
Mutations in the Serine/Threonine Kinase BRAF: Oncogenic Drivers in Solid Tumors


Abstract
Simple Summary
Abstract
1. Introduction
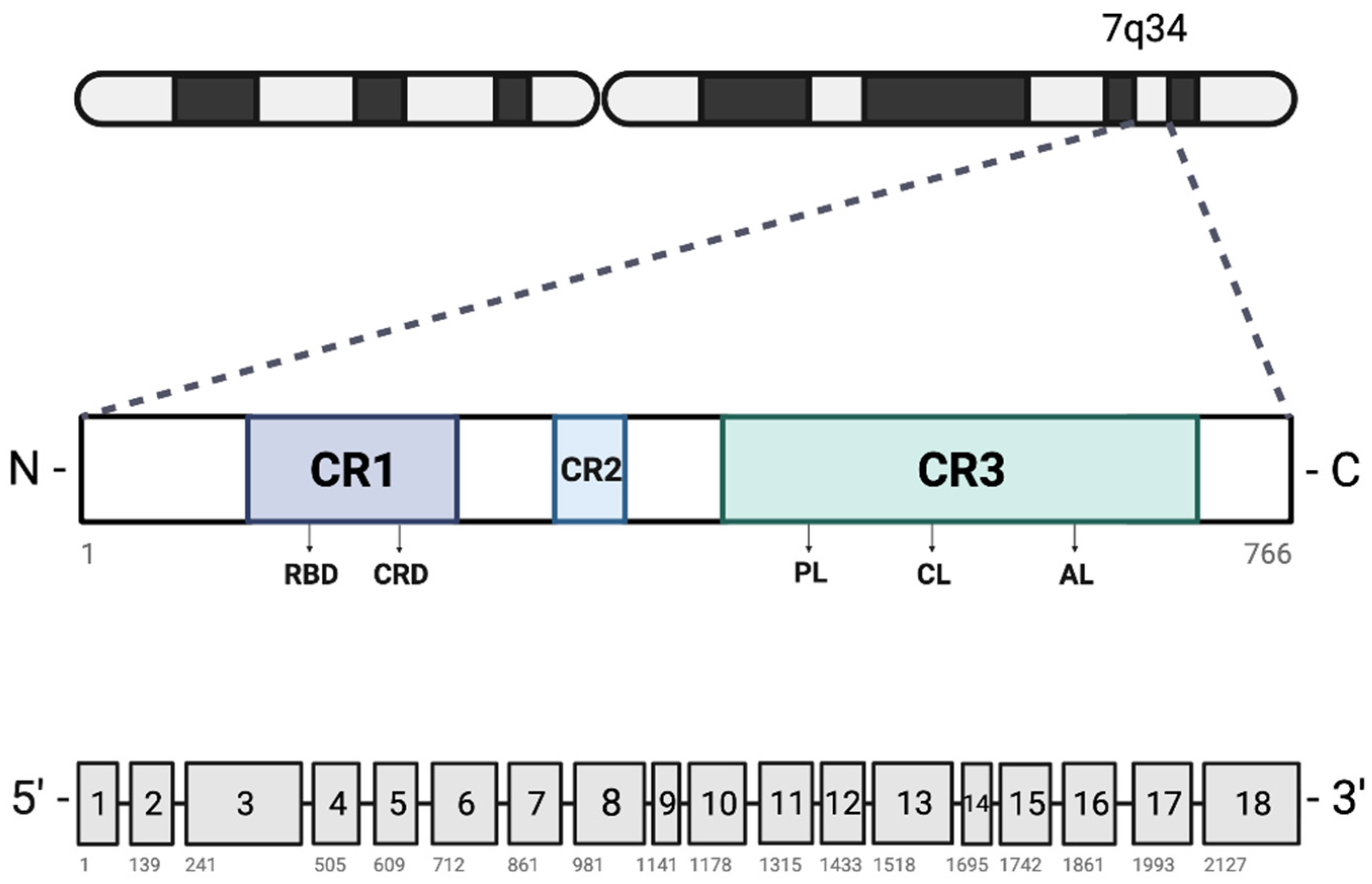
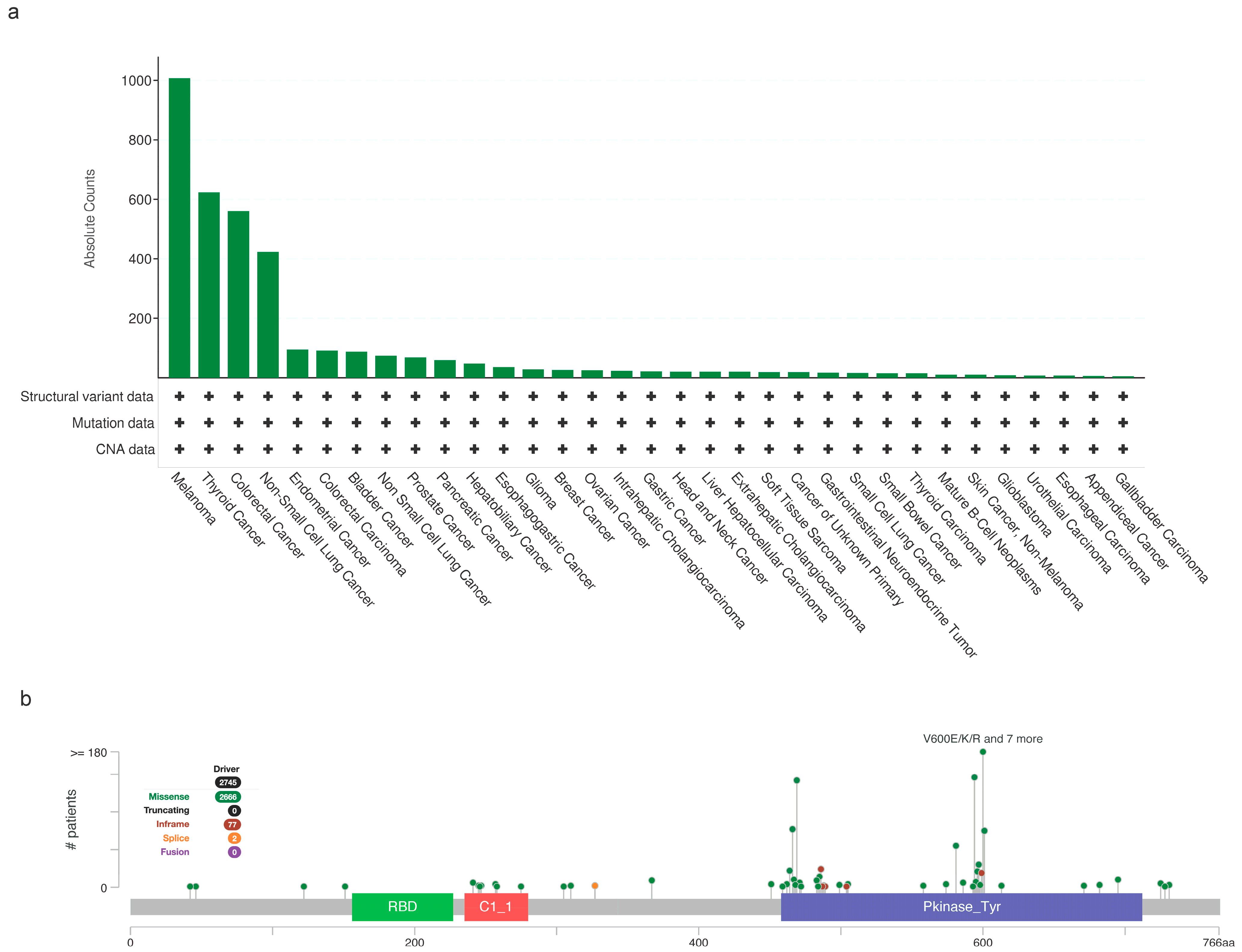
2. BRAF Mutations in Cancer
2.1. Melanoma
2.2. Thyroid
2.3. Lung Cancer
2.4. Colorectal
2.5. Other Cancers
3. Diagnostic Approaches for BRAF Mutations
3.1. Detection of Mutations
3.2. Associated Biomarkers
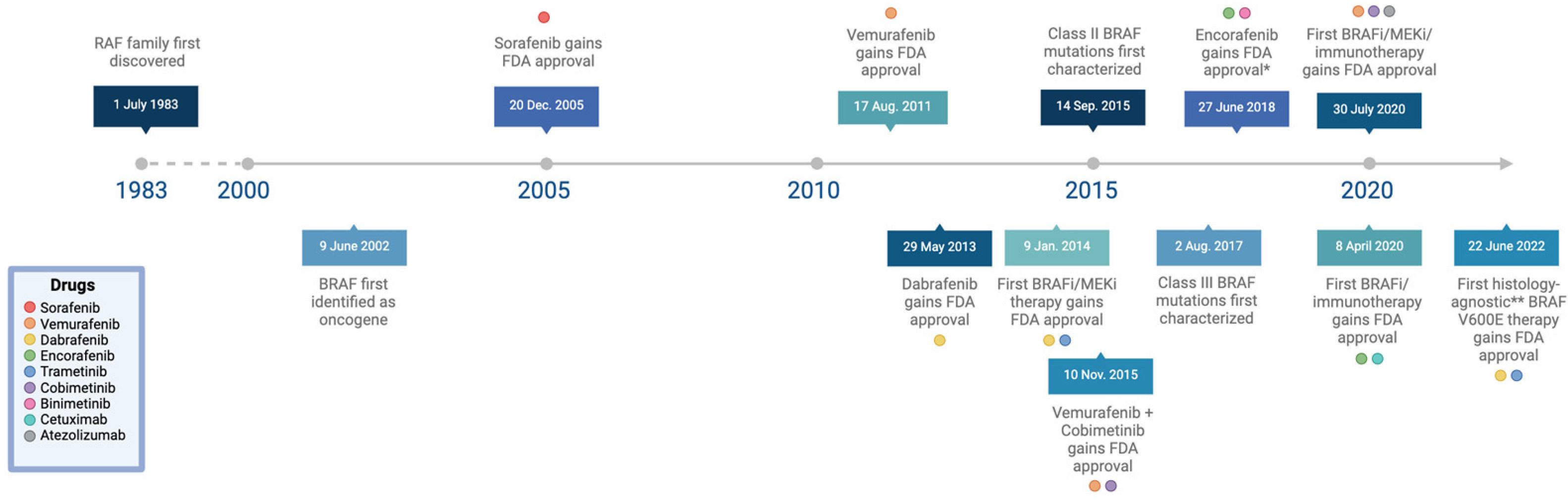
4. Targeted Therapies for BRAF-Mutant Cancers
4.1. First-Generation BRAF Inhibitors
4.2. Second-Generation BRAF Inhibitors
4.3. Combination Therapies
4.4. Third-Generation BRAF Inhibitors
4.5. Current Efforts in Targeting Resistance to Therapy
5. Resistance Mechanisms and Overcoming Challenges
5.1. BRAF and the MAPK Signaling Pathway
5.2. MEK/ERK-Dependent Resistance
5.3. MEK/ERK-Independent Resistance
5.4. Potential New Targets for Overcoming Mechanisms of Resistance
6. Clinical Implications and Patient Outcomes
7. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes. Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Haser, W.; Haystead, T.A.; Vincent, L.A.; Roberts, T.M.; Sturgill, T.W. Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992, 257, 1404–1407. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Van Aelst, L.; Barr, M.; Marcus, S.; Polverino, A.; Wigler, M. Complex formation between RAS and RAF and other protein kinases. Proc. Natl. Acad. Sci. USA 1993, 90, 6213–6217. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993, 74, 205–214. [Google Scholar] [CrossRef]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Zhang, X.F.; Settleman, J.; Kyriakis, J.M.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Sullivan, R.J.; Yaeger, R. Molecular Pathways and Mechanisms of BRAF in Cancer Therapy. Clin. Cancer Res. 2022, 28, 4618–4628. [Google Scholar] [CrossRef]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Meikle, S.; Yazici, Z.; Eulitz, M.; Kolch, W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 2002, 21, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Lee, J.; Guan, K.L. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Tecentriq [Package Insert]; Genentech, Inc.: South San Francisco, CA, USA, 2023.
