, J.F.; Raha, K.; Rendina, A.R.; et al. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.Y.; Wang de, G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983. [Google Scholar] [CrossRef]
- Lea, M.A.; Guzman, Y.; Desbordes, C. Inhibition of Growth by Combined Treatment with Inhibitors of Lactate Dehydrogenase and either Phenformin or Inhibitors of 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase 3. Anticancer Res. 2016, 36, 1479–1488. [Google Scholar] [PubMed]
- Todenhofer, T.; Seiler, R.; Stewart, C.; Moskalev, I.; Gao, J.; Ladhar, S.; Kamjabi, A.; Al Nakouzi, N.; Hayashi, T.; Choi, S.; et al. Selective Inhibition of the Lactate Transporter MCT4 Reduces Growth of Invasive Bladder Cancer. Mol. Cancer Ther. 2018, 17, 2746–2755. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell. Physiol. Biochem. 2017, 41, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Loschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rodel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, I.; Ferreira, C.; Moreira, D.; Kluck, G.E.G.; Barbosa, A.M.; Torrado, E.; Dinis-Oliveira, R.J.; Goncalves, L.G.; Beauparlant, C.J.; Droit, A.; et al. The Absence of HIF-1alpha Increases Susceptibility to Leishmania donovani Infection via Activation of BNIP3/mTOR/SREBP-1c Axis. Cell Rep. 2020, 30, 4052–4064 e4057. [Google Scholar] [CrossRef]
- Teng, Q.; Huang, W.; Collette, T.W.; Ekman, D.R.; Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics Off. J. Metabolomic Soc. 2009, 5, 199–208. [Google Scholar] [CrossRef]
- Amaro, F.; Pisoeiro, C.; Valente, M.J.; Bastos, M.L.; Guedes de Pinho, P.; Carvalho, M.; Pinto, J. Sunitinib versus Pazopanib Dilemma in Renal Cell Carcinoma: New Insights into the In Vitro Metabolic Impact, Efficacy, and Safety. Int. J. Mol. Sci. 2022, 23, 9898. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef]
- Jacob, D.; Deborde, C.; Lefebvre, M.; Maucourt, M.; Moing, A. NMRProcFlow: A graphical and interactive tool dedicated to 1D spectra processing for NMR-based metabolomics. Metabolomics Off. J. Metabolomic Soc. 2017, 13, 36. [Google Scholar] [CrossRef]
- Bloemberg, T.G.; Gerretzen, J.; Wouters, H.J.P.; Gloerich, J.; van Dael, M.; Wessels, H.J.C.T.; van den Heuvel, L.P.; Eilers, P.H.C.; Buydens, L.M.C.; Wehrens, R. Improved parametric time warping for proteomics. Chemom. Intell. Lab. Syst. 2010, 104, 65–74. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 12 July 2023).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Cham, Switzerland, 2016; pp. 189–201. [Google Scholar]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2021, 79, 82–104. [Google Scholar] [CrossRef]
- von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef]
- Lugones, Y.; Loren, P.; Salazar, L.A. Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules 2022, 12, 1365. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Melnikova, M.; van Roermund, J.G.H.; Romano, A.; Albers, P.; Thomale, J.; Schulz, W.A.; Niegisch, G.; Hoffmann, M.J. Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2018, 19, 590. [Google Scholar] [CrossRef]
- Vallo, S.; Michaelis, M.; Rothweiler, F.; Bartsch, G.; Gust, K.M.; Limbart, D.M.; Rodel, F.; Wezel, F.; Haferkamp, A.; Cinatl, J., Jr. Drug-Resistant Urothelial Cancer Cell Lines Display Diverse Sensitivity Profiles to Potential Second-Line Therapeutics. Transl. Oncol. 2015, 8, 210–216. [Google Scholar] [CrossRef]
- Pinto-Leite, R.; Carreira, I.; Melo, J.; Ferreira, S.I.; Ribeiro, I.; Ferreira, J.; Filipe, M.; Bernardo, C.; Arantes-Rodrigues, R.; Oliveira, P.; et al. Genomic characterization of three urinary bladder cancer cell lines: Understanding genomic types of urinary bladder cancer. Tumour Biol. 2014, 35, 4599–4617. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.P.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017, 14, 215–229. [Google Scholar] [CrossRef]
- Silva, A.; Félix, A.; Cerqueira, M.; Gonçalves, C.S.; Sampaio-Marques, B.; Longatto-Filho, A.; Baltazar, F.; Afonso, J. Effects of Lactate Transport Inhibition by AZD3965 in Muscle-Invasive Urothelial Bladder Cancer. Pharmaceutics 2023, 15, 2688. [Google Scholar] [CrossRef]
