

Mechanisms of Resistance to Small Molecules in Acute Myeloid Leukemia


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. FLT3 Inhibitors
2.1. Overview of FLT3 Inhibitors
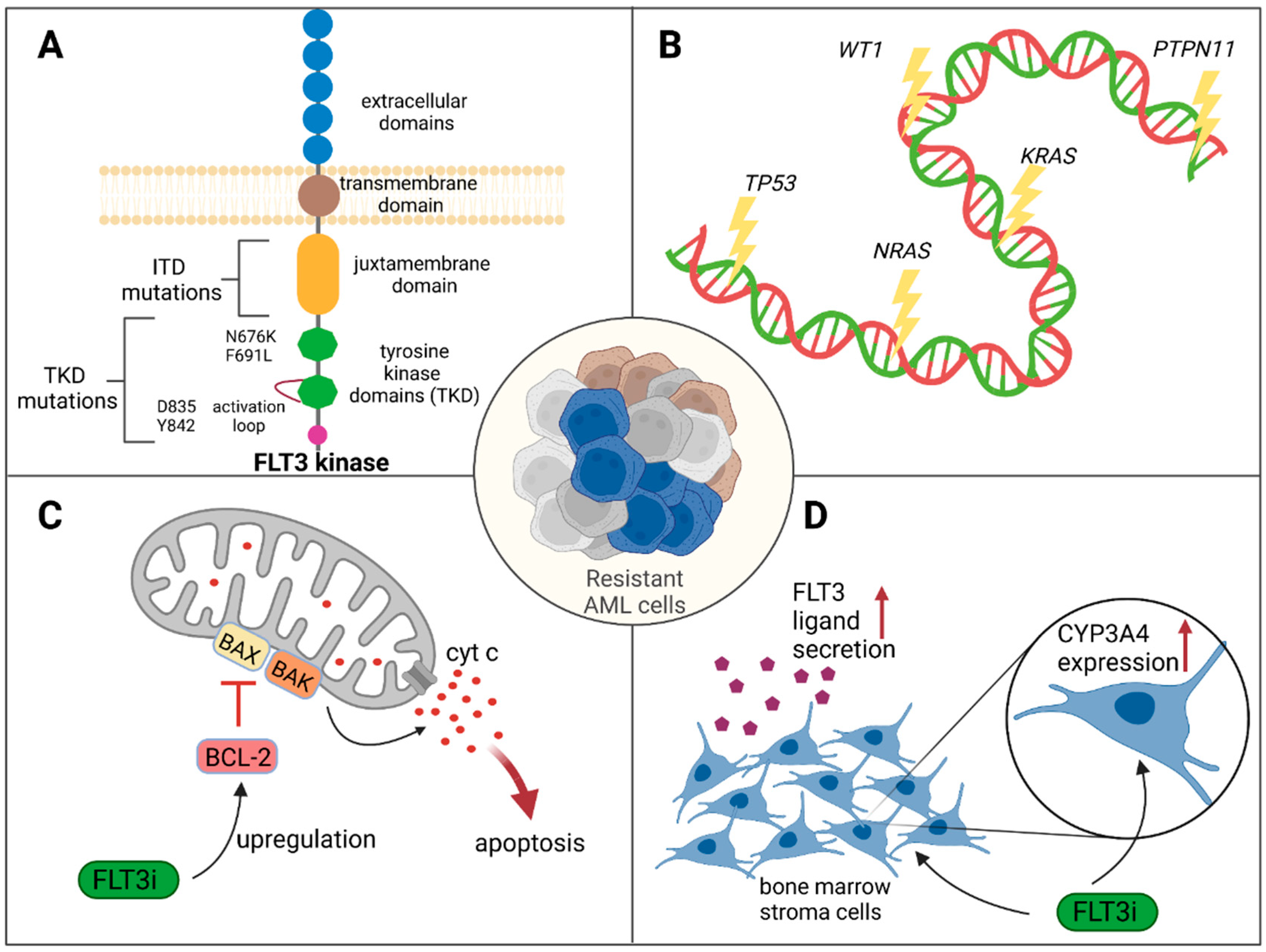
2.2. Mechanisms of Resistance to FLT3 Inhibition
2.2.1. Genetic Mechanisms Causing Resistance
2.2.2. Nongenetic Mechanisms Causing Resistance
2.3. Overcoming Resistance to FLT3 Inhibitors
3. IDH Inhibitors
3.1. Overview of IDH Inhibitors
3.2. Mechanisms of Resistance to IDH Inhibition
3.2.1. Genetic Mechanisms Causing Resistance
3.2.2. Nongenetic Mechanisms Causing Resistance
3.3. Overcoming Resistance
4. BCL2 Inhibitors
4.1. Pharmacologic Inhibition of BCL2
4.2. Mechanisms of Resistance
4.2.1. Genetics of Response and Resistance to Venetoclax
4.2.2. Other Mechanisms of Resistance to Venetoclax
4.3. Overcoming Resistance to BCL2 Inhibition
5. Other Small Molecules for the Treatment of AML
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Rai, K.R.; Holland, J.F.; Glidewell, O.J.; Weinberg, V.; Brunner, K.; Obrecht, J.P.; Preisler, H.D.; Nawabi, I.W.; Prager, D.; Carey, R.W.; et al. Treatment of acute myelocytic leukemia: A study by cancer and leukemia group B. Blood 1981, 58, 1203–1212. [Google Scholar] [CrossRef]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline Dose Intensification in Acute Myeloid Leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Rosnet, O.; Buhring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996, 10, 238–248. [Google Scholar]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Agnès, F.; Shamoon, B.; Dina, C.; Rosnet, O.; Birnbaum, D.; Galibert, F. Genomic structure of the downstream part of the human FLT3 gene: Exon/intron structure conservation among genes encoding receptor tyrosine kinases (RTK) of subclass III. Gene 1994, 145, 283–288. [Google Scholar] [CrossRef]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Fröhling, S.; Schlenk, R.F.; Breitruck, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Döhner, H.; Döhner, K. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML Study Group Ulm. Blood 2002, 100, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Sawyers, C.L. Finding the next Gleevec: FLT3 targeted kinase inhibitor therapy for acute myeloid leukemia. Cancer Cell 2002, 1, 413–415. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Rollig, C.; Wollmer, E.; Wasch, R.; Bornhauser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Röllig, C.; Serve, H.; Noppeney, R.; Hanoun, M.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; Krämer, A.; et al. Sorafenib or placebo in patients with newly diagnosed acute myeloid leukaemia: Long-term follow-up of the randomized controlled SORAML trial. Leukemia 2021, 35, 2517–2525. [Google Scholar] [CrossRef]