- Zelboraf [Package Insert]; Hoffmann La Roche: Basel, Switzerland, 2017.
- Mekinist [Package Insert]; Novartis Pharmaceuticals: Basel, Switzerland, 2023.
- Braftovi [Package Insert]; Array Biopharma Inc.: Boulder, CO, USA, 2023.
- Nexavar [Package Insert]; Bayer Healthcare Pharmaceuticals LLC: Berlin, Germany, 2023.
- Tafinlar [Package Insert]; Novartis Pharmaceuticals: Basel, Switzerland, 2023.
- Erbitux [Package Insert]; ImClone Systems Incorporated: Bridgewater, NJ, USA, 2021.
- Rapp, U.R.; Goldsborough, M.D.; Mark, G.E.; Bonner, T.I.; Groffen, J.; Reynolds, F.H., Jr.; Stephenson, J.R. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4218–4222. [Google Scholar] [CrossRef]
- Yao, H.; Sun, Q.; Zhu, J. Identification and Characterization of Small-Molecule Inhibitors to Selectively Target the DFG-in over the DFG-out Conformation of the B-Raf Kinase V600E Mutant in Colorectal Cancer. Arch. Pharm. 2016, 349, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Dhomen, N.; Marais, R. New insight into BRAF mutations in cancer. Curr. Opin. Genet. Dev. 2007, 17, 31–39. [Google Scholar] [CrossRef]
- Marchetti, A.; Felicioni, L.; Malatesta, S.; Grazia Sciarrotta, M.; Guetti, L.; Chella, A.; Viola, P.; Pullara, C.; Mucilli, F.; Buttitta, F. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 3574–3579. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Malapelle, U.; Troncone, G. BRAF Mutations in Lung Cancer. Acta Cytol. 2019, 63, 247–250. [Google Scholar] [CrossRef]
- Schirripa, M.; Biason, P.; Lonardi, S.; Pella, N.; Pino, M.S.; Urbano, F.; Antoniotti, C.; Cremolini, C.; Corallo, S.; Pietrantonio, F.; et al. Class 1, 2, and 3 BRAF-Mutated Metastatic Colorectal Cancer: A Detailed Clinical, Pathologic, and Molecular Characterization. Clin. Cancer Res. 2019, 25, 3954–3961. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Sonawane, P.; Kumar, A.; Singh, H.; Naumovich, V.; Pathak, P.; Grishina, M.; Khalilullah, H.; Jaremko, M.; Emwas, A.H.; et al. Challenges and Opportunities in the Crusade of BRAF Inhibitors: From 2002 to 2022. ACS Omega 2023, 8, 27819–27844. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF new world. Crit. Rev. Oncol. Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef] [PubMed]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF Mutations Classes I, II, and III in NSCLC Patients Included in the SLLIP Trial: The Need for a New Pre-Clinical Treatment Rationale. Cancers 2019, 11, 1381. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Malapelle, U.; Andre, F.; Paz-Ares, L.; Schuler, M.; Thomas, D.M.; Vainer, G.; Yoshino, T.; Rolfo, C. Practical Considerations for the Use of Circulating Tumor DNA in the Treatment of Patients With Cancer: A Narrative Review. JAMA Oncol. 2022, 8, 1830–1839. [Google Scholar] [CrossRef]
- Smiech, M.; Leszczynski, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Ihle, M.A.; Fassunke, J.; Konig, K.; Grunewald, I.; Schlaak, M.; Kreuzberg, N.; Tietze, L.; Schildhaus, H.U.; Buttner, R.; Merkelbach-Bruse, S. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer 2014, 14, 13. [Google Scholar] [CrossRef]
- Bradish, J.R.; Cheng, L. Molecular pathology of malignant melanoma: Changing the clinical practice paradigm toward a personalized approach. Hum. Pathol. 2014, 45, 1315–1326. [Google Scholar] [CrossRef]
- Ribas, A.; Flaherty, K.T. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat. Rev. Clin. Oncol. 2011, 8, 426–433. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.; Haidar, A.; Anghel, R.M. Treating malignant melanoma when a rare BRAF V600M mutation is present: Case report and literature review. Rom. J. Intern. Med. 2018, 56, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Hanrahan, A.J.; Chen, Z.; Rosen, N.; Solit, D.B. BRAF—A tumour-agnostic drug target with lineage-specific dependencies. Nat. Rev. Clin. Oncol. 2024, 21, 224–247. [Google Scholar] [CrossRef]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Elisei, R.; Ugolini, C.; Viola, D.; Lupi, C.; Biagini, A.; Giannini, R.; Romei, C.; Miccoli, P.; Pinchera, A.; Basolo, F. BRAF(V600E) mutation and outcome of patients with papillary thyroid carcinoma: A 15-year median follow-up study. J. Clin. Endocrinol. Metab. 2008, 93, 3943–3949. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Rosenbaum, E.; Clark, D.P.; Zeiger, M.A.; Umbricht, C.B.; Tufano, R.P.; Sidransky, D.; Westra, W.H. Mutational analysis of BRAF in fine needle aspiration biopsies of the thyroid: A potential application for the preoperative assessment of thyroid nodules. Clin. Cancer Res. 2004, 10, 2761–2765. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Vasko, V.; Tallini, G.; Larin, A.; Wu, G.; Udelsman, R.; Ringel, M.D.; Ladenson, P.W.; Sidransky, D. BRAF T1796A transversion mutation in various thyroid neoplasms. J. Clin. Endocrinol. Metab. 2004, 89, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Trovisco, V.; Soares, P.; Sobrinho-Simoes, M. B-RAF mutations in the etiopathogenesis, diagnosis, and prognosis of thyroid carcinomas. Hum. Pathol. 2006, 37, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Knauf, J.A.; Ma, X.; Smith, E.P.; Zhang, L.; Mitsutake, N.; Liao, X.H.; Refetoff, S.; Nikiforov, Y.E.; Fagin, J.A. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005, 65, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Mesa, C., Jr.; Mirza, M.; Mitsutake, N.; Sartor, M.; Medvedovic, M.; Tomlinson, C.; Knauf, J.A.; Weber, G.F.; Fagin, J.A. Conditional activation of RET/PTC3 and BRAFV600E in thyroid cells is associated with gene expression profiles that predict a preferential role of BRAF in extracellular matrix remodeling. Cancer Res. 2006, 66, 6521–6529. [Google Scholar] [CrossRef] [PubMed]