- Shang, D.; Wu, J.; Guo, L.; Xu, Y.; Liu, L.; Lu, J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int. J. Oncol. 2017, 50, 1848–1856. [Google Scholar] [CrossRef]
- Yan, C.; Yang, F.; Zhou, C.; Chen, X.; Han, X.; Liu, X.; Ma, H.; Zheng, W. MCT1 promotes the cisplatin-resistance by antagonizing Fas in epithelial ovarian cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 2710–2718. [Google Scholar] [PubMed]
- Zhang, X.Y.; Zhang, M.; Cong, Q.; Zhang, M.X.; Zhang, M.Y.; Lu, Y.Y.; Xu, C.J. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.Z.; Qu, Y.Q.; Liang, A.B.; Deng, A.M.; Zhang, W.J.; Xiu, B.; Wang, H.; Wang, H. Expression of CD147 in advanced non-small cell lung cancer correlated with cisplatin-based chemotherapy resistance. Neoplasma 2011, 58, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The SLC16 gene family-structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Wilson, M.C.; Heddle, C.; Brown, M.H.; Barclay, A.N.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef]
- Sawayama, H.; Ogata, Y.; Ishimoto, T.; Mima, K.; Hiyoshi, Y.; Iwatsuki, M.; Baba, Y.; Miyamoto, Y.; Yoshida, N.; Baba, H. Glucose transporter 1 regulates the proliferation and cisplatin sensitivity of esophageal cancer. Cancer Sci. 2019, 110, 1705–1714. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, M.L.; Fan, J. Inhibition of GLUT-1 expression and the PI3K/Akt pathway to enhance the chemosensitivity of laryngeal carcinoma cells in vitro. Onco Targets Ther. 2018, 11, 7865–7872. [Google Scholar] [CrossRef]
- Li, S.; Yang, X.; Wang, P.; Ran, X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell Physiol. Biochem. 2013, 32, 624–634. [Google Scholar] [CrossRef]
- Li, H.; Fu, L.; Liu, B.; Lin, X.; Dong, Q.; Wang, E. Ajuba overexpression regulates mitochondrial potential and glucose uptake through YAP/Bcl-xL/GLUT1 in human gastric cancer. Gene 2019, 693, 16–24. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Ling, H.H.; Chiang, M.C.; Chung, C.H.; Lee, W.Y.; Chu, C.Y.; Wu, Y.C.; Chen, C.H.; Lai, Y.W.; Tsai, I.L.; et al. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019, 9, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The progression from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and pyruvate metabolism. Exp. Cell Res. 2015, 335, 91–98. [Google Scholar] [CrossRef]
- Le Calve, B.; Rynkowski, M.; Le Mercier, M.; Bruyere, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Kuang, R.; Jahangiri, A.; Mascharak, S.; Nguyen, A.; Chandra, A.; Flanigan, P.M.; Yagnik, G.; Wagner, J.R.; De Lay, M.; Carrera, D.; et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI insight 2017, 2, e88815. [Google Scholar] [CrossRef]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From Warburg effect to Reverse Warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef]
- Ma, L.; Zong, X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front. Oncol 2020, 10, 5. [Google Scholar] [CrossRef]
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys. Res. Commun. 2018, 503, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.-M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021, 46, 102065. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Hong, S.Y.; Xun, Y.; Liu, C.Q.; Sun, J.X.; Xu, J.Z.; Xu, M.Y.; An, Y.; He, D.; Xia, Q.D.; et al. Characterization of the Lipid Metabolism in Bladder Cancer to Guide Clinical Therapy. J. Oncol. 2022, 2022, 7679652. [Google Scholar] [CrossRef]
- Jin, L.; Chun, J.; Pan, C.; Li, D.; Lin, R.; Alesi, G.N.; Wang, X.; Kang, H.B.; Song, L.; Wang, D.; et al. MAST1 Drives Cisplatin Resistance in Human Cancers by Rewiring cRaf-Independent MEK Activation. Cancer Cell 2018, 34, 315–330 e317. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Chen, X.; Liu, B.; Li, P.; Cao, W. Omega-3 Polyunsaturated Fatty Acids Enhance Cisplatin Efficacy in Gastric Cancer Cells by Inducing Apoptosis via ADORA1. Anticancer Agents Med. Chem. 2016, 16, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. Onco Targets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Tan, Y.; Li, J.; Zhao, G.; Huang, K.C.; Cardenas, H.; Wang, Y.; Matei, D.; Cheng, J.X. Metabolic reprogramming from glycolysis to fatty acid uptake and beta-oxidation in platinum-resistant cancer cells. Nat. Commun. 2022, 13, 4554. [Google Scholar] [CrossRef]
