
Venetoclax Resistance in Acute Myeloid Leukemia


Abstract
Simple Summary
Abstract
1. Introduction
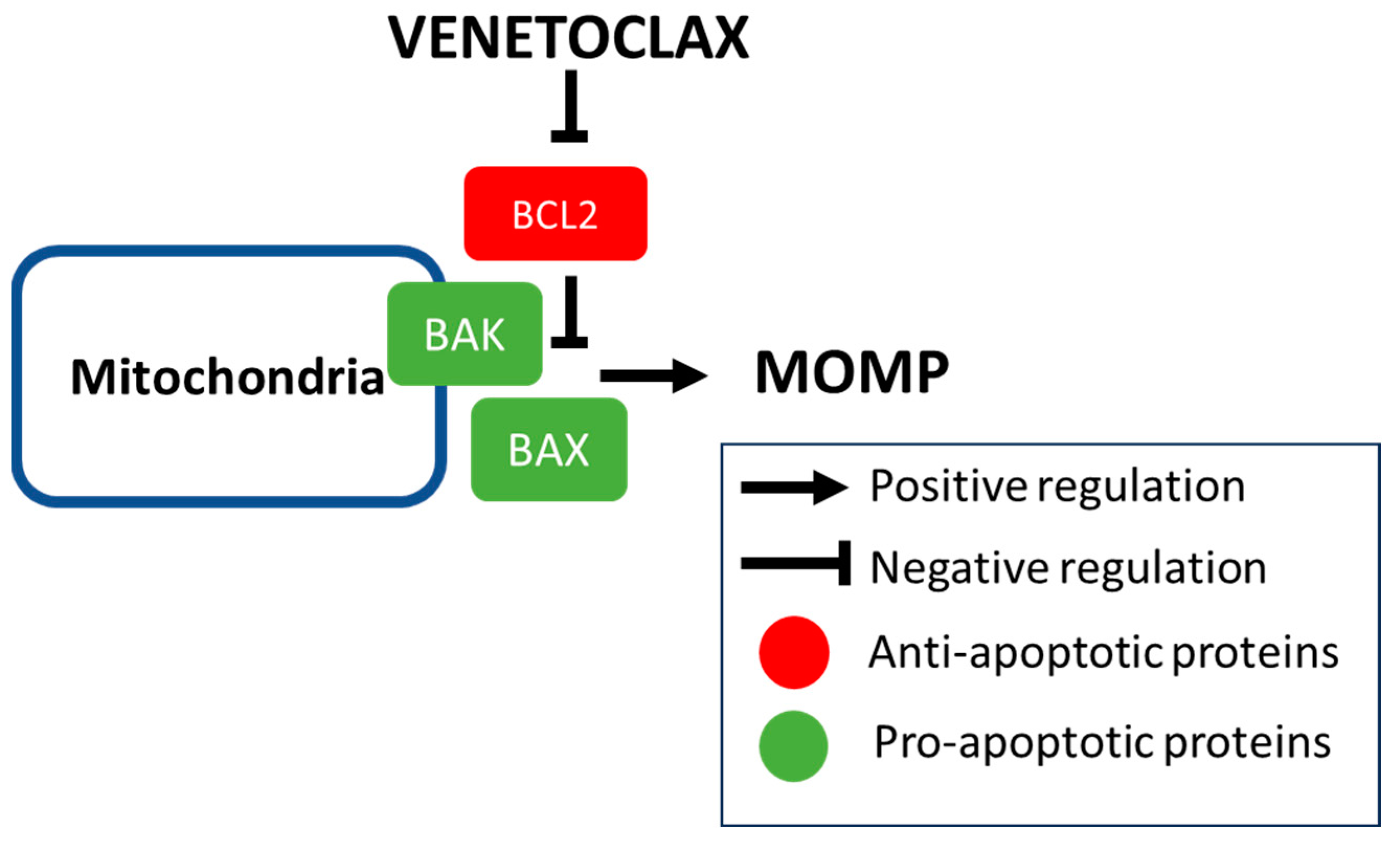
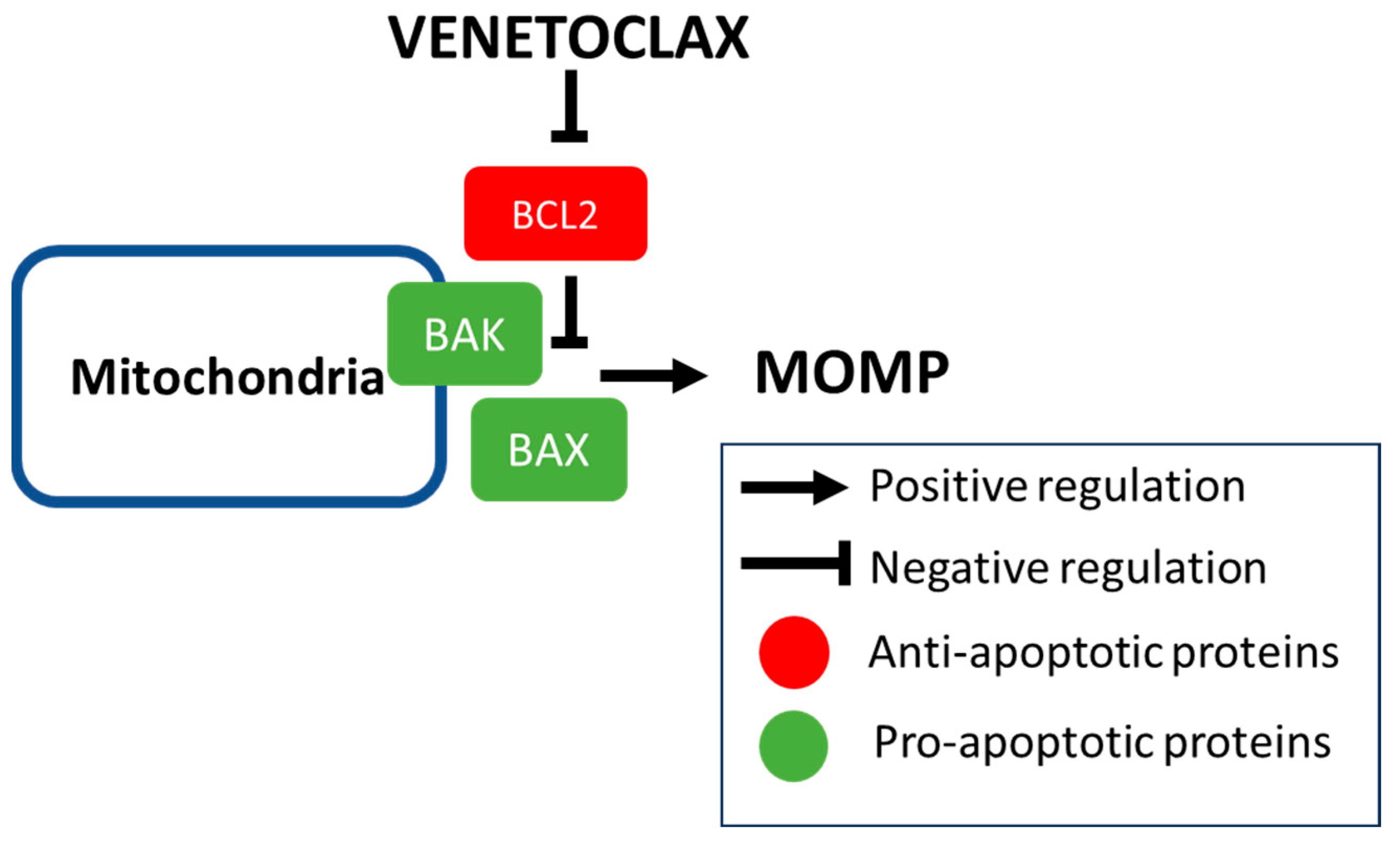
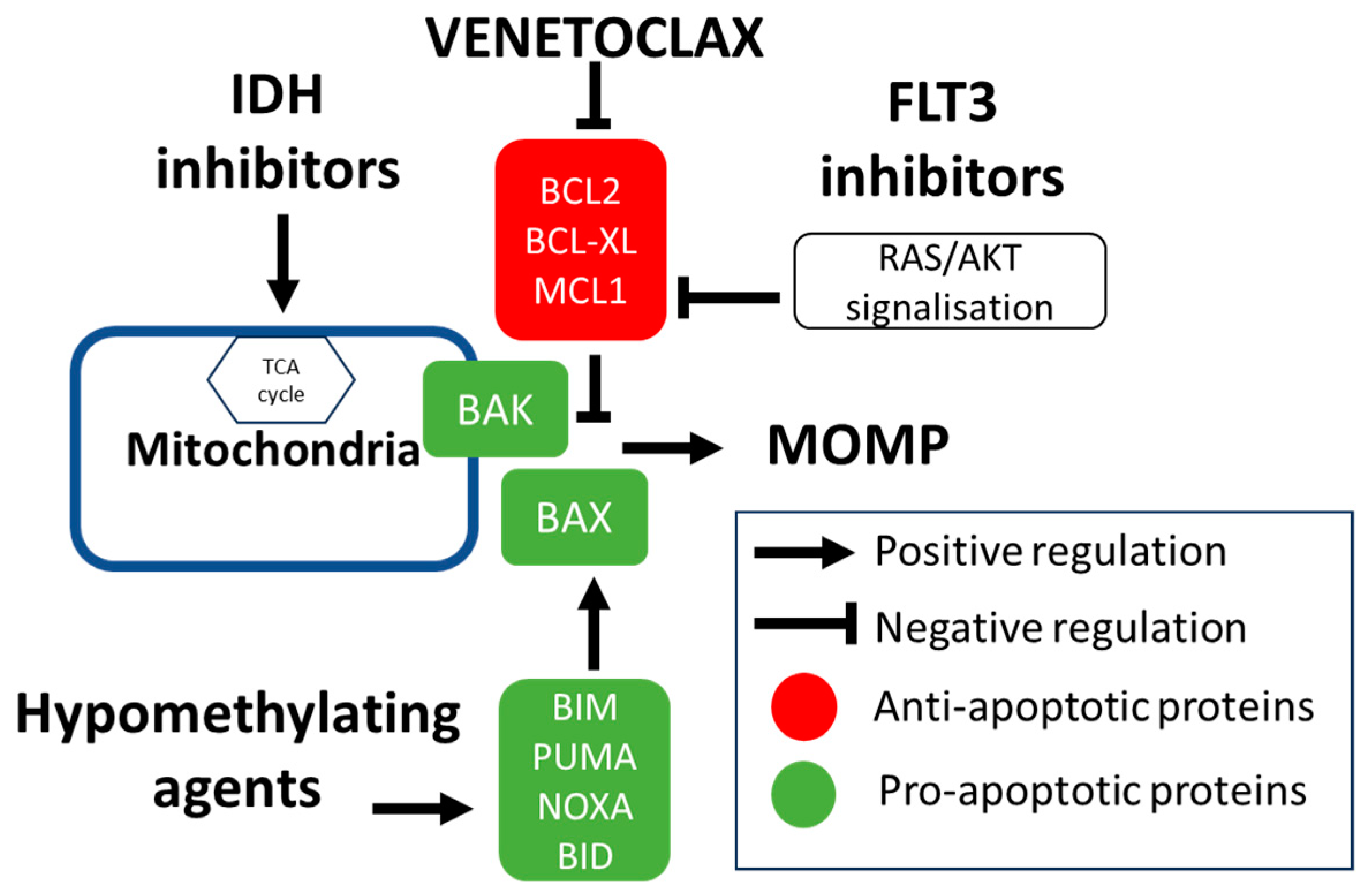
2. Rationale of BCL2 Inhibition
2.1. Mechanism of Intrinsic Apoptosis and Developmlent of BH3 Mimetics
