
Activation of Epstein–Barr Virus’ Lytic Cycle in Nasopharyngeal Carcinoma Cells by NEO212, a Conjugate of Perillyl Alcohol and Temozolomide

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pharmacological Agents
2.2. Cell Culture
2.3. Primary Cells (BM-NPC)
2.4. Immunoblots
2.5. Colony Formation Assays
2.6. Transfection with siRNA
2.7. Animal Model
2.8. Calculation of Drug Combination Effects
2.9. Other Statistical Analyses
3. Results
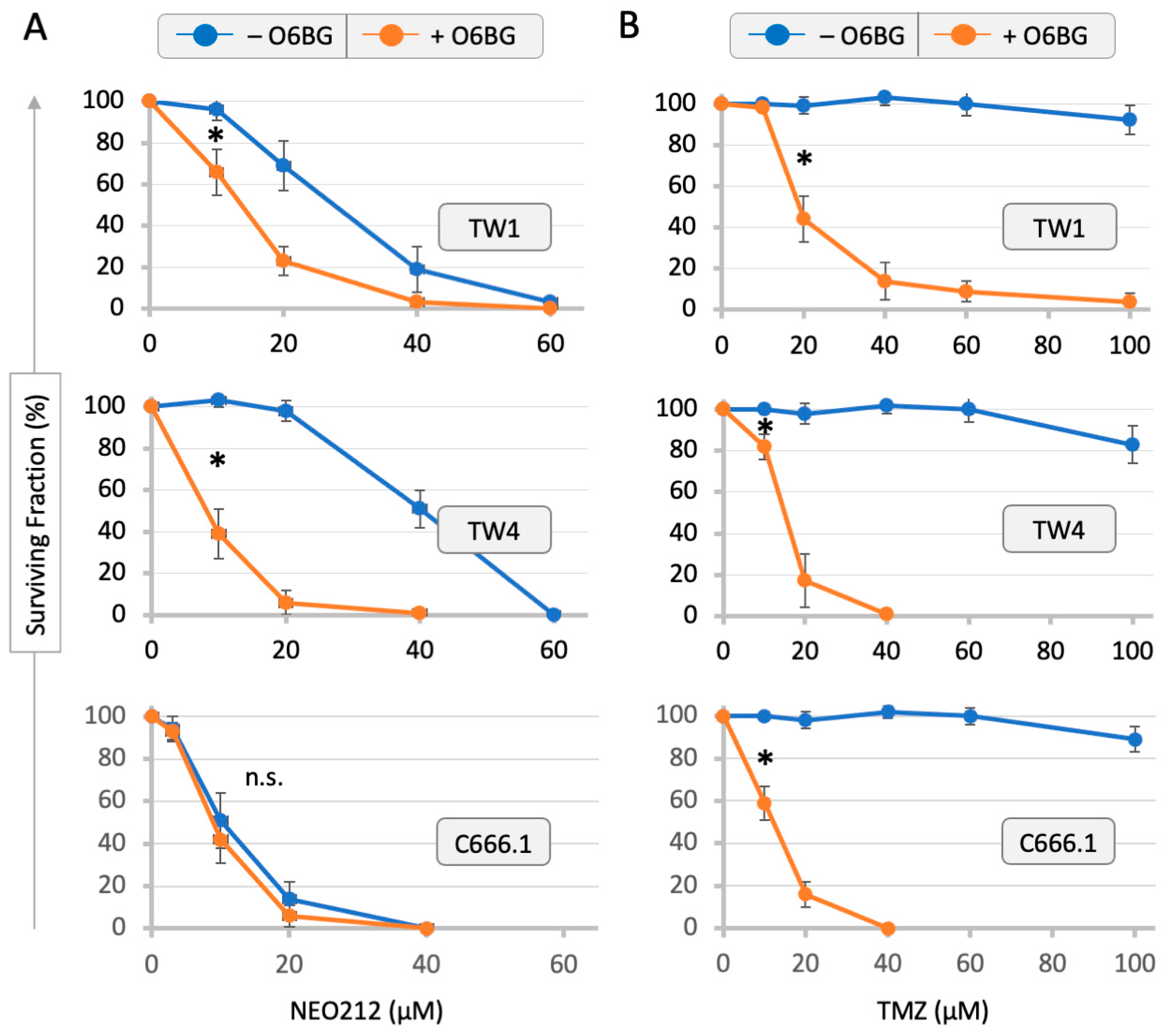
3.1. Drug Sensitivity of NPC Cells and Correlation with MGMT Levels
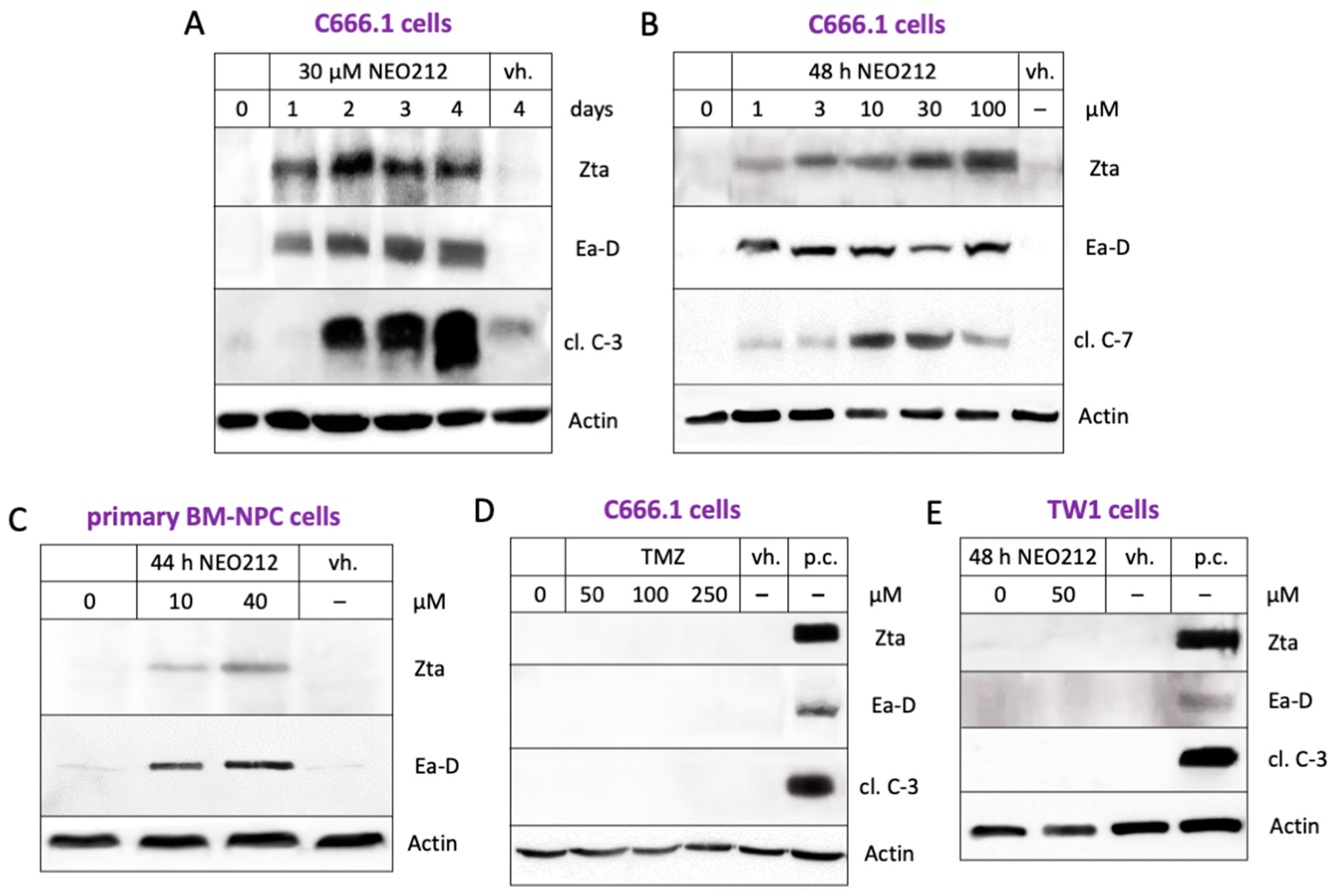
3.2. NEO212 Triggers Immediate-Early- and Early-Stage Lytic Viral Protein Production
3.3. NEO212 Exerts Therapeutic Activity and Triggers the Lytic Cycle In Vivo
3.4. CHOP Is Required for the NEO212-Induced Lytic Cycle
3.5. ROS Are Involved in the NEO212-Induced Lytic Cycle
3.6. NEO212 Sensitizes C666.1 Cells to Ganciclovir
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- zur Hausen, H.; Schulte-Holthausen, H.; Klein, G.; Henle, W.; Henle, G.; Clifford, P.; Santesson, L. Ebv DNA in biopsies of burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 1970, 228, 1056–1058. [Google Scholar] [CrossRef]
- Su, Z.Y.; Siak, P.Y.; Leong, C.O.; Cheah, S.C. The role of epstein-barr virus in nasopharyngeal carcinoma. Front. Microbiol. 2023, 14, 1116143. [Google Scholar] [CrossRef]
- Münz, C. Epstein Barr Virus Volume—One Herpes Virus: Many Diseases; Springer International Publishing: Cham, Switzerland, 2015; Volume 391, p. 505. [Google Scholar]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Dorothea, M.; Hui, K.F.; Chiang, A.K.S. Lytic induction therapy against epstein-barr virus-associated malignancies: Past, present, and future. Cancers 2020, 12, 2142. [Google Scholar] [CrossRef] [PubMed]
- Stoker, S.D.; Novalic, Z.; Wildeman, M.A.; Huitema, A.D.; Verkuijlen, S.A.; Juwana, H.; Greijer, A.E.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P. Epstein-barr virus-targeted therapy in nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Yiu, S.P.T.; Tam, K.P.; Chiang, A.K.S. Viral-targeted strategies against ebv-associated lymphoproliferative diseases. Front. Oncol. 2019, 9, 81. [Google Scholar] [CrossRef]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular basis of epstein-barr virus latency establishment and lytic reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef]
- Amon, W.; Farrell, P.J. Reactivation of epstein-barr virus from latency. Rev. Med. Virol. 2005, 15, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.L.; Mak, N.K.; Lo, K.W. Targeting epstein-barr virus in nasopharyngeal carcinoma. Front. Oncol. 2020, 10, 600. [Google Scholar] [CrossRef]