- Larson, R.A.; Mandrekar, S.J.; Huebner, L.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: The Alliance CALGB 10603/RATIFY trial. Leukemia 2021, 35, 2539–2551. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Erba, H.P.; Montesinos, P.; Kim, H.J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus chemotherapy in newly diagnosed patients with FLT3-internal-tandem-duplication-positive acute myeloid leukaemia (QuANTUM-First): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Sharzehi, S.; Pittsenbarger, J.; Bottomly, D.; Tognon, C.E.; McWeeney, S.K.; Druker, B.J.; Traer, E. A noncanonical FLT3 gatekeeper mutation disrupts gilteritinib binding and confers resistance. Am. J. Hematol. 2021, 96, E226–E229. [Google Scholar] [CrossRef]
- Williams, A.B.; Nguyen, B.; Li, L.; Brown, P.; Levis, M.; Leahy, D.; Small, D. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia 2013, 27, 48–55. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Strang, E.; Theis, F.; Jahn, N.; Panina, E.; Blatte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Sharzehi, S.; Joshi, S.K.; Pittsenbarger, J.; Tyner, J.W.; Traer, E. The FLT3 F691L Gatekeeper Mutation Promotes Clinical Resistance to Gilteritinib + Venetoclax (GILT + VEN) in AML. Blood 2021, 138, 2235. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Alfayez, M.; DiNardo, C.D.; Borthakur, G.; Kadia, T.M.; Konopleva, M.Y.; Loghavi, S.; Kanagal-Shamanna, R.; Patel, K.P.; Jabbour, E.J.; et al. Outcomes with sequential FLT3-inhibitor-based therapies in patients with AML. J. Hematol. Oncol. 2020, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 inhibitors. Blood Cancer Discov. 2021, 2, 125–134. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J.; et al. Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia 2021, 35, 2017–2029. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Breitenbuecher, F.; Markova, B.; Kasper, S.; Carius, B.; Stauder, T.; Böhmer, F.D.; Masson, K.; Rönnstrand, L.; Huber, C.; Kindler, T.; et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood 2009, 113, 4063–4073. [Google Scholar] [CrossRef]
- Kohl, T.M.; Hellinger, C.; Ahmed, F.; Buske, C.; Hiddemann, W.; Bohlander, S.K.; Spiekermann, K. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia 2007, 21, 1763–1772. [Google Scholar] [CrossRef]
- Hunter, H.M.; Pallis, M.; Seedhouse, C.H.; Grundy, M.; Gray, C.; Russell, N.H. The expression of P-glycoprotein in AML cells with FLT3 internal tandem duplications is associated with reduced apoptosis in response to FLT3 inhibitors. Br. J. Haematol. 2004, 127, 26–33. [Google Scholar] [CrossRef]
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Dumas, P.Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef]
- Young, D.J.; Nguyen, B.; Li, L.; Higashimoto, T.; Levis, M.J.; Liu, J.O.; Small, D. A Method for Overcoming Plasma Protein Inhibition of Tyrosine Kinase Inhibitors. Blood Cancer Discov. 2021, 2, 532–547. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, P.; Wang, Y.; Shen, Y. Sitravatinib as a potent FLT3 inhibitor can overcome gilteritinib resistance in acute myeloid leukemia. Biomark. Res. 2023, 11, 8. [Google Scholar] [CrossRef]
- Yamaura, T.; Nakatani, T.; Uda, K.; Ogura, H.; Shin, W.; Kurokawa, N.; Saito, K.; Fujikawa, N.; Date, T.; Takasaki, M.; et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood 2018, 131, 426–438. [Google Scholar] [CrossRef]