2.2. Main Clinical Studies Using Venetoclax in AML
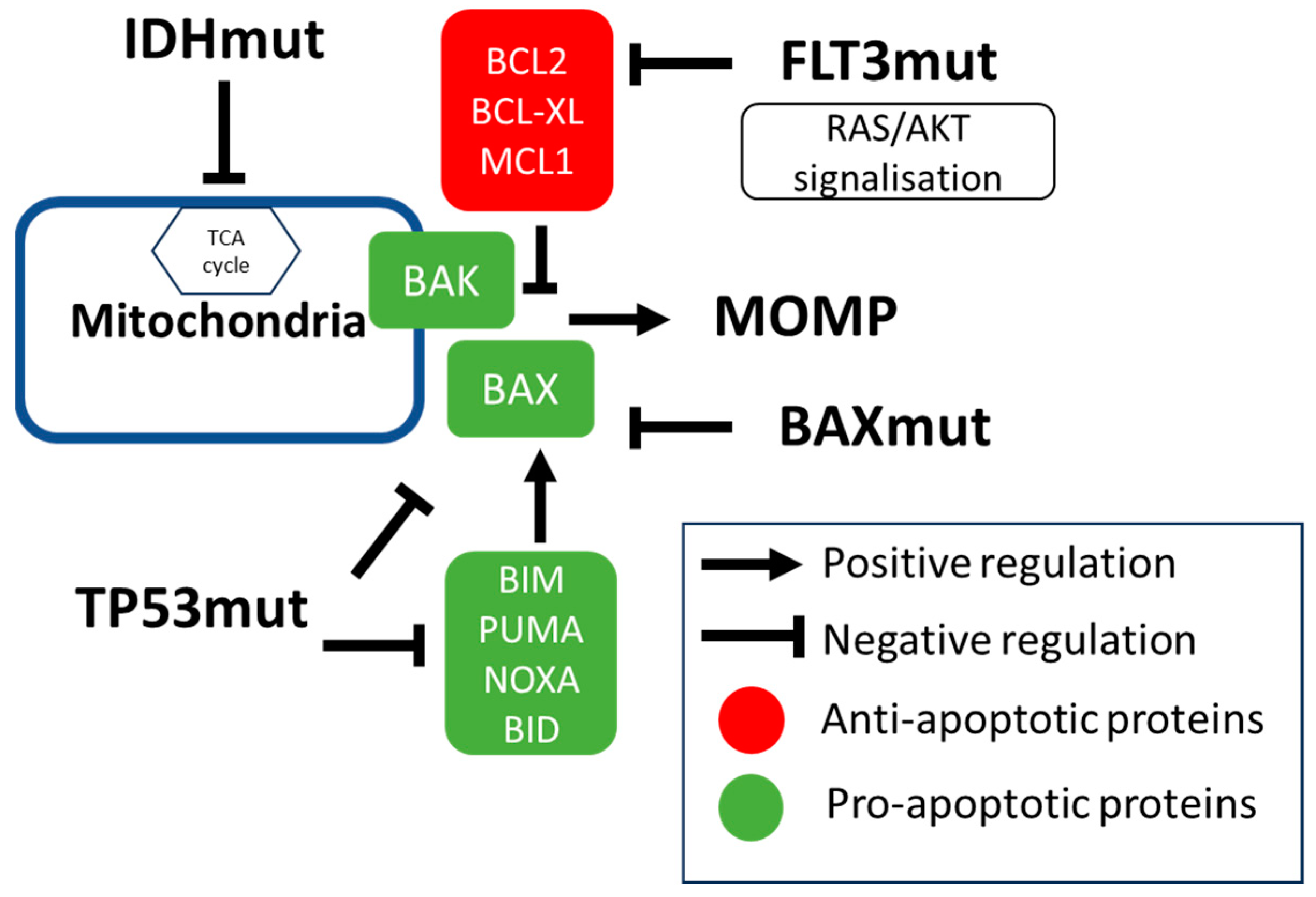
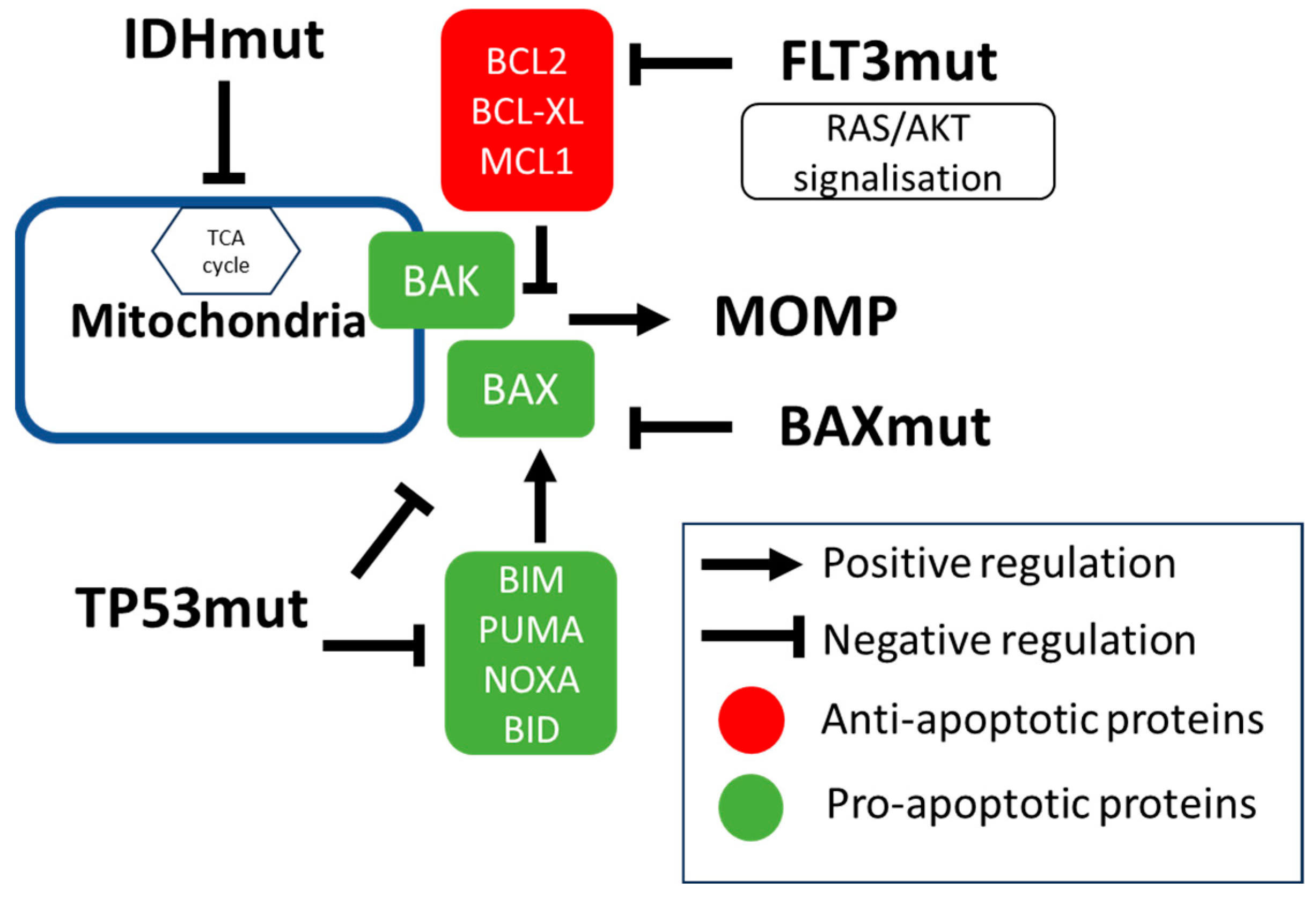
3. Molecular Factors Modulating Venetoclax Efficacy
3.1. IDH Mutations
3.2. TP53 Mutation
3.3. Signaling Mutations (Including FLT3 Mutations)
3.4. Secondary Type Mutations
3.5. Mutations in Apoptotic Genes
4. Non-Genetic Factors Driving Venetoclax Efficacy
4.1. Regulators of Intrinsic Apoptosis
- (a)
- Increase in anti-apoptotic proteins.
- BCL2
- MCL1
- BCLXL and BCL2A1
- (b)
- Decrease in pro-apoptotic signals.