- Bellevicine, C.; Migliatico, I.; Sgariglia, R.; Nacchio, M.; Vigliar, E.; Pisapia, P.; Iaccarino, A.; Bruzzese, D.; Fonderico, F.; Salvatore, D.; et al. Evaluation of BRAF, RAS, RET/PTC, and PAX8/PPARg alterations in different Bethesda diagnostic categories: A multicentric prospective study on the validity of the 7-gene panel test in 1172 thyroid FNAs deriving from different hospitals in South Italy. Cancer Cytopathol. 2020, 128, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bellevicine, C.; Sgariglia, R.; Malapelle, U.; Vigliar, E.; Nacchio, M.; Ciancia, G.; Eszlinger, M.; Paschke, R.; Troncone, G. Young investigator challenge: Can the Ion AmpliSeq Cancer Hotspot Panel v2 be used for next-generation sequencing of thyroid FNA samples? Cancer Cytopathol. 2016, 124, 776–784. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: A meta-analysis. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, E.S.; Kim, Y.S. Clinicopathologic significance of BRAF V600E mutation in papillary carcinomas of the thyroid: A meta-analysis. Cancer 2007, 110, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G.; et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–470, discussion 470–461. [Google Scholar] [CrossRef]
- Jo, Y.S.; Li, S.; Song, J.H.; Kwon, K.H.; Lee, J.C.; Rha, S.Y.; Lee, H.J.; Sul, J.Y.; Kweon, G.R.; Ro, H.K.; et al. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2006, 91, 3667–3670. [Google Scholar] [CrossRef]
- Kim, J.; Giuliano, A.E.; Turner, R.R.; Gaffney, R.E.; Umetani, N.; Kitago, M.; Elashoff, D.; Hoon, D.S. Lymphatic mapping establishes the role of BRAF gene mutation in papillary thyroid carcinoma. Ann. Surg. 2006, 244, 799–804. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, W.B.; Rhee, Y.S.; Song, J.Y.; Kim, J.M.; Gong, G.; Lee, S.; Kim, S.Y.; Kim, S.C.; Hong, S.J.; et al. The BRAF mutation is useful for prediction of clinical recurrence in low-risk patients with conventional papillary thyroid carcinoma. Clin. Endocrinol. 2006, 65, 364–368. [Google Scholar] [CrossRef]
- Namba, H.; Nakashima, M.; Hayashi, T.; Hayashida, N.; Maeda, S.; Rogounovitch, T.I.; Ohtsuru, A.; Saenko, V.A.; Kanematsu, T.; Yamashita, S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 2003, 88, 4393–4397. [Google Scholar] [CrossRef] [PubMed]
- Trovisco, V.; Soares, P.; Preto, A.; de Castro, I.V.; Lima, J.; Castro, P.; Maximo, V.; Botelho, T.; Moreira, S.; Meireles, A.M.; et al. Type and prevalence of BRAF mutations are closely associated with papillary thyroid carcinoma histotype and patients’ age but not with tumour aggressiveness. Virchows Arch. 2005, 446, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Quiros, R.M.; Gattuso, P.; Ain, K.B.; Prinz, R.A. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003, 63, 4561–4567. [Google Scholar] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef]
- Harada, G.; Yang, S.R.; Cocco, E.; Drilon, A. Rare molecular subtypes of lung cancer. Nat. Rev. Clin. Oncol. 2023, 20, 229–249. [Google Scholar] [CrossRef]
- Brustugun, O.T.; Khattak, A.M.; Tromborg, A.K.; Beigi, M.; Beiske, K.; Lund-Iversen, M.; Helland, A. BRAF-mutations in non-small cell lung cancer. Lung Cancer 2014, 84, 36–38. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Schrock, A.B.; Li, S.D.; Frampton, G.M.; Suh, J.; Braun, E.; Mehra, R.; Buck, S.C.; Bufill, J.A.; Peled, N.; Karim, N.A.; et al. Pulmonary Sarcomatoid Carcinomas Commonly Harbor Either Potentially Targetable Genomic Alterations or High Tumor Mutational Burden as Observed by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2017, 12, 932–942. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Gautschi, O.; Pauli, C.; Strobel, K.; Hirschmann, A.; Printzen, G.; Aebi, S.; Diebold, J. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J. Thorac. Oncol. 2012, 7, e23–e24. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Michielin, O.; Zimmermann, S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J. Clin. Oncol. 2013, 31, e341–e344. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; O’Shaughnessy, J.A.; Cowey, C.L.; Konduri, K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer 2014, 85, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Siano, M.; Joerger, M.; Rodriguez, R.; Muller, J.; Fruh, M. Response to dabrafenib after progression on vemurafenib in a patient with advanced BRAF V600E-mutant bronchial adenocarcinoma. Lung Cancer 2015, 87, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Cropet, C.; Montane, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAF(V600) and BRAF(nonV600) mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.J.; Barlesi, F.; Baik, C.; et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients With BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Pisapia, P.; Sgariglia, R.; Vigliar, E.; Biglietto, M.; Carlomagno, C.; Giuffre, G.; Bellevicine, C.; Troncone, G. Less frequently mutated genes in colorectal cancer: Evidences from next-generation sequencing of 653 routine cases. J. Clin. Pathol. 2016, 69, 767–771. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Mahoney, M.R.; Sargent, D.J.; Nelson, G.D.; Alberts, S.R.; Sinicrope, F.A.; Goldberg, R.M.; Limburg, P.J.; Thibodeau, S.N.; Grothey, A.; et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J. Natl. Cancer Inst. 2014, 106, dju106. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Cocco, E.; Benhamida, J.; Middha, S.; Zehir, A.; Mullaney, K.; Shia, J.; Yaeger, R.; Zhang, L.; Wong, D.; Villafania, L.; et al. Colorectal Carcinomas Containing Hypermethylated MLH1 Promoter and Wild-Type BRAF/KRAS Are Enriched for Targetable Kinase Fusions. Cancer Res. 2019, 79, 1047–1053. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Kopetz, S.; Tie, J.; Gibbs, P.; Jiang, Z.Q.; Lieu, C.H.; Agarwal, A.; Maru, D.M.; Sieber, O.; Desai, J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011, 117, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Farina-Sarasqueta, A.; van Lijnschoten, G.; Moerland, E.; Creemers, G.J.; Lemmens, V.; Rutten, H.J.T.; van den Brule, A.J.C. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann. Oncol. 2010, 21, 2396–2402. [Google Scholar] [CrossRef]
- Tabernero, J.; Ros, J.; Elez, E. The Evolving Treatment Landscape in BRAF-V600E-Mutated Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book. 2022, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Subbiah, V.; Lockhart, A.C.; Blay, J.Y.; Puzanov, I.; Chau, I.; Raje, N.S.; Wolf, J.; Erinjeri, J.P.; Torrisi, J.; et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018, 4, 384–388. [Google Scholar] [CrossRef]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef]
- Horbinski, C.; Nikiforova, M.N.; Hagenkord, J.M.; Hamilton, R.L.; Pollack, I.F. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol. 2012, 14, 777–789. [Google Scholar] [CrossRef]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Guerreiro Stucklin, A.; Zhukova, N.; Arnoldo, A.; Ryall, S.; et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J. Clin. Oncol. 2017, 35, 2934–2941. [Google Scholar] [CrossRef]
- Behling, F.; Schittenhelm, J. Oncogenic BRAF Alterations and Their Role in Brain Tumors. Cancers 2019, 11, 794. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Iaccarino, A.; Sgariglia, R.; Nacchio, M.; Russo, G.; Gragnano, G.; Malapelle, U.; Troncone, G. BRAF: A Two-Faced Janus. Cells 2020, 9, 2549. [Google Scholar] [CrossRef]
- Martins-de-Barros, A.V.; Anjos, R.S.D.; Silva, C.C.G.; Silva, E.; Araujo, F.; Carvalho, M.V. Diagnostic accuracy of immunohistochemistry compared with molecular tests for detection of BRAF V600E mutation in ameloblastomas: Systematic review and meta-analysis. J. Oral. Pathol. Med. 2022, 51, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The Current State of Molecular Testing in the BRAF-Mutated Melanoma Landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Ong, E.; Huang, H.J.; McPhaul, L.W.; Yoon, S.; Janku, F.; Gianoukakis, A.G. Ultrasensitive detection of BRAF V600E mutations in circulating tumor DNA of patients with metastatic thyroid cancer. Endocrine 2022, 76, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Polivka, J.; Huang, H.J.; Treskova, I.; Pivovarcikova, K.; Fikrle, T.; Woznica, V.; Dustin, D.J.; Call, S.G.; Meric-Bernstam, F.; et al. Ultrasensitive detection of BRAF mutations in circulating tumor DNA of non-metastatic melanoma. ESMO Open 2022, 7, 100357. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Subbiah, V. Precision oncology for biliary tract tumors: It’s written in blood! Ann. Oncol. 2022, 33, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Iyer, P.C.; Cote, G.J.; Hai, T.; Gule-Monroe, M.; Bui-Griffith, J.; Williams, M.D.; Hess, K.; Hofmann, M.C.; Dadu, R.; Zafereo, M.; et al. Circulating BRAF V600E Cell-Free DNA as a Biomarker in the Management of Anaplastic Thyroid Carcinoma. JCO Precis. Oncol. 2018, 2, PO.18.00173. [Google Scholar] [CrossRef] [PubMed]
- Passiglia, F.; Malapelle, U.; Normanno, N.; Pinto, C. Optimizing diagnosis and treatment of EGFR exon 20 insertions mutant NSCLC. Cancer Treat. Rev. 2022, 109, 102438. [Google Scholar] [CrossRef]