- Pinninti, S.; Hough-Telford, C.; Pati, S.; Boppana, S. Cytomegalovirus and epstein-barr virus infections. Pediatr. Rev. 2016, 37, 223–234. [Google Scholar] [CrossRef]
- Zamora, M.R. DNA viruses (cmv, ebv, and the herpesviruses). Semin. Respir. Crit. Care Med. 2011, 32, 454–470. [Google Scholar] [CrossRef]
- Poole, C.L.; James, S.H. Antiviral therapies for herpesviruses: Current agents and new directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef]
- Feng, W.H.; Israel, B.; Raab-Traub, N.; Busson, P.; Kenney, S.C. Chemotherapy induces lytic ebv replication and confers ganciclovir susceptibility to ebv-positive epithelial cell tumors. Cancer Res. 2002, 62, 1920–1926. [Google Scholar]
- Moore, S.M.; Cannon, J.S.; Tanhehco, Y.C.; Hamzeh, F.M.; Ambinder, R.F. Induction of epstein-barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrob. Agents Chemother. 2001, 45, 2082–2091. [Google Scholar] [CrossRef]
- Crumpacker, C.S. Ganciclovir. N. Engl. J. Med. 1996, 335, 721–729. [Google Scholar] [CrossRef]
- Faulds, D.; Heel, R.C. Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs 1990, 39, 597–638. [Google Scholar] [CrossRef]
- Novalic, Z.; Verkuijlen, S.; Verlaan, M.; Eersels, J.L.H.; de Greeuw, I.; Molthoff, C.F.M.; Middeldorp, J.M.; Greijer, A.E. Cytolytic virus activation therapy and treatment monitoring for epstein-barr virus associated nasopharyngeal carcinoma in a mouse tumor model. J. Med. Virol. 2017, 89, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.A.; Novalic, Z.; Verkuijlen, S.A.; Juwana, H.; Huitema, A.D.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P.; Greijer, A.E. Cytolytic virus activation therapy for epstein-barr virus-driven tumors. Clin. Cancer Res. 2012, 18, 5061–5070. [Google Scholar] [CrossRef] [PubMed]
- Daibata, M.; Bandobashi, K.; Kuroda, M.; Imai, S.; Miyoshi, I.; Taguchi, H. Induction of lytic epstein-barr virus (ebv) infection by synergistic action of rituximab and dexamethasone renders ebv-positive lymphoma cells more susceptible to ganciclovir cytotoxicity in vitro and in vivo. J. Virol. 2005, 79, 5875–5879. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent ebv and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Kuo, Y.C.; Huang, Y.; Huang, Y.C.; Lui, K.W.; Chang, K.P.; Lin, T.L.; Fan, H.C.; Lin, A.C.; Hsieh, C.H.; et al. Application of a patient-derived xenograft model in cytolytic viral activation therapy for nasopharyngeal carcinoma. Oncotarget 2015, 6, 31323–31334. [Google Scholar] [CrossRef][Green Version]