4.2. Differentiation Status
4.3. Metabolism and Beyond
5. Overcoming Resistance with New Therapies
5.1. Hypomethylating Agents
5.2. Innovative BH3 Mimetics
- MCL1
- Dual BCL2 and BCLXL inhibitors
5.3. Tyrosine Kinase Inhibitor (TKI)
5.4. FLT3 Inhibitors
5.5. IDH Inhibitors
5.6. Future Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, With Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients With Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Pratz, K.W.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Garcia, J.S.; Fiedler, W.; Yamamoto, K.; Wang, J.; Yoon, S.-S.; Wolach, O.; et al. Long-Term Follow-up of the Phase 3 Viale-a Clinical Trial of Venetoclax Plus Azacitidine for Patients with Untreated Acute Myeloid Leukemia Ineligible for Intensive Chemotherapy. Blood 2022, 140, 529–531. [Google Scholar] [CrossRef]
- Maiti, A.; Rausch, C.R.; Cortes, J.E.; Pemmaraju, N.; Daver, N.G.; Ravandi, F.; Garcia-Manero, G.; Borthakur, G.; Naqvi, K.; Ohanian, M.; et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica 2021, 106, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Carter, B.Z.; Andreeff, M.; Konopleva, M.Y. SOHO State of the Art Updates and Next Questions|Beyond BCL-2 Inhibition in Acute Myeloid Leukemia: Other Approaches to Leverage the Apoptotic Pathway. Clin. Lymphoma Myeloma Leuk. 2022, 22, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Garciaz, S.; Saillard, C.; Hicheri, Y.; Hospital, M.-A.; Vey, N. Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance. Cancers 2021, 13, 5608. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.P.; Flanagan, L.; Rodrigues, D.A.; Chonghaile, T.N. The path to venetoclax resistance is paved with mutations, metabolism, and more. Sci. Transl. Med. 2022, 14, eabo6891. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ye, H. Progress in understanding the mechanisms of resistance to BCL-2 inhibitors. Exp. Hematol. Oncol. 2022, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Stubbins, R.J.; Maksakova, I.A.; Sanford, D.S.; Rouhi, A.; Kuchenbauer, F. Mitochondrial metabolism: Powering new directions in acute myeloid leukemia. Leuk. Lymphoma 2021, 62, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Ong, F.; Kim, K.; Konopleva, M.Y. Venetoclax resistance: Mechanistic insights and future strategies. Cancer Drug Resist. 2022, 5, 380–400. [Google Scholar] [CrossRef]
- Garciaz, S.; Miller, T.; Collette, Y.; Vey, N. Targeting regulated cell death pathways in acute myeloid leukemia. Cancer Drug Resist. 2023, 6, 151–168. [Google Scholar] [CrossRef]
- Griffioen, M.S.; de Leeuw, D.C.; Janssen, J.J.W.M.; Smit, L. Targeting Acute Myeloid Leukemia with Venetoclax; Biomarkers for Sensitivity and Rationale for Venetoclax-Based Combination Therapies. Cancers 2022, 14, 3456. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: A single-centre, phase 2 trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Garciaz, S.; Hospital, M.-A.; Alary, A.-S.; Saillard, C.; Hicheri, Y.; Mohty, B.; Rey, J.; D’incan, E.; Charbonnier, A.; Villetard, F.; et al. Azacitidine Plus Venetoclax for the Treatment of Relapsed and Newly Diagnosed Acute Myeloid Leukemia Patients. Cancers 2022, 14, 2025. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Saillard, C.; Mohty, B.; Hicheri, Y.; Villetard, F.; Maisano, V.; Charbonnier, A.; Rey, J.; D‘incan, E.; Rouzaud, C.; et al. Azacitidine–venetoclax versus azacitidine salvage treatment for primary induction failure or first relapsed acute myeloid leukaemia patients. Eur. J. Haematol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Todisco, E.; Papayannidis, C.; Fracchiolla, N.; Petracci, E.; Zingaretti, C.; Vetro, C.; Martelli, M.P.; Zappasodi, P.; Di Renzo, N.; Gallo, S.; et al. AVALON: The Italian cohort study on real-life efficacy of hypomethylating agents plus venetoclax in newly diagnosed or relapsed/refractory patients with acute myeloid leukemia. Cancer 2023, 129, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Shimony, S.; Bewersdorf, J.P.; Shallis, R.M.; Liu, Y.; Schaefer, E.J.; Zeidan, A.M.; Goldberg, A.D.; Stein, E.M.; Marcucci, G.; Lindsley, R.C.; et al. Hypomethylating agents plus venetoclax compared with intensive induction chemotherapy regimens in molecularly defined secondary AML. Leukemia 2024. [Google Scholar] [CrossRef]
- Döhner, H.; Pratz, K.W.; DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Recher, C.; Schuh, A.C.; Babu, S.; Dail, M.; et al. ELN Risk Stratification Is Not Predictive of Outcomes for Treatment-Naïve Patients with Acute Myeloid Leukemia Treated with Venetoclax and Azacitidine. Blood 2022, 140, 1441–1444. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Wilmink, J.W. IDH1/2 Mutations in Cancer Stem Cells and Their Implications for Differentiation Therapy. J. Histochem. Cytochem. 2022, 70, 83–97. [Google Scholar] [CrossRef]
- Pollyea, D.A.; DiNardo, C.D.; Arellano, M.L.; Pigneux, A.; Fiedler, W.; Konopleva, M.; Rizzieri, D.A.; Smith, B.D.; Shinagawa, A.; Lemoli, R.M.; et al. Impact of Venetoclax and Azacitidine in Treatment-Naïve Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin. Cancer Res. 2022, 28, 2753–2761. [Google Scholar] [CrossRef]
- Hammond, D.; Loghavi, S.; Wang, S.A.; Konopleva, M.Y.; Kadia, T.M.; Daver, N.G.; Ohanian, M.; Issa, G.C.; Alvarado, Y.; Short, N.J.; et al. Response patterns and impact of MRD in patients with IDH1/2-mutated AML treated with venetoclax and hypomethylating agents. Blood Cancer J. 2023, 13, 148. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 2021, 218, e20200924. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Fleming, S.; Tsai, X.C.-H.; Morris, R.; Hou, H.-A.; Wei, A.H. TP53 status and impact on AML prognosis within the ELN 2022 risk classification. Blood 2023, 142, 2029–2033. [Google Scholar] [CrossRef] [PubMed]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; Kooy, M.v.M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Iqbal, S.; Huang, J.; Renard, C.; Lin, J.; Pan, Y.; Williamson, M.; Ramsingh, G. Clinical characteristics and overall survival among acute myeloid leukemia patients with TP53 gene mutation or chromosome 17p deletion. Am. J. Hematol. 2023, 98, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Iqbal, S.; Renard, C.; Chan, R.J.; Hasegawa, K.; Hu, H.; Tse, P.; Yan, J.; Zoratti, M.J.; Xie, F.; et al. Treatment outcomes for newly diagnosed, treatment-naïve TP53-mutated acute myeloid leukemia: A systematic review and meta-analysis. J. Hematol. Oncol. 2023, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53 -mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Pratz, K.W.; Wei, A.H.; Pullarkat, V.; Jonas, B.A.; Recher, C.; Babu, S.; Schuh, A.C.; Dail, M.; Sun, Y.; et al. Outcomes in Patients with Poor-Risk Cytogenetics with or without TP53 Mutations Treated with Venetoclax and Azacitidine. Clin. Cancer Res. 2022, 28, 5272–5279. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.; Chew, E.; Flensburg, C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.; et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021, 137, 2721–2735. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci. Transl. Med. 2019, 11, eaax2863. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Ishizawa, J.; Ayoub, E.; Montoya, R.H.; Ostermann, L.B.; Muftuoglu, M.; Ruvolo, V.R.; Patsilevas, T.; Scruggs, D.A.; Khazaei, S.; et al. Enhanced TP53 reactivation disrupts MYC transcriptional program and overcomes venetoclax resistance in acute myeloid leukemias. Sci. Adv. 2023, 9, eadh1436. [Google Scholar] [CrossRef]