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours—Molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Le, A.T.; Sheren, J.; Nijmeh, H.; Gowan, K.; Jones, K.L.; Varella-Garcia, M.; Aisner, D.L.; Doebele, R.C. Comparison of Molecular Testing Modalities for Detection of ROS1 Rearrangements in a Cohort of Positive Patient Samples. J. Thorac. Oncol. 2018, 13, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Aypar, U.; Rosen, E.Y.; Mata, D.A.; Benayed, R.; Mullaney, K.; Jayakumaran, G.; Zhang, Y.; Frosina, D.; Drilon, A.; et al. A Performance Comparison of Commonly Used Assays to Detect RET Fusions. Clin. Cancer Res. 2021, 27, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Offin, M.; Brannon, A.R.; Chang, J.; Chow, A.; Delasos, L.; Girshman, J.; Wilkins, O.; McCarthy, C.G.; Makhnin, A.; et al. MET Exon 14-altered Lung Cancers and MET Inhibitor Resistance. Clin. Cancer Res. 2021, 27, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Hondelink, L.M.; Solleveld-Westerink, N.; Uljee, S.M.; Ruano, D.; Cleton-Jansen, A.M.; von der Thusen, J.H.; Ramai, S.R.S.; Postmus, P.E.; Graadt van Roggen, J.F.; et al. Optimizing Mutation and Fusion Detection in NSCLC by Sequential DNA and RNA Sequencing. J. Thorac. Oncol. 2020, 15, 1000–1014. [Google Scholar] [CrossRef]
- Desmeules, P.; Boudreau, D.K.; Bastien, N.; Boulanger, M.C.; Bosse, Y.; Joubert, P.; Couture, C. Performance of an RNA-Based Next-Generation Sequencing Assay for Combined Detection of Clinically Actionable Fusions and Hotspot Mutations in NSCLC. JTO Clin. Res. Rep. 2022, 3, 100276. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef]
- Capper, D.; Preusser, M.; Habel, A.; Sahm, F.; Ackermann, U.; Schindler, G.; Pusch, S.; Mechtersheimer, G.; Zentgraf, H.; von Deimling, A. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011, 122, 11–19. [Google Scholar] [CrossRef]
- Al-Kateb, H.; Nguyen, T.T.; Steger-May, K.; Pfeifer, J.D. Identification of major factors associated with failed clinical molecular oncology testing performed by next generation sequencing (NGS). Mol. Oncol. 2015, 9, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Rose Brannon, A.; Jayakumaran, G.; Diosdado, M.; Patel, J.; Razumova, A.; Hu, Y.; Meng, F.; Haque, M.; Sadowska, J.; Murphy, B.J.; et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat. Commun. 2021, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Sun, W.; Madan, R.; Lamsal, K.; Schmitt, S.; Godwin, A.K.; Kasi, A. Microsatellite Instability with BRAF V600E Associated with Delayed Presentation but Poor Survival in Stage III Colorectal Cancer. Fortune J. Health Sci. 2023, 6, 167–173. [Google Scholar] [CrossRef]
- Malapelle, U.; Rossi, G.; Pisapia, P.; Barberis, M.; Buttitta, F.; Castiglione, F.; Cecere, F.L.; Grimaldi, A.M.; Iaccarino, A.; Marchetti, A.; et al. BRAF as a positive predictive biomarker: Focus on lung cancer and melanoma patients. Crit. Rev. Oncol. Hematol. 2020, 156, 103118. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, N.; Martinez-Useros, J.; Li, W.; Orta, A.; Perez, N.; Carames, C.; Hernandez, T.; Moreno, I.; Serrano, G.; Garcia-Foncillas, J. KRAS and BRAF Mutations as Prognostic and Predictive Biomarkers for Standard Chemotherapy Response in Metastatic Colorectal Cancer: A Single Institutional Study. Cells 2020, 9, 219. [Google Scholar] [CrossRef]
- van der Kooij, M.K.; Wetzels, M.; Aarts, M.J.B.; van den Berkmortel, F.; Blank, C.U.; Boers-Sonderen, M.J.; Dierselhuis, M.P.; de Groot, J.W.B.; Hospers, G.A.P.; Piersma, D.; et al. Age Does Matter in Adolescents and Young Adults versus Older Adults with Advanced Melanoma; A National Cohort Study Comparing Tumor Characteristics, Treatment Pattern, Toxicity and Response. Cancers 2020, 12, 2072. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Subbiah, V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: From melanoma to tissue-agnostic therapy. ESMO Open 2023, 8, 100788. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Hall-Jackson, C.A.; Eyers, P.A.; Cohen, P.; Goedert, M.; Boyle, F.T.; Hewitt, N.; Plant, H.; Hedge, P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999, 6, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L., Jr.; Levy, R.M. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Bayer; Amgen. Study of BAY43-9006 in Patients with Unresectable and/or Metastatic Renal Cell Cancer; Bayer: Bayer Leverkusen, Germany; Amgen: Southend Oaks, CA, USA, 2006. [Google Scholar]
- Tabernero, J.; Garcia-Carbonero, R.; Cassidy, J.; Sobrero, A.; Van Cutsem, E.; Kohne, C.H.; Tejpar, S.; Gladkov, O.; Davidenko, I.; Salazar, R.; et al. Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: The RESPECT trial. Clin. Cancer Res. 2013, 19, 2541–2550. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef]
- Boussemart, L.; Routier, E.; Mateus, C.; Opletalova, K.; Sebille, G.; Kamsu-Kom, N.; Thomas, M.; Vagner, S.; Favre, M.; Tomasic, G.; et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: A study of 42 patients. Ann. Oncol. 2013, 24, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Cotellic [Package Insert], Genentech, Inc.: South San Francisco, CA, USA, 2015.