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class i histone deacetylases by romidepsin potently induces epstein-barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef]
- Jones, K.; Nourse, J.; Corbett, G.; Gandhi, M.K. Sodium valproate in combination with ganciclovir induces lysis of ebv-infected lymphoma cells without impairing ebv-specific t-cell immunity. Int. J. Lab. Hematol. 2010, 32, e169–e174. [Google Scholar] [CrossRef]
- Liu, S.F.; Wang, H.; Li, Z.J.; Deng, X.Y.; Xiang, H.; Tao, Y.G.; Li, W.; Tang, M.; Cao, Y. Aspirin induces lytic cytotoxicity in epstein-barr virus-positive cells. Eur. J. Pharmacol. 2008, 589, 8–13. [Google Scholar] [CrossRef]
- Perrine, S.P.; Hermine, O.; Small, T.; Suarez, F.; O’Reilly, R.; Boulad, F.; Fingeroth, J.; Askin, M.; Levy, A.; Mentzer, S.J.; et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with epstein-barr virus-associated lymphoid malignancies. Blood 2007, 109, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Da Fonseca, C.O.; Schönthal, A.H. Perillyl alcohol and its drug-conjugated derivatives as potential novel methods of treating brain metastases. Int. J. Mol. Sci. 2016, 17, 1463. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B. Temozolomide, procarbazine and nitrosoureas in the therapy of malignant gliomas: Update of mechanisms, drug resistance and therapeutic implications. J. Clin. Med. 2023, 12, 7442. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. Mgmt: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair. 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, S.H.; Kim, T.M.; Choi, S.H.; Park, S.H.; Heo, D.S.; Kim, I.H.; Jung, H.W. The value of temozolomide in combination with radiotherapy during standard treatment for newly diagnosed glioblastoma. J. Neurooncol. 2013, 112, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Crowell, P.L.; Elson, C.E. Isoprenoids, Health and Disease. In Neutraceuticals and Functional Foods; Wildman, R.E.C., Ed.; CRC Press: Boca Raton, FL, USA, 2001; pp. 31–54. [Google Scholar]
- Bailey, H.H.; Levy, D.; Harris, L.S.; Schink, J.C.; Foss, F.; Beatty, P.; Wadler, S. A phase II trial of daily perillyl alcohol in patients with advanced ovarian cancer: Eastern Cooperative Oncology Group Study E2E96. Gynecol. Oncol. 2002, 85, 464–468. [Google Scholar]
- Liu, G.; Oettel, K.; Bailey, H.; Van Ummersen, L.; Tutsch, K.; Staab, M.J.; Horvath, D.; Alberti, D.; Arzoomanian, R.; Rezazadeh, H.; et al. Phase II trial of perillyl alcohol (NSC 641066) administered daily in patients with metastatic androgen independent prostate cancer. Investig. New Drugs 2003, 21, 367–372. [Google Scholar] [CrossRef]