- Garciaz, S.; Hospital, M.-A. FMS-Like Tyrosine Kinase 3 Inhibitors in the Treatment of Acute Myeloid Leukemia: An Update on the Emerging Evidence and Safety Profile. OncoTargets Ther. 2023, 16, 31–45. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Konopleva, M.; Thirman, M.J.; Pratz, K.W.; Garcia, J.S.; Recher, C.; Pullarkat, V.; Kantarjian, H.M.; DiNardo, C.D.; Dail, M.; Duan, Y.; et al. Impact of F LT3 Mutation on Outcomes after Venetoclax and Azacitidine for Patients with Treatment-Naïve Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 2744–2752. [Google Scholar] [CrossRef]
- Stahl, M.; Menghrajani, K.; Derkach, A.; Chan, A.; Xiao, W.; Glass, J.; King, A.C.; Daniyan, A.F.; Famulare, C.; Cuello, B.M.; et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021, 5, 1552–1564. [Google Scholar] [CrossRef]
- Bataller, A.; Bazinet, A.; DiNardo, C.D.; Maiti, A.; Borthakur, G.; Daver, N.G.; Short, N.J.; Jabbour, E.J.; Issa, G.C.; Pemmaraju, N.; et al. Prognostic risk signature in patients with acute myeloid leukemia treated with hypomethylating agents and venetoclax. Blood Adv. 2023, 8, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Jahn, E.; Saadati, M.; Fenaux, P.; Gobbi, M.; Roboz, G.J.; Bullinger, L.; Lutsik, P.; Riedel, A.; Plass, C.; Jahn, N.; et al. Clinical impact of the genomic landscape and leukemogenic trajectories in non-intensively treated elderly acute myeloid leukemia patients. Leukemia 2023, 37, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- McCarter, J.G.W.; Nemirovsky, D.; Famulare, C.A.; Farnoud, N.; Mohanty, A.S.; Stone-Molloy, Z.S.; Chervin, J.; Ball, B.J.; Epstein-Peterson, Z.D.; Arcila, M.E.; et al. Interaction between myelodysplasia-related gene mutations and ontogeny in acute myeloid leukemia. Blood Adv. 2023, 7, 5000–5013. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Doyel, M.; Bouligny, I.M.; Murray, G.; Patel, T.; Boron, J.; Tran, V.; Gor, J.; Hang, Y.; Ho, T.; Zacholski, K.; et al. The impact of secondary-type mutations in newly diagnosed acute myeloid leukemia treated with venetoclax and decitabine or azacitidine. J. Clin. Oncol. 2023, 41, e19048. [Google Scholar] [CrossRef]
- Gangat, N.; Johnson, I.; McCullough, K.; Farrukh, F.; Al-Kali, A.; Alkhateeb, H.; Begna, K.; Mangaonkar, A.; Litzow, M.; Hogan, W.; et al. Molecular predictors of response to venetoclax plus hypomethylating agent in treatment-naïve acute myeloid leukemia. Haematologica 2022, 107, 2501–2505. [Google Scholar] [CrossRef]
- Rahmani, N.E.; Ramachandra, N.; Sahu, S.; Gitego, N.; Lopez, A.; Pradhan, K.; Bhagat, T.D.; Gordon-Mitchell, S.; Pena, B.R.; Kazemi, M.; et al. ASXL1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J. 2021, 11, 157. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Caprioli, C.; Lussana, F.; Salmoiraghi, S.; Cavagna, R.; Buklijas, K.; Elidi, L.; Zanghi’, P.; Michelato, A.; Delaini, F.; Oldani, E.; et al. Clinical significance of chromatin-spliceosome acute myeloid leukemia: A report from the Northern Italy Leukemia Group (NILG) randomized trial 02/06. Haematologica 2020, 106, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, I.; Wojtuszkiewicz, A.; Meggendorfer, M.; Hutter, S.; Baer, C.; Heymans, M.; Valk, P.J.M.; Kern, W.; Haferlach, C.; Janssen, J.J.W.M.; et al. Splicing factor gene mutations in acute myeloid leukemia offer additive value if incorporated in current risk classification. Blood Adv. 2021, 5, 3254–3265. [Google Scholar] [CrossRef] [PubMed]
- Senapati, J.; Urrutia, S.; Loghavi, S.; Short, N.J.; Issa, G.C.; Maiti, A.; Abbas, H.A.; Daver, N.G.; Pemmaraju, N.; Pierce, S.; et al. Venetoclax Abrogates the Prognostic Impact of Splicing Factor Gene Mutations in Newly Diagnosed Acute Myeloid Leukemia. Blood 2023, 142, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Loghavi, S.; Furudate, K.; Montalban-Bravo, G.; Maiti, A.; Kadia, T.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Sasaki, K.; et al. Impact of splicing mutations in acute myeloid leukemia treated with hypomethylating agents combined with venetoclax. Blood Adv. 2021, 5, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Berton, G. Poor Prognosis of SRSF2 Gene Mutations in Patients Treated with Venetoclax-Azacitidine (VEN-AZA) for Newly Diagnosed Acute Myeloid Leukemia. a Multicentric Real-Life Study of 117 Patients. In Proceedings of the 65th ASH Annual Meeting & Exposition, ASH, San Diego, CA, USA, 9–12 December 2023; Available online: https://ash.confex.com/ash/2023/webprogram/Paper180733.html (accessed on 24 December 2023).
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Lew, T.E.; Dengler, M.A.; Thompson, E.R.; Lin, V.S.; Chen, X.; Nguyen, T.; Panigrahi, A.; Handunnetti, S.M.; Carney, D.A.; et al. Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood 2022, 139, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Brown, F.C.; Chua, C.C.; Dengler, M.A.; Pomilio, G.; Anstee, N.S.; Litalien, V.; Thompson, E.; Morley, T.; MacRaild, S.; et al. Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia. Blood 2023, 141, 634–644. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Delia, D.; Aiello, A.; Soligo, D.; Fontanella, E.; Melani, C.; Pezzella, F.; Pierotti, M.A.; Della Porta, G. bcl-2 proto-oncogene expression in normal and neoplastic human myeloid cells. Blood 1992, 79, 1291–1298. [Google Scholar] [CrossRef]
- Karakas, T.; Maurer, U.; Weidmann, E.; Miething, C.C.; Hoelzer, D.; Bergmann, L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998, 9, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.J.F.; Zhu, F.; Dashevsky, O.; Mizuno, R.; Lai, J.X.; Hackett, L.; Ryan, C.E.; Collins, M.C.; Iorgulescu, J.B.; Guièze, R.; et al. Hyperphosphorylation of BCL-2 family proteins underlies functional resistance to venetoclax in lymphoid malignancies. J. Clin. Investig. 2023, 133, e170169. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Zhang, Q.; Ruvolo, V.; Kuruvilla, V.M.; Wang, X.; Mak, D.H.; Battula, V.L.; Konopleva, M.; et al. Maximal Activation of Apoptosis Signaling by Cotargeting Antiapoptotic Proteins in BH3 Mimetic–Resistant AML and AML Stem Cells. Mol. Cancer Ther. 2022, 21, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Ryan, J.; Yecies, D.; Opferman, J.T.; Letai, A. MCL-1–dependent leukemia cells are more sensitive to chemotherapy than BCL-2–dependent counterparts. J. Cell Biol. 2009, 187, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef]
- Kasper, S.; Breitenbuecher, F.; Heidel, F.; Hoffarth, S.; Markova, B.; Schuler, M.; Fischer, T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012, 2, e60. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD–specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed]