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef]
- Bertazza, L.; Barollo, S.; Radu, C.M.; Cavedon, E.; Simioni, P.; Faggian, D.; Plebani, M.; Pelizzo, M.R.; Rubin, B.; Boscaro, M.; et al. Synergistic antitumour activity of RAF265 and ZSTK474 on human TT medullary thyroid cancer cells. J. Cell Mol. Med. 2015, 19, 2244–2252. [Google Scholar] [CrossRef]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef]
- Jin, T.; Lavoie, H.; Sahmi, M.; David, M.; Hilt, C.; Hammell, A.; Therrien, M. RAF inhibitors promote RAS-RAF interaction by allosterically disrupting RAF autoinhibition. Nat. Commun. 2017, 8, 1211. [Google Scholar] [CrossRef]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, E.J.; Kugel, C.H., 3rd; Vido, M.J.; Berger, A.C.; Purwin, T.J.; Goldberg, A.; Davies, M.A.; Schiewer, M.J.; Knudsen, K.E.; Bollag, G.; et al. Response and Resistance to Paradox-Breaking BRAF Inhibitor in Melanomas In Vivo and Ex Vivo. Mol. Cancer Ther. 2018, 17, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Bonfill-Teixidor, E.; Iurlaro, R.; Handl, C.; Wichmann, J.; Arias, A.; Cuartas, I.; Emmenegger, J.; Romagnani, A.; Mangano, L.; Lorber, T.; et al. Activity and Resistance of a Brain-Permeable Paradox Breaker BRAF Inhibitor in Melanoma Brain Metastasis. Cancer Res. 2022, 82, 2552–2564. [Google Scholar] [CrossRef]
- Wichmann, J.; Rynn, C.; Friess, T.; Petrig-Schaffland, J.; Kornacker, M.; Handl, C.; Emmenegger, J.; Eckmann, J.; Herting, F.; Frances, N.; et al. Preclinical Characterization of a Next-Generation Brain Permeable, Paradox Breaker BRAF Inhibitor. Clin. Cancer Res. 2022, 28, 770–780. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Pan, Y.L. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Chen, W.; Park, J.I. Tumor Cell Resistance to the Inhibition of BRAF and MEK1/2. Int. J. Mol. Sci. 2023, 24, 14837. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: Current status in clinical trials. Genes. Dis. 2023, 10, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef]
- Johnson, D.B.; Childress, M.A.; Chalmers, Z.R.; Frampton, G.M.; Ali, S.M.; Rubinstein, S.M.; Fabrizio, D.; Ross, J.S.; Balasubramanian, S.; Miller, V.A.; et al. BRAF internal deletions and resistance to BRAF/MEK inhibitor therapy. Pigment. Cell Melanoma Res. 2018, 31, 432–436. [Google Scholar] [CrossRef]
- Foster, S.A.; Whalen, D.M.; Ozen, A.; Wongchenko, M.J.; Yin, J.; Yen, I.; Schaefer, G.; Mayfield, J.D.; Chmielecki, J.; Stephens, P.J.; et al. Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell 2016, 29, 477–493. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, Y.; Yao, X. Resistance mechanism of the oncogenic beta3-alphaC deletion mutation in BRAF kinase to dabrafenib and vemurafenib revealed by molecular dynamics simulations and binding free energy calculations. Chem. Biol. Drug Des. 2019, 93, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yue, Q.; Ma, J.; Liu, Y.; Zhao, T.; Guo, W.; Zhu, G.; Guo, S.; Wang, S.; Gao, T.; et al. POU4F1 promotes the resistance of melanoma to BRAF inhibitors through MEK/ERK pathway activation and MITF up-regulation. Cell Death Dis. 2020, 11, 451. [Google Scholar] [CrossRef]
- Vidarsdottir, L.; Azimi, A.; Das, I.; Sigvaldadottir, I.; Suryo Rahmanto, A.; Petri, A.; Kauppinen, S.; Ingvar, C.; Jonsson, G.; Olsson, H.; et al. PTENP1-AS contributes to BRAF inhibitor resistance and is associated with adverse clinical outcome in stage III melanoma. Sci. Rep. 2021, 11, 11023. [Google Scholar] [CrossRef] [PubMed]
- Malindi, Z.; Barth, S.; Abrahamse, H. The Potential of Antibody Technology and Silver Nanoparticles for Enhancing Photodynamic Therapy for Melanoma. Biomedicines 2022, 10, 2158. [Google Scholar] [CrossRef] [PubMed]
- Biteghe, F.A.N.; Chalomie, N.E.T.; Mungra, N.; Vignaux, G.; Gao, N.; Vergeade, A.; Okem, A.; Naran, K.; Ndong, J.C.; Barth, S. Antibody-Based Immunotherapy: Alternative Approaches for the Treatment of Metastatic Melanoma. Biomedicines 2020, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351 Pt 2, 289–305. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [PubMed]
- NCC MERP. Statement on Medication Error Rates [WWW Document]. National Coordinating Council for Medication Error Reporting and Prevention. 2008. Available online: https://www.nccmerp.org/statement-medication-error-rates (accessed on 24 January 2024).