- Meadows, S.M.; Mulkerin, D.; Berlin, J.; Bailey, H.; Kolesar, J.; Warren, D.; Thomas, J.P. Phase II Trial of Perillyl Alcohol in Patients with Metastatic Colorectal Cancer. J. Gastrointest. Cancer 2002, 32, 125–128. [Google Scholar] [CrossRef]
- Chen, T.C.; da Fonseca, C.O.; Schönthal, A.H. Intranasal perillyl alcohol for glioma therapy: Molecular mechanisms and clinical development. Int. J. Mol. Sci. 2018, 19, 3905. [Google Scholar] [CrossRef]
- Schönthal, A.H.; Peereboom, D.M.; Wagle, N.; Lai, R.; Mathew, A.J.; Hurth, K.M.; Simmon, V.F.; Howard, S.P.; Taylor, L.P.; Chow, F.; et al. Phase i trial of intranasal neo100, highly purified perillyl alcohol, in adult patients with recurrent glioblastoma. Neurooncol. Adv. 2021, 3, vdab005. [Google Scholar]
- Schönthal, A.H.; Swenson, S.; Bonney, P.A.; Wagle, N.; Simmon, V.F.; Mathew, A.J.; Hurth, K.M.; Chen, T.C. Detection of perillyl alcohol and its metabolite perillic acid in postsurgical glioblastoma tissue after intranasal administration of neo100: Illustrative case. J. Neurosurg. Case Lessons 2022, 4, CASE22215. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Barath, M.; Sharma, N.; Hofman, F.M.; Schönthal, A.H. A novel temozolomide-perillyl alcohol conjugate exhibits superior activity against breast cancer cells in vitro and intracranial triple-negative tumor growth in vivo. Mol. Cancer Ther. 2014, 13, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Nguyen, J.; Jhaveri, N.; Rosenstein-Sisson, R.; Hofman, F.M.; Schönthal, A.H. A novel temozolomide analog, neo212, with enhanced activity against mgmt-positive melanoma in vitro and in vivo. Cancer Lett. 2015, 358, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Wetzel, S.J.; Singh, A.; Nguyen, J.; Hofman, F.M.; Schönthal, A.H. Chemotherapeutic effect of a novel temozolomide analog on nasopharyngeal carcinoma in vitro and in vivo. J. Biomed. Sci. 2015, 22, 71–80. [Google Scholar] [CrossRef]
- Chen, T.C.; Minea, R.O.; Swenson, S.; Yang, Z.; Thein, T.Z.; Schonthal, A.H. Neo212, a perillyl alcohol-temozolomide conjugate, triggers macrophage differentiation of acute myeloid leukemia cells and blocks their tumorigenicity. Cancers 2022, 14, 6065. [Google Scholar] [CrossRef]
- Cho, H.Y.; Wang, W.; Jhaveri, N.; Lee, D.J.; Sharma, N.; Dubeau, L.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Neo212, temozolomide conjugated to perillyl alcohol, is a novel drug for effective treatment of a broad range of temozolomide-resistant gliomas. Mol. Cancer Ther. 2014, 13, 2004–2017. [Google Scholar] [CrossRef]