- Ippolito, L.; Comito, G.; Parri, M.; Iozzo, M.; Duatti, A.; Virgilio, F.; Lorito, N.; Bacci, M.; Pardella, E.; Sandrini, G.; et al. Lactate Rewires Lipid Metabolism and Sustains a Metabolic-Epigenetic Axis in Prostate Cancer. Cancer Res. 2022, 82, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.R.; Wang, J.; Wang, Z.J.; Xi, M.J.; Xia, B.H.; Deng, K.; Yang, J.L. Lipid metabolic reprogramming in tumor microenvironment: From mechanisms to therapeutics. J. Hematol. Oncol. 2023, 16, 103. [Google Scholar] [CrossRef]
- Perez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hee, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Z.; Yang, Z.; Guo, L.; Zhang, Y.; Deng, H.; Wang, C.; Feng, M. A Glutamine-Rich Carrier Efficiently Delivers Anti-CD47 siRNA Driven by a “Glutamine Trap” To Inhibit Lung Cancer Cell Growth. Mol. Pharm. 2018, 15, 3032–3045. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283 e212. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Pompella, A.; De Tata, V.; Paolicchi, A.; Zunino, F. Expression of gamma-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem. Pharmacol. 2006, 71, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Jang, M.; Song, M.J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCalpha Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef]
- Elda Valenti, G.; Tasso, B.; Traverso, N.; Domenicotti, C.; Marengo, B. Glutathione in cancer progression and chemoresistance: An update. Redox Exp. Med. 2023, 2023, e220023. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Shirato, A.; Kikugawa, T.; Miura, N.; Tanji, N.; Takemori, N.; Higashiyama, S.; Yokoyama, M. Cisplatin resistance by induction of aldo-keto reductase family 1 member C2 in human bladder cancer cells. Oncol. Lett. 2014, 7, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, X.; Fu, J.; Xu, W.; Yuan, J. The Role of Tumour Metabolism in Cisplatin Resistance. Front. Mol. Biosci. 2021, 8, 691795. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Bibby, B.A.; Dunne, M.R.; Kennedy, S.A.; Davern, M.B.; Kennedy, B.N.; Maher, S.G.; O’Sullivan, J. Characterisation of an Isogenic Model of Cisplatin Resistance in Oesophageal Adenocarcinoma Cells. Pharmaceuticals 2019, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, W.; Zhang, Y.; Wu, S.; Liu, Y.; Deng, X.; Xie, L.; Yang, J.; Yu, H.; Su, J.; et al. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int. J. Oncol. 2018, 53, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Brunelli, L.; Affatato, R.; Chila, R.; Verza, M.; Indraccolo, S.; Falcetta, F.; Fratelli, M.; Fruscio, R.; Pastorelli, R.; et al. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther. Adv. Med. Oncol. 2019, 11, 1758835919839543. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, J.; Barbosa-Matos, C.; Silvestre, R.; Pereira-Vieira, J.; Gonçalves, S.M.; Mendes-Alves, C.; Parpot, P.; Pinto, J.; Carapito, Â.; Guedes de Pinho, P.; et al. Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect. Cancers 2024, 16, 1418. https://doi.org/10.3390/cancers16071418
Afonso J, Barbosa-Matos C, Silvestre R, Pereira-Vieira J, Gonçalves SM, Mendes-Alves C, Parpot P, Pinto J, Carapito Â, Guedes de Pinho P, et al. Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect. Cancers. 2024; 16(7):1418. https://doi.org/10.3390/cancers16071418
Chicago/Turabian StyleAfonso, Julieta, Catarina Barbosa-Matos, Ricardo Silvestre, Joana Pereira-Vieira, Samuel Martins Gonçalves, Camille Mendes-Alves, Pier Parpot, Joana Pinto, Ângela Carapito, Paula Guedes de Pinho, and et al. 2024. "Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect" Cancers 16, no. 7: 1418. https://doi.org/10.3390/cancers16071418
APA StyleAfonso, J., Barbosa-Matos, C., Silvestre, R., Pereira-Vieira, J., Gonçalves, S. M., Mendes-Alves, C., Parpot, P., Pinto, J., Carapito, Â., Guedes de Pinho, P., Santos, L., Longatto-Filho, A., & Baltazar, F. (2024). Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect. Cancers, 16(7), 1418. https://doi.org/10.3390/cancers16071418