- Satta, T.; Li, L.; Chalasani, S.L.; Hu, X.; Nkwocha, J.; Sharma, K.; Kmieciak, M.; Rahmani, M.; Zhou, L.; Grant, S. Dual mTORC1/2 Inhibition Synergistically Enhances AML Cell Death in Combination with the BCL2 Antagonist Venetoclax. Clin. Cancer Res. 2023, 29, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Bouligny, I.M.; Maher, K.R.; Grant, S. Augmenting Venetoclax Activity Through Signal Transduction in AML. J. Cell. Signal. 2023, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Target. Ther. 2022, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.M.; Abbott, D.; Amaya, M.; McMahon, C.; Schwartz, M.; Rosser, J.; Sato, A.; Schowinsky, J.T.; Inguva, A.; Minhajuddin, M.; et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021, 5, 5565–5573. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Leppä, A.-M.; Pölönen, P.; Kontro, M.; Dufva, O.; Deb, D.; Yadav, B.; Brück, O.; Kumar, A.; Everaus, H.; et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 2020, 105, 708–720. [Google Scholar] [CrossRef] [PubMed]
- White, B.S.; Khan, S.A.; Mason, M.J.; Ammad-Ud-Din, M.; Potdar, S.; Malani, D.; Kuusanmäki, H.; Druker, B.J.; Heckman, C.; Kallioniemi, O.; et al. Bayesian multi-source regression and monocyte-associated gene expression predict BCL-2 inhibitor resistance in acute myeloid leukemia. npj Precis. Oncol. 2021, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Kurtz, S.E.; Eide, C.A.; Kaempf, A.; Long, N.; Bottomly, D.; Nikolova, O.; Druker, B.J.; McWeeney, S.K.; Chang, B.H.; Tyner, J.W.; et al. Associating drug sensitivity with differentiation status identifies effective combinations for acute myeloid leukemia. Blood Adv. 2022, 6, 3062–3067. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Dufva, O.; Vähä-Koskela, M.; Leppä, A.-M.; Huuhtanen, J.; Vänttinen, I.; Nygren, P.; Klievink, J.; Bouhlal, J.; Pölönen, P.; et al. Erythroid/megakaryocytic differentiation confers BCL-XL dependency and venetoclax resistance in acute myeloid leukemia. Blood 2023, 141, 1610–1625. [Google Scholar] [CrossRef]
- Brown, F.C.; Wei, A.H. Is BCL-xL the Achilles’ heel of AEL and AMKL? Blood 2023, 141, 1505–1506. [Google Scholar] [CrossRef]
- Bisaillon, R.; Moison, C.; Thiollier, C.; Krosl, J.; Bordeleau, M.-E.; Lehnertz, B.; Lavallée, V.-P.; MacRae, T.; Mayotte, N.; Labelle, C.; et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2020, 34, 63–74. [Google Scholar] [CrossRef]
- Carter, J.L.; Su, Y.; Qiao, X.; Zhao, J.; Wang, G.; Howard, M.; Edwards, H.; Bao, X.; Li, J.; Hüttemann, M.; et al. Acquired resistance to venetoclax plus azacitidine in acute myeloid leukemia: In vitro models and mechanisms. Biochem. Pharmacol. 2023, 216, 115759. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Romanovsky, E.; Kluck, K.; Ourailidis, I.; Menzel, M.; Beck, S.; Ball, M.; Kazdal, D.; Christopoulos, P.; Schirmacher, P.; Stiewe, T.; et al. Homogenous TP53mut-associated tumor biology across mutation and cancer types revealed by transcriptome analysis. Cell Death Discov. 2023, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral role of Noxa in p53-mediated apoptotic response. Minerva Anestesiol. 2003, 17, 2233–2238. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef]
- Odinius, T.O.; Buschhorn, L.; Wagner, C.; Hauch, R.T.; Dill, V.; Dechant, M.; Buck, M.C.; Shoumariyeh, K.; Moog, P.; Schwaab, J.; et al. Comprehensive characterization of central BCL-2 family members in aberrant eosinophils and their impact on therapeutic strategies. J. Cancer Res. Clin. Oncol. 2022, 148, 331–340. [Google Scholar] [CrossRef]
- Stubbins, R.J.; Karsan, A. Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 2021, 11, 193. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nat. Cell Biol. 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Waclawiczek, A.; Leppä, A.-M.; Renders, S.; Stumpf, K.; Reyneri, C.; Betz, B.; Janssen, M.; Shahswar, R.; Donato, E.; Karpova, D.; et al. Combinatorial BCL2 Family Expression in Acute Myeloid Leukemia Stem Cells Predicts Clinical Response to Azacitidine/Venetoclax. Cancer Discov. 2023, 13, 1408–1427. [Google Scholar] [CrossRef]
- Collignon, A.; Hospital, M.A.; Montersino, C.; Courtier, F.; Charbonnier, A.; Saillard, C.; D’incan, E.; Mohty, B.; Guille, A.; Adelaïde, J.; et al. A chemogenomic approach to identify personalized therapy for patients with relapse or refractory acute myeloid leukemia: Results of a prospective feasibility study. Blood Cancer J. 2020, 10, 64. [Google Scholar] [CrossRef]
- Garciaz, S.; Guirguis, A.A.; Müller, S.; Brown, F.C.; Chan, Y.-C.; Motazedian, A.; Rowe, C.L.; Kuzich, J.A.; Chan, K.L.; Tran, K.; et al. Pharmacologic Reduction of Mitochondrial Iron Triggers a Noncanonical BAX/BAK-Dependent Cell Death. Cancer Discov. 2021, 12, 774–791. [Google Scholar] [CrossRef]
- Nakao, F.; Setoguchi, K.; Semba, Y.; Yamauchi, T.; Nogami, J.; Sasaki, K.; Imanaga, H.; Terasaki, T.; Miyazaki, M.; Hirabayashi, S.; et al. Targeting a mitochondrial E3 ubiquitin ligase complex to overcome AML cell-intrinsic Venetoclax resistance. Leukemia 2023, 37, 1028–1038. [Google Scholar] [CrossRef]
- AHuang, A.S.; Chin, H.S.; Reljic, B.; Djajawi, T.M.; Tan, I.K.L.; Gong, J.-N.; Stroud, D.A.; Huang, D.C.S.; van Delft, M.F.; Dewson, G. Mitochondrial E3 ubiquitin ligase MARCHF5 controls BAK apoptotic activity independently of BH3-only proteins. Cell Death Differ. 2022, 30, 632–646. [Google Scholar] [CrossRef]
- Jang, J.E.; Hwang, D.Y.; Eom, J.-I.; Cheong, J.-W.; Jeung, H.-K.; Cho, H.; Chung, H.; Kim, J.S.; Min, Y.H. DRP1 Inhibition Enhances Venetoclax-Induced Mitochondrial Apoptosis in TP53-Mutated Acute Myeloid Leukemia Cells through BAX/BAK Activation. Cancers 2023, 15, 745. [Google Scholar] [CrossRef] [PubMed]
- Hurrish, K.H.; Qiao, X.; Li, X.; Su, Y.; Carter, J.; Ma, J.; Kalpage, H.A.; Hüttemann, M.; Edwards, H.; Wang, G.; et al. Co-targeting of HDAC, PI3K, and Bcl-2 results in metabolic and transcriptional reprogramming and decreased mitochondrial function in acute myeloid leukemia. Biochem. Pharmacol. 2022, 205, 115283. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Culp-Hill, R.; Stevens, B.M.; Jones, C.L.; Pei, S.; Dzieciatkowska, M.; Minhajuddin, M.; Jordan, C.T.; D’alessandro, A. Therapy-Resistant Acute Myeloid Leukemia Stem Cells Are Resensitized to Venetoclax + Azacitidine by Targeting Fatty Acid Desaturases 1 and 2. Metabolites 2023, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.H.; Morales, C.; Rodriguez, I.R.; Valerio, M.; Guo, J.; Chen, M.-H.; Wu, X.; Horne, D.; Gandhi, V.; Chen, L.S.; et al. Synergy of Venetoclax and 8-Chloro-Adenosine in AML: The Interplay of rRNA Inhibition and Fatty Acid Metabolism. Cancers 2022, 14, 1446. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.; Steen, T.V.; Espinoza, I.; Venkatapoorna, C.M.K.; Hu, Z.; Silva, F.M.; Regan, K.; Cuyàs, E.; Meng, X.W.; Verdura, S.; et al. Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death Dis. 2021, 12, 977. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Khokhlatchev, A.V.; Sharma, A.; Deering, T.G.; Shaw, J.J.P.; Costa-Pinheiro, P.; Golla, U.; Annageldiyev, C.; Cabot, M.C.; Conaway, M.R.; Tan, S.; et al. Ceramide nanoliposomes augment the efficacy of venetoclax and cytarabine in models of acute myeloid leukemia. FASEB J. 2022, 36, e22514. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.C.; Pope, V.S.; Tea, M.N.; Li, M.; Nwosu, G.O.; Nguyen, T.M.; Wallington-Beddoe, C.T.; Moretti, P.A.B.; Anderson, D.; Creek, D.J.; et al. Ceramide-induced integrated stress response overcomes Bcl-2 inhibitor resistance in acute myeloid leukemia. Blood 2022, 139, 3737–3751. [Google Scholar] [CrossRef] [PubMed]