- Xie, L.; Song, X.; Guo, W.; Wang, X.; Wei, L.; Li, Y.; Lv, L.; Wang, W.; Chen, T.C.; Song, X. Therapeutic effect of tmz-poh on human nasopharyngeal carcinoma depends on reactive oxygen species accumulation. Oncotarget 2015, 7, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.T.; Chan, W.Y.; Chen, W.; Huang, H.M.; Wu, H.C.; Hsu, M.M.; Chuang, S.M.; Wang, C.C. Characterization of seven newly established nasopharyngeal carcinoma cell lines. Lab. Investig. 1993, 68, 716–727. [Google Scholar]
- Cheung, S.T.; Huang, D.P.; Hui, A.B.; Lo, K.W.; Ko, C.W.; Tsang, Y.S.; Wong, N.; Whitney, B.M.; Lee, J.C. Nasopharyngeal carcinoma cell line (c666-1) consistently harbouring epstein-barr virus. Int. J. Cancer 1999, 83, 121–126. [Google Scholar] [CrossRef]
- Silva-Hirschberg, C.; Hartman, H.; Stack, S.; Swenson, S.; Minea, R.O.; Davitz, M.A.; Chen, T.C.; Schönthal, A.H. Cytotoxic impact of a perillyl alcohol-temozolomide conjugate, neo212, on cutaneous t-cell lymphoma in vitro. Ther. Adv. Med. Oncol. 2019, 11, 1758835919891567. [Google Scholar] [CrossRef]
- Greco, W.R.; Bravo, G.; Parsons, J.C. The search for synergy: A critical review from a response surface perspective. Pharmacol. Rev. 1995, 47, 331–385. [Google Scholar] [PubMed]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of chop-induced apoptosis in er stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef]
- Gegotek, A.; Skrzydlewska, E. Ascorbic acid as antioxidant. Vitam. Horm. 2023, 121, 247–270. [Google Scholar]
- Wong, S.; Kirkland, J.L.; Schwanz, H.A.; Simmons, A.L.; Hamilton, J.A.; Corkey, B.E.; Guo, W. Effects of thiol antioxidant beta-mercaptoethanol on diet-induced obese mice. Life Sci. 2014, 107, 32–41. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: Ros as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Al-Mozaini, M.; Bodelon, G.; Karstegl, C.E.; Jin, B.; Al-Ahdal, M.; Farrell, P.J. Epstein-barr virus bart gene expression. J. Gen. Virol. 2009, 90, 307–316. [Google Scholar] [CrossRef]
- Lin, W.; Yip, Y.L.; Jia, L.; Deng, W.; Zheng, H.; Dai, W.; Ko, J.M.Y.; Lo, K.W.; Chung, G.T.Y.; Yip, K.Y.; et al. Establishment and characterization of new tumor xenografts and cancer cell lines from ebv-positive nasopharyngeal carcinoma. Nat. Commun. 2018, 9, 4663. [Google Scholar] [CrossRef]
- Ke, X.; Yang, Y.C.; Hong, S.L. Ebv-lmp1-targeted dnazyme restrains nasopharyngeal carcinoma growth in a mouse c666-1 xenograft model. Med. Oncol. 2011, 28 (Suppl. 1), S326–S332. [Google Scholar] [CrossRef]