- Levis, M.J.; Smith, C.C.; Perl, A.E.; Schiller, G.J.; Fathi, A.T.; Roboz, G.J.; Wang, E.S.; Altman, J.K.; Ando, M.; Suzuki, T.; et al. Phase 1 first-in-human study of irreversible FLT3 inhibitor FF-10101-01 in relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2021, 39 (Suppl. 15), 7008. [Google Scholar] [CrossRef]
- Singh Mali, R.; Zhang, Q.; DeFilippis, R.A.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021, 106, 1034–1046. [Google Scholar] [CrossRef]
- Brinton, L.T.; Zhang, P.; Williams, K.; Canfield, D.; Orwick, S.; Sher, S.; Wasmuth, R.; Beaver, L.; Cempre, C.; Skinner, J.; et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 139. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Li, L.; Nguyen, B.; Seo, J.; Wu, M.; Seale, T.; Levis, M.; Duffield, A.; Hu, Y.; Small, D. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct. Target. Ther. 2021, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.; Schmidt, C.; Bruch, P.M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Liu, Q.; Nelson, E.; Kung, A.L.; Christie, A.L.; Bronson, R.; Sattler, M.; Sanda, T.; Zhao, Z.; Hur, W.; et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: Synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia 2012, 26, 2233–2244. [Google Scholar] [CrossRef]
- Weisberg, E.; Liu, Q.; Zhang, X.; Nelson, E.; Sattler, M.; Liu, F.; Nicolais, M.; Zhang, J.; Mitsiades, C.; Smith, R.W.; et al. Selective Akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS ONE 2013, 8, e56473. [Google Scholar] [CrossRef]
- Kapoor, S.; Natarajan, K.; Baldwin, P.R.; Doshi, K.A.; Lapidus, R.G.; Mathias, T.J.; Scarpa, M.; Trotta, R.; Davila, E.; Kraus, M.; et al. Concurrent Inhibition of Pim and FLT3 Kinases Enhances Apoptosis of FLT3-ITD Acute Myeloid Leukemia Cells through Increased Mcl-1 Proteasomal Degradation. Clin. Cancer Res. 2018, 24, 234–247. [Google Scholar] [CrossRef]
- Traer, E.; Martinez, J.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Kovacsovics, T.; Tyner, J.W.; Wong, M.; Druker, B.J. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016, 76, 6471–6482. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, G.; Zhang, H.; Basyal, M.; Ly, C.; Yuan, B.; Ruvolo, V.; Piya, S.; Bhattacharya, S.; Zhang, Q.; et al. Concomitant targeting of FLT3 and BTK overcomes FLT3 inhibitor resistance in acute myeloid leukemia through the inhibition of autophagy. Haematologica 2023, 108, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Wagner, K.; Damm, F.; Göhring, G.; Görlich, K.; Heuser, M.; Schäfer, I.; Ottmann, O.; Lübbert, M.; Heit, W.; Kanz, L.; et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J. Clin. Oncol. 2010, 28, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef]
- De Botton, S.; Fenaux, P.; Yee, K.; Récher, C.; Wei, A.H.; Montesinos, P.; Taussig, D.C.; Pigneux, A.; Braun, T.; Curti, A.; et al. Olutasidenib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-mutated AML. Blood Adv. 2023, 7, 3117–3127. [Google Scholar] [CrossRef]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, E.S.; et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: Phase 1 results of a phase 1/2 trial. Lancet Haematol. 2023, 10, e46–e58. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Tallman, M.S.; de Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Zeidan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): A single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Morita, K.; DiNardo, C.D.; Furudate, K.; Tanaka, T.; Yan, Y.; Patel, K.P.; MacBeth, K.J.; Wu, B.; Liu, G.; et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat. Commun. 2021, 12, 2607. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Bensi, L.; Longo, R.; Vecchi, A.; Messora, C.; Garagnani, L.; Bernardi, S.; Tamassia, M.G.; Sacchi, S. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 1995, 80, 98–102. [Google Scholar]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers Of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, E202–E205. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, P.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef]
- Pollyea, D.A.; DiNardo, C.D.; Arellano, M.L.; Pigneux, A.; Fiedler, W.; Konopleva, M.; Rizzieri, D.A.; Smith, B.D.; Shinagawa, A.; Lemoli, R.M.; et al. Impact of Venetoclax and Azacitidine in Treatment-Naïve Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin. Cancer Res. 2022, 28, 2753–2761. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Pillai, R.; Sanchez, J.F.; Mei, M.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Salhotra, A.; et al. Association of leukemia genetics with response to venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Am. J. Hematol. 2019, 94, E253–E255. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Wang, Y.W.; Tsai, C.H.; Lin, C.C.; Tien, F.M.; Chen, Y.W.; Lin, H.Y.; Yao, M.; Lin, Y.C.; Lin, C.T.; Cheng, C.L.; et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann. Hematol. 2020, 99, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.C.; Roberts, A.W.; Reynolds, J.; Fong, C.Y.; Ting, S.B.; Salmon, J.M.; MacRaild, S.; Ivey, A.; Tiong, I.S.; Fleming, S.; et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined With Modified Intensive Chemotherapy. J. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, M.S.; de Leeuw, D.C.; Janssen, J.; Smit, L. Targeting Acute Myeloid Leukemia with Venetoclax; Biomarkers for Sensitivity and Rationale for Venetoclax-Based Combination Therapies. Cancers 2022, 14, 3456. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, R.; Moison, C.; Thiollier, C.; Krosl, J.; Bordeleau, M.E.; Lehnertz, B.; Lavallee, V.P.; MacRae, T.; Mayotte, N.; Labelle, C.; et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2020, 34, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakauchi, Y.; Kohnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef]
- Tantawy, S.I.; Sarkar, A.; Hubner, S.; Tan, Z.; Wierda, W.G.; Eldeib, A.; Zhang, S.; Kornblau, S.; Gandhi, V. Mechanisms of MCL-1 Protein Stability Induced by MCL-1 Antagonists in B-Cell Malignancies. Clin. Cancer Res. 2023, 29, 446–457. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A Phase Ib/II Study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-Mutated Myeloid Malignancies. Blood Cancer Discov. 