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.; Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert, E.R.; et al. Pevonedistat and azacitidine upregulate NOXA (PMAIP1) to increase sensitivity to venetoclax in preclinical models of acute myeloid leukemia. Haematologica 2022, 107, 825–835. [Google Scholar] [CrossRef]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.V.; Jitkova, Y.; et al. Venetoclax enhances T cell-mediated anti-leukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and re-sensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica 2022, 107, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Levitz, D.; Saunthararajah, Y.; Fedorov, K.; Shapiro, L.C.; Mantzaris, I.; Shastri, A.; Kornblum, N.; Sica, R.A.; Shah, N.; Konopleva, M.; et al. A Metabolically Optimized, Noncytotoxic Low-Dose Weekly Decitabine/Venetoclax in MDS and AML. Clin. Cancer Res. 2023, 29, 2774–2780. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef]
- Ravandi, F.; Abuasab, T.; Valero, Y.A.; Issa, G.C.; Islam, R.; Short, N.J.; Yilmaz, M.; Jain, N.; Masarova, L.; Kornblau, S.M.; et al. Phase 2 study of ASTX727 (cedazuridine/decitabine) plus venetoclax (ven) in patients with relapsed/refractory acute myeloid leukemia (AML) or previously untreated, elderly patients (pts) unfit for chemotherapy. J. Clin. Oncol. 2022, 40, 7037. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Ayoub, E.; Ostermann, L.B.; Huang, X.; Loghavi, S.; Boettcher, S.; Nishida, Y.; Ruvolo, V.; et al. Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics. Blood Cancer J. 2023, 13, 57. [Google Scholar] [CrossRef]
- Teh, T.-C.; Nguyen, N.-Y.; Moujalled, D.M.; Segal, D.; Pomilio, G.; Rijal, S.; Jabbour, A.; Cummins, K.; Lackovic, K.; Blombery, P.; et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018, 32, 303–312. [Google Scholar] [CrossRef]
- Saxena, K.; Carter, B.Z.; Konopleva, M. EXABS-147-AML How Do We Overcome Resistance to Venetoclax. Clin. Lymphoma Myeloma Leuk. 2022, 22, S55–S57. [Google Scholar] [CrossRef]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Forsberg, M.; Konopleva, M. SOHO State of the Art Updates and Next Questions: Understanding and Overcoming Venetoclax Resistance in Hematologic Malignancies. Clin. Lymphoma Myeloma Leuk. 2024, 24, 1–14. [Google Scholar] [CrossRef]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef] [PubMed]
- Balachander, S.B.; Criscione, S.W.; Byth, K.F.; Cidado, J.; Adam, A.; Lewis, P.; Macintyre, T.; Wen, S.; Lawson, D.; Burke, K.; et al. AZD4320, A Dual Inhibitor of Bcl-2 and Bcl-xL, Induces Tumor Regression in Hematologic Cancer Models without Dose-limiting Thrombocytopenia. Clin. Cancer Res. 2020, 26, 6535–6549. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef]
- Patel, H.; Periyasamy, M.; Sava, G.P.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; Martin, M.S.; Pop-Damkov, P.; Su, N.; Franklin, V.N.R.; Chilamakuri, C.S.R.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef]
- He, L.; Arnold, C.; Thoma, J.; Rohde, C.; Kholmatov, M.; Garg, S.; Hsiao, C.; Viol, L.; Zhang, K.; Sun, R.; et al. CDK7/12/13 inhibition targets an oscillating leukemia stem cell network and synergizes with venetoclax in acute myeloid leukemia. EMBO Mol. Med. 2022, 14, e14990. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; Graber, C.; Flückiger, L.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 8092. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Jalte, M.; Abbassi, M.; Belghiti, H.D.; Ahakoud, M.; Bekkari, H.; EL Mouhi, H. FLT3 Mutations in Acute Myeloid Leukemia: Unraveling the Molecular Mechanisms and Implications for Targeted Therapies. Cureus 2023, 15, e45765. [Google Scholar] [CrossRef]
- Janssen, M.; Schmidt, C.; Bruch, P.-M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef]
- Short, N.; Macaron, W.; Dinardo, C.; Daver, N.; Yilmaz, M.; Borthakur, G.; Montalban-Bravo, G.; Garcia-Manero, G.; Issa, G.; Sasaki, K.; et al. P485: Azacitidine, venetoclax and gilteritinib for patients with newly diagnosed flt3-mutated acute myeloid leukemia: A subgroup analysis from a phase II study. Hemasphere 2023, 7, e535260f. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Short, N.J.; Reville, P.; Konopleva, M.; Kadia, T.; DiNardo, C.; Borthakur, G.; Pemmaraju, N.; Maiti, A.; et al. Hypomethylating agent and venetoclax with FLT3 inhibitor “triplet” therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J. 2022, 12, 77. [Google Scholar] [CrossRef]
- Yilmaz, M.; Muftuoglu, M.; Kantarjian, H.M.; Dinardo, C.D.; Kadia, T.M.; Konopleva, M.; Borthakur, G.; Pemmaraju, N.; Short, N.J.; Valero, Y.A.; et al. Quizartinib (QUIZ) with decitabine (DAC) and venetoclax (VEN) is active in patients (pts) with FLT3-ITD mutated acute myeloid leukemia (AML): A phase I/II clinical trial. J. Clin. Oncol. 2022, 40, 7036. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine inIDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A Phase Ib/II Study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-Mutated Myeloid Malignancies. Blood Cancer Discov. 2023, 4, 276–293. [Google Scholar] [CrossRef]
- Venugopal, S.; Takahashi, K.; Daver, N.; Maiti, A.; Borthakur, G.; Loghavi, S.; Short, N.J.; Ohanian, M.; Masarova, L.; Issa, G.; et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. 2022, 12, 10. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Daver, N.G.; Dail, M.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.L.; Kelly, K.R.; Vey, N.; Assouline, S.; Roboz, G.J.; Paolini, S.; et al. Venetoclax and idasanutlin in relapsed/refractory AML: A nonrandomized, open-label phase 1b trial. Blood 2023, 141, 1265–1276. [Google Scholar] [CrossRef]
- Senapati, J.; Muftuoglu, M.; Ishizawa, J.; Abbas, H.A.; Loghavi, S.; Borthakur, G.; Yilmaz, M.; Issa, G.C.; Dara, S.I.; Basyal, M.; et al. A Phase I study of Milademetan (DS3032b) in combination with low dose cytarabine with or without venetoclax in acute myeloid leukemia: Clinical safety, efficacy, and correlative analysis. Blood Cancer J. 2023, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: A phase 1, dose-finding and expansion study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.A.; Wang, E.S. Menin Inhibitors in Acute Myeloid Leukemia—What Does the Future Hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef]
- Jabbour, E.; Searle, E.; Abdul-Hay, M.; Abedin, S.; Aldoss, I.; Piérola, A.A.; Alonso-Dominguez, J.M.; Chevallier, P.; Cost, C.; Daskalakis, N.; et al. A First-in-Human Phase 1 Study of the Menin-KMT2A (MLL1) Inhibitor JNJ-75276617 in Adult Patients with Relapsed/Refractory Acute Leukemia Harboring KMT2A or NPM1 Alterations. Blood 2023, 142, 57. [Google Scholar] [CrossRef]
- Gilead Announces Partial Clinical Hold for Magrolimab Studies in AML. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2023/8/gilead-announces-partial-clinical-hold-for-magrolimab-studies-in-aml (accessed on 28 January 2024).