- Fitzsimmons, L.; Kelly, G.L. Ebv and apoptosis: The viral master regulator of cell fate? Viruses 2017, 9, 339. [Google Scholar] [CrossRef]
- Messick, T.E.; Smith, G.R.; Soldan, S.S.; McDonnell, M.E.; Deakyne, J.S.; Malecka, K.A.; Tolvinski, L.; van den Heuvel, A.P.J.; Gu, B.W.; Cassel, J.A.; et al. Structure-based design of small-molecule inhibitors of ebna1 DNA binding blocks epstein-barr virus latent infection and tumor growth. Sci. Transl. Med. 2019, 11, eaau5612. [Google Scholar] [CrossRef]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of epstein-barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef]
- Altmann, M.; Pich, D.; Ruiss, R.; Wang, J.; Sugden, B.; Hammerschmidt, W. Transcriptional activation by ebv nuclear antigen 1 is essential for the expression of ebv’s transforming genes. Proc. Natl. Acad. Sci. USA 2006, 103, 14188–14193. [Google Scholar] [CrossRef]
- Tempera, I.; De Leo, A.; Kossenkov, A.V.; Cesaroni, M.; Song, H.; Dawany, N.; Showe, L.; Lu, F.; Wikramasinghe, P.; Lieberman, P.M. Identification of mef2b, ebf1, and il6r as direct gene targets of epstein-barr virus (ebv) nuclear antigen 1 critical for ebv-infected b-lymphocyte survival. J. Virol. 2016, 90, 345–355. [Google Scholar] [CrossRef]
- Taylor, G.M.; Raghuwanshi, S.K.; Rowe, D.T.; Wadowsky, R.M.; Rosendorff, A. Endoplasmic reticulum stress causes ebv lytic replication. Blood 2011, 118, 5528–5539. [Google Scholar] [CrossRef]
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes khsv and ebv lytic cycle by activating jnk and autophagy. Sci. Rep. 2017, 7, 13052. [Google Scholar] [CrossRef]
- Lee, J.; Kosowicz, J.G.; Hayward, S.D.; Desai, P.; Stone, J.; Lee, J.M.; Liu, J.O.; Ambinder, R.F. Pharmacologic activation of lytic epstein-barr virus gene expression without virion production. J. Virol. 2019, 93, e00998-19. [Google Scholar] [CrossRef]
- Hoji, A.; Xu, S.; Bilben, H.; Rowe, D.T. Calcium mobilization is responsible for thapsigargin induced epstein barr virus lytic reactivation in in vitro immortalized lymphoblstoid cell lines. Heliyon 2018, 4, e00917. [Google Scholar] [CrossRef]
- Wu, F.Y.; Chen, H.; Wang, S.E.; ApRhys, C.M.; Liao, G.; Fujimuro, M.; Farrell, C.J.; Huang, J.; Hayward, S.D.; Hayward, G.S. Ccaat/enhancer binding protein alpha interacts with zta and mediates zta-induced p21(cip-1) accumulation and g(1) cell cycle arrest during the epstein-barr virus lytic cycle. J. Virol. 2003, 77, 1481–1500. [Google Scholar] [CrossRef]