2023, 4, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Smith, B.D.; Fiedler, W.; Sekeres, M.A.; Montesinos, P.; Leber, B.; Merchant, A.; Papayannidis, C.; Pérez-Simón, J.A.; Hoang, C.J.; et al. Clinical benefit of glasdegib plus low-dose cytarabine in patients with de novo and secondary acute myeloid leukemia: Long-term analysis of a phase II randomized trial. Ann. Hematol. 2021, 100, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Faramand, R.; Carter-Cooper, B.; Tolu, S.; Ford, L.A.; Lapidus, R.G.; Wetzler, M.; Wang, E.S.; Etemadi, A.; Griffiths, E.A. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am. J. Hematol. 2015, 90, E77–E79. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Valencia, A.; Masala, E.; Rossi, A.; Martino, A.; Sanna, A.; Buchi, F.; Canzian, F.; Cilloni, D.; Gaidano, V.; Voso, M.T.; et al. Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 2014, 28, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, Q.; Li, S.; Wang, X. Resistance to Hypomethylating Agents in Myelodysplastic Syndrome and Acute Myeloid Leukemia From Clinical Data and Molecular Mechanism. Front. Oncol. 2021, 11, 706030. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahme, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myelodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef]
- Erba, H.P.; Fathi, A.T.; Issa, G.C.; Altman, J.K.; Montesinos, P.; Patnaik, M.M.; Foran, J.M.; De Botton, S.; Baer, M.R.; Schiller, G.J.; et al. Update on a Phase 1/2 First-in-Human Study of the Menin-KMT2A (MLL) Inhibitor Ziftomenib (KO-539) in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2022, 140 (Suppl. 1), 153–156. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef]
- Erbani, J.; Tay, J.; Barbier, V.; Levesque, J.P.; Winkler, I.G. Acute Myeloid Leukemia Chemo-Resistance Is Mediated by E-selectin Receptor CD162 in Bone Marrow Niches. Front. Cell Dev. Biol. 2020, 8, 668. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; Magnani, J.L.; Thackray, H.M.; Feldman, E.J.; O’Dwyer, M.E.; et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood 2022, 139, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Abe, A.; Imagama, S.; Nomura, Y.; Tanizaki, R.; Minami, Y.; Hayakawa, F.; Ito, Y.; Katsumi, A.; Yamamoto, K.; et al. BCR-ABL-independent and RAS / MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur. J. Haematol. 2010, 84, 229–238. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, T.J.L.; Damm, F.; Bullinger, L.; Frick, M. Mechanisms of Resistance to Small Molecules in Acute Myeloid Leukemia. Cancers 2023, 15, 4573. https://doi.org/10.3390/cancers15184573
Lang TJL, Damm F, Bullinger L, Frick M. Mechanisms of Resistance to Small Molecules in Acute Myeloid Leukemia. Cancers. 2023; 15(18):4573. https://doi.org/10.3390/cancers15184573
Chicago/Turabian StyleLang, Tonio Johannes Lukas, Frederik Damm, Lars Bullinger, and Mareike Frick. 2023. "Mechanisms of Resistance to Small Molecules in Acute Myeloid Leukemia" Cancers 15, no. 18: 4573. https://doi.org/10.3390/cancers15184573
APA StyleLang, T. J. L., Damm, F., Bullinger, L., & Frick, M. (2023). Mechanisms of Resistance to Small Molecules in Acute Myeloid Leukemia. Cancers, 15(18), 4573. https://doi.org/10.3390/cancers15184573