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2021, 138, 371. [Google Scholar] [CrossRef]
- Daver, N. Pivekimab Sunirine (PVEK, IMGN632), a CD123-Targeting Antibody-Drug Conjugate, in Combination with Azacitidine and Venetoclax in Patients with Newly Diagnosed Acute Myeloid Leukemia. In Proceedings of the 65th ASH Annual Meeting & Exposition, ASH, San Diego, CA, USA, 9–12 December 2023; Available online: https://ash.confex.com/ash/2023/webprogram/Paper173413.html (accessed on 28 January 2024).
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef]
- Winters, A.C.; Minhajuddin, M.; Stevens, B.M.; Major, A.; Bosma, G.; Abbott, D.; Miltgen, N.; Yuan, J.; Treece, A.L.; Siegele, B.J.; et al. Multi-gene measurable residual disease assessed by digital polymerase chain reaction has clinical and biological utility in acute myeloid leukemia patients receiving venetoclax/azacitidine. Haematologica 2023. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Wang, S.A.; Jorgensen, J.; Kadia, T.M.; Daver, N.G.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; Borthakur, G.; et al. Prognostic value of measurable residual disease after venetoclax and decitabine in acute myeloid leukemia. Blood Adv. 2021, 5, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Othman, J.; Tiong, I.S.; O’Nions, J.; Dennis, M.; Mokretar, K.; Ivey, A.; Austin, M.J.; Latif, A.-L.; Amer, M.; Chan, W.Y.; et al. Molecular MRD is strongly prognostic in patients with NPM1-mutated AML receiving venetoclax-based nonintensive therapy. Blood 2023, 143, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Single-Cell Multi-Omics|Mission Bio. Available online: https://missionbio.com/capabilities/dna-protein/?gad_source=1&gclid=CjwKCAiArfauBhApEiwAeoB7qKxJJBI0pFtA47lwoW5BNyLcHWRO6uvfse-fZ_60Tdbf8DUtfQN5QhoCOXEQAvD_BwE (accessed on 27 February 2024).
- Garciaz, S.; Bertoli, S.; Sallman, D.A.; Decroocq, J.; Dumas, P.-Y.; Belhabri, A.; Orvain, C.; Requena, G.A.; Simand, C.; Laribi, K.; et al. Acute Myeloid Leukemia Patients Who Stopped Venetoclax or/and Azacytidine for Other Reasons Than Progression Have a Prolonged Treatment Free Remission and Overall Survival. a Filo Study. Blood 2023, 142, 161. [Google Scholar] [CrossRef]
- Chua, C.C.; Hammond, D.; Kent, A.; Tiong, I.S.; Konopleva, M.Y.; Pollyea, D.A.; DiNardo, C.D.; Wei, A.H. Treatment-free remission after ceasing venetoclax-based therapy in patients with acute myeloid leukemia. Blood Adv. 2022, 6, 3879–3883. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Phase; Name; ID | Study Population | Venetoclax Doses | Other Agent Administration Regimens | Response Rate | Survival Rate |
|---|---|---|---|---|---|
| I; (M14-358); NCT02203773 | ND AML ineligible for chemotherapy | 400–800–1200 mg D1 to D28 | AZA 75 mg/m2, D1 to D7 or DEC 20 mg/m2, D1 to D5 | 37% (CR), 68% (ORR), 83% (cCR) | 17.5 months (median OS) |
| II; NCT03404193 | ND patients with AML > 60 years | 400 mg D1 to D28 (D1 to D21 if blasts < 5%) | DEC 20 mg/m2, D1 to D10 (induction) DEC 20 mg/m2, D1 to D5 (consolidation) | 63% (CR + CRi), 74% (ORR) | 18.1 months (median OS) |
| III; (VIALE-A); NCT02993523 | ND AML ineligible for chemotherapy | 400 mg D1 to D28 vs. placebo | AZA 75 mg/m2, D1 to D7 | 36.7% vs. 17.9% (CR), 64.7% vs. 22.8% (CR + CRi) | 14.7 months vs. 9.6 months (median OS) |
| Treatment | Study ID | Phase | Number of Patients | Response | Treatment |
|---|---|---|---|---|---|
| Quizartinib (FLT3 inhibitor) | NCT03661307 | I/II | 28 (23 R/R AML FLT3mut) | 78% achieved cCR (3 CR, 15 CRi) | [147] |
| Ivosidenib (IDH1 inhibitor) | NCT03471260 | Ib | 31 (all R/R, IDHmut) | 94% had a CR + CRh + CRi + PR + MLFS | [149] |
| Magrolimab (anti CD47) | NCT04435691 | I/II | 60 (41 ND, 29) | ORR [CR, CRi, MLFS] Frontline = 80% | [159] |
| Pivekimab sunirine (PVEK, anti CD123) | NCT04086264 | I/II | 50 ND patients | CR rate was 52% (26/50), and cCR rate was 66% (33/50) | [160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garciaz, S.; Hospital, M.-A.; Collette, Y.; Vey, N. Venetoclax Resistance in Acute Myeloid Leukemia. Cancers 2024, 16, 1091. https://doi.org/10.3390/cancers16061091
Garciaz S, Hospital M-A, Collette Y, Vey N. Venetoclax Resistance in Acute Myeloid Leukemia. Cancers. 2024; 16(6):1091. https://doi.org/10.3390/cancers16061091
Chicago/Turabian StyleGarciaz, Sylvain, Marie-Anne Hospital, Yves Collette, and Norbert Vey. 2024. "Venetoclax Resistance in Acute Myeloid Leukemia" Cancers 16, no. 6: 1091. https://doi.org/10.3390/cancers16061091
APA StyleGarciaz, S., Hospital, M.-A., Collette, Y., & Vey, N. (2024). Venetoclax Resistance in Acute Myeloid Leukemia. Cancers, 16(6), 1091. https://doi.org/10.3390/cancers16061091