- Wu, F.Y.; Wang, S.E.; Chen, H.; Wang, L.; Hayward, S.D.; Hayward, G.S. Ccaat/enhancer binding protein alpha binds to the epstein-barr virus (ebv) zta protein through oligomeric interactions and contributes to cooperative transcriptional activation of the zta promoter through direct binding to the zii and ziiib motifs during induction of the ebv lytic cycle. J. Virol. 2004, 78, 4847–4865. [Google Scholar] [PubMed]
- Huang, J.; Liao, G.; Chen, H.; Wu, F.Y.; Hutt-Fletcher, L.; Hayward, G.S.; Hayward, S.D. Contribution of c/ebp proteins to epstein-barr virus lytic gene expression and replication in epithelial cells. J. Virol. 2006, 80, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Shirley, C.M.; Chen, J.; Shamay, M.; Li, H.; Zahnow, C.A.; Hayward, S.D.; Ambinder, R.F. Bortezomib induction of c/ebpbeta mediates epstein-barr virus lytic activation in burkitt lymphoma. Blood 2011, 117, 6297–6303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.C.; Dong, S.H.; Liu, Z.S.; Liu, S.; Zhang, C.C.; Liang, X.Z. Regulation of gammaherpesvirus lytic replication by endoplasmic reticulum stress-induced transcription factors atf4 and chop. J. Biol. Chem. 2018, 293, 2801–2814. [Google Scholar] [CrossRef]
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in ebv lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729. [Google Scholar] [CrossRef]
- Lassoued, S.; Gargouri, B.; El Feki Ael, F.; Attia, H.; Van Pelt, J. Transcription of the epstein-barr virus lytic cycle activator bzlf-1 during oxidative stress induction. Biol. Trace Elem. Res. 2010, 137, 13–22. [Google Scholar] [CrossRef]
- Zhao, L.; Xie, F.; Wang, T.T.; Liu, M.Y.; Li, J.L.; Shang, L.; Deng, Z.X.; Zhao, P.X.; Ma, X.M. Chlorpyrifos induces the expression of the epstein-barr virus lytic cycle activator bzlf-1 via reactive oxygen species. Oxid. Med. Cell Longev. 2015, 2015, 309125. [Google Scholar] [CrossRef]
- Huang, S.Y.; Fang, C.Y.; Wu, C.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Reactive oxygen species mediate epstein-barr virus reactivation by n-methyl-n’-nitro-n-nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [PubMed]
- Osipova-Goldberg, H.I.; Turchanowa, L.V.; Adler, B.; Pfeilschifter, J.M. H2O2 inhibits bcr-dependent immediate early induction of ebv genes in burkitt’s lymphoma cells. Free Radic. Biol. Med. 2009, 47, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Lam, B.H.; Ho, D.N.; Tsao, S.W.; Chiang, A.K. Bortezomib and saha synergistically induce ros-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of epstein-barr virus. Mol. Cancer Ther. 2013, 12, 747–758. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Xu, Q.; Holbrook, N.J. Induction of the mammalian stress response gene gadd153 by oxidative stress: Role of ap-1 element. Biochem. J. 1996, 314 Pt 2, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.R.; Nakamura, M.; Okura, T.; Takata, Y.; Watanabe, S.; Yang, Z.H.; Liu, J.; Kitami, Y.; Hiwada, K. Mechanism of oxidative stress-induced gadd153 gene expression in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2002, 290, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, L.; Huang, X.; Li, Y.; Zou, T.; Zhuo, X.; Chen, Y.; Liu, Y.; Tang, Y. Radiotherapy-induced dysphagia and its impact on quality of life in patients with nasopharyngeal carcinoma. Strahlenther. Onkol. 2019, 195, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, F.; Min, X.; Zhang, Q.; Shen, L.J.; Jiang, Y.; Yan, J. Toxicities of chemoradiotherapy and radiotherapy in nasopharyngeal carcinoma: An updated meta-analysis. J. Int. Med. Res. 2019, 47, 2832–2847. [Google Scholar] [CrossRef]
- Guan, S.; Wei, J.; Huang, L.; Wu, L. Chemotherapy and chemo-resistance in nasopharyngeal carcinoma. Eur. J. Med. Chem. 2020, 207, 112758. [Google Scholar] [CrossRef]
- Morales-Sanchez, A.; Fuentes-Panana, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef]
- Ameya, G.; Birri, D.J. The molecular mechanisms of virus-induced human cancers. Microb. Pathog. 2023, 183, 106292. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman-Houstman, H.; Swenson, S.; Minea, R.O.; Sinha, U.K.; Chiang, M.-F.; Chen, T.C.; Schönthal, A.H. Activation of Epstein–Barr Virus’ Lytic Cycle in Nasopharyngeal Carcinoma Cells by NEO212, a Conjugate of Perillyl Alcohol and Temozolomide. Cancers 2024, 16, 936. https://doi.org/10.3390/cancers16050936
Hartman-Houstman H, Swenson S, Minea RO, Sinha UK, Chiang M-F, Chen TC, Schönthal AH. Activation of Epstein–Barr Virus’ Lytic Cycle in Nasopharyngeal Carcinoma Cells by NEO212, a Conjugate of Perillyl Alcohol and Temozolomide. Cancers. 2024; 16(5):936. https://doi.org/10.3390/cancers16050936
Chicago/Turabian StyleHartman-Houstman, Hannah, Steve Swenson, Radu O. Minea, Uttam K. Sinha, Ming-Fu Chiang, Thomas C. Chen, and Axel H. Schönthal. 2024. "Activation of Epstein–Barr Virus’ Lytic Cycle in Nasopharyngeal Carcinoma Cells by NEO212, a Conjugate of Perillyl Alcohol and Temozolomide" Cancers 16, no. 5: 936. https://doi.org/10.3390/cancers16050936
APA StyleHartman-Houstman, H., Swenson, S., Minea, R. O., Sinha, U. K., Chiang, M.-F., Chen, T. C., & Schönthal, A. H. (2024). Activation of Epstein–Barr Virus’ Lytic Cycle in Nasopharyngeal Carcinoma Cells by NEO212, a Conjugate of Perillyl Alcohol and Temozolomide. Cancers, 16(5), 936. https://doi.org/10.3390/cancers16050936