- Garutti, M.; Bergnach, M.; Polesel, J.; Palmero, L.; Pizzichetta, M.A.; Puglisi, F. BRAF and MEK Inhibitors and Their Toxicities: A Meta-Analysis. Cancers 2022, 15, 141. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRAF Inhibitors | |||
| Drug | Inhibitor Type | Approved to Treat | Date approved by the FDA |
| Sorafenib (Nexlavar) Also: Sorafenib tosylate | 1st-gen BRAFi | Advanced renal cell carcinoma Unresectable hepatocellular carcinoma Locally recurrent or metastatic, progressive, differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment | 20 December 2005 |
| Vemurafenib (Zelboraf) Also: PLX4032 | 2nd-gen BRAFi | Unresectable or metastatic melanoma with BRAF V600E mutation Erdheim–Chester disease with BRAF V600 mutation | 17 August 2011 |
| Dabrafenib (Tafinlar) Also: GSK-2118436 | 2nd-gen BRAFi | Unresectable or metastatic melanoma with BRAF V600E mutation | 29 May 2013 |
| Encorafenib (Braftovi) Also: LGX818 | 2nd-gen BRAFi | (Not approved for use as a single agent) | 27 June 2018 |
| Combination Therapies | |||
| Drug | Inhibitor types | Approved to treat | Date approved by the FDA |
| Dabrafenib + Trametinib (Tafinlar + Mekinist) Also: GSK-2118436 + GSK-1120212 | Dabrafenib: BRAFi Trametinib: MEK1/2i | Unresectable or metastatic melanoma with BRAF V600E/K NSCLC with BRAF V600E mutation and involvement of the lymph node(s), following complete resection Locally advanced or metastatic anaplastic thyroid cancer with a BRAF V600E mutation Unresectable or metastatic solid tumors with BRAF V600E mutation and no satisfactory alternative treatment options Pediatric patients 1 year of age and older with low-grade glioma with a BRAF V600E mutation | 9 January 2014 |
| Vemurafenib + Cobimetinib (Zelboraf + Cotellic) Also: PLX4032 + GDC-0973 | Vemurafenib: BRAFi Cobimetinib: MEK1/2i | Unresectable or metastatic melanoma with BRAF V600E/K | 10 November 2015 |
| Encorafenib + Binimetinib (Braftovi + Mektovi) Also: LGX818 + MEK162 | Encorafenib: BRAFi Binimetinib: MEK1/2i | Unresectable or metastatic melanoma with BRAF V600E/K Metastatic NSCLC with BRAF V600E | 27 June 2018 |
| Encorafenib + Cetuximab (Braftovi + Erbitux) Also: LGX818 + Encorafenib | Encorafenib: BRAFi Cetuximab: monoclonal antibody, EGFR antagonist | Metastatic CRC with a BRAF V600E mutation (after prior therapy) | 8 April 2020 |
| Vemurafenib + Cobimetinib + Atezolizumab (Zelboraf + Cotellic + Tecentriq) Also: PLX4032 + GDC-0973 + RG7446 | Vemurafenib: BRAFi Cobimetinib: MEK1/2i Atezolizumab: PD-L1 blocking antibody | Unresectable or metastatic melanoma with BRAF V600 | 30 July 2020 |
| Drug(s) | G3–G5 | Any Grade |
|---|---|---|
| Vemurafenib (Zelboraf) | 51% | 94% |
| Dabrafenib (Tafinlar) | 50% | 85% |
| Encorafenib (Braftovi) | 68% | 99% |
| Dabrafenib + Trametinib (Tafinlar + Mekinist) | 43% | 95% |
| Vemurafenib + Cobimetinib (Zelboraf + Cotellic) | 72% | 98% |
| Encorafenib + Binimetinib (Braftovi + Mektovi) | 68% | 98% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roa, P.; Bremer, N.V.; Foglizzo, V.; Cocco, E. Mutations in the Serine/Threonine Kinase BRAF: Oncogenic Drivers in Solid Tumors. Cancers 2024, 16, 1215. https://doi.org/10.3390/cancers16061215
Roa P, Bremer NV, Foglizzo V, Cocco E. Mutations in the Serine/Threonine Kinase BRAF: Oncogenic Drivers in Solid Tumors. Cancers. 2024; 16(6):1215. https://doi.org/10.3390/cancers16061215
Chicago/Turabian StyleRoa, Paola, Nicole Virginia Bremer, Valentina Foglizzo, and Emiliano Cocco. 2024. "Mutations in the Serine/Threonine Kinase BRAF: Oncogenic Drivers in Solid Tumors" Cancers 16, no. 6: 1215. https://doi.org/10.3390/cancers16061215
APA StyleRoa, P., Bremer, N. V., Foglizzo, V., & Cocco, E. (2024). Mutations in the Serine/Threonine Kinase BRAF: Oncogenic Drivers in Solid Tumors. Cancers, 16(6), 1215. https://doi.org/10.3390/cancers16061215