1. Introduction
Among the extensively studied epigenetic modifications, DNA methylation is considered the most somatically heritable and stable mark [
1,
2]. The significance of DNA methylation in governing gene expression through epigenetic mechanisms is well-documented. Nevertheless, methylation also plays a pivotal role in numerous other scenarios, including X-chromosome inactivation, maintaining genomic stability, and regulating genomic imprinting [
3,
4]. Across this extensive array of functions, DNA methylation imparts an additional level of plasticity and dynamicity to genetic regulation, while still allowing for the stable transmission of a specific epigenotype during the process of cell replication. Given that the fundamental DNA sequence remains nearly uniform in all mammalian cells, the epigenetic profile is critical in moulding distinct cell lineages, making a substantial contribution to the diverse phenotypes observed [
5]. Disruptions in the regulation of DNA methylation are associated with the pathogenesis of several diseases, especially cancer. Changes in DNA methylation, such as genome-wide hypomethylation and promoter hypermethylation of tumour-suppressor genes (TSGs), contribute to tumour development and progression [
6,
7,
8]. This scope of global hypomethylation and promoter hypermethylation intensifies as the tumour progresses to metastasis, further amplifying the bimodal characteristics of the tumour DNA methylome [
9]. Growing evidence from recent studies suggests a further defined role for site-specific DNA methylation changes acting as “epigenetic drivers” and potential markers in different cancer types that enable cancer growth [
10,
11,
12,
13].
Transcriptional regulation of a specific gene is influenced by DNA methylation, which modulates the recruitment of methyl CpG binding domain (MBD) proteins and transcriptional repressors. Conventionally, the lack of DNA methylation in gene promoter regions rich in CpG sites is linked to an ideal chromatin organisation that promotes active gene transcription. DNA hypermethylation of TSGs such as
CDKN2A and
CDKN2B has been demonstrated to result in their transcriptional silencing across various cancers [
14]. A high level of methylation results in a condensed heterochromatin structure, leading to the subsequent inhibition of transcription [
2,
15,
16]. The silencing of TSGs by hypermethylation contributes to cancer initiation, progression, and metastasis [
17]. Conversely, global hypomethylation is recognised for its role in promoting gene expression changes, providing cancer cells with a selective advantage. This phenomenon affects genomic domains and contributes to genomic instability in cancer cells [
18]. These fundamental concepts form the foundation of our current comprehension of how DNA methylation relates to gene expression. This mechanism of transcriptional silencing has been extensively investigated and is supported by a significant body of evidence accumulated over recent decades. While substantial evidence supports the classical paradigm, we and others have now established that promoter DNA methylation could be a mechanism of the paradoxical activation of genes in different contexts, such as metastasis and development [
10,
19,
20]. In these scenarios, instances of active transcription from hypermethylated gene promoters have been documented, indicating an alternative role for DNA methylation as a facilitator of transcription. This paradoxical activity may be caused due to the binding of repressive transcription factors, distal element interactions, or alternative promoter expression. These possible molecular mechanisms of gene activation have been discussed in detail in our previous work [
19].
Although not extensively studied,
EBF3 is known to play roles in the differentiation and migration of various cell types.
EBF3 is primarily known for its critical role in B-cell development; it acts as a master regulator in the commitment of progenitor cells to the B-cell lineage [
21]. Dysregulation of
EBF3 expression can have significant consequences, leading to developmental abnormalities or disease conditions, particularly in cases where EBF3 acts as a master regulator, such as in B-cell development and neuronal differentiation. Moreover, it has been detected as a potential tumour suppressor in brain, breast, colorectal, gastric, liver, bone tumours, and acute myeloid leukaemia [
10,
21,
22].
EBF3 is often classically shown to be downregulated or mutated in certain malignancies, suggesting a potential tumour suppressor role [
21]. Contrary to earlier findings, we identified
EBF3 as a putative epigenetic driver of metastasis in melanoma [
10] and other cancer types [
23], displaying the phenomenon of hypermethylation causing gene activation. Thus,
EBF3 in the context of melanoma presents a favourable framework to establish the mechanism of paradoxical gene expression. Hence, the work described in this study focuses on investigating
EBF3 as an example.
Despite the remarkable discoveries made in the field of DNA methylation and gene expression, it is important to note their limitations regarding the lack of validation of direct causality between promoter DNA methylation and ensuing gene expression. To advance and evaluate the viability of a novel concept, it is crucial to investigate this causality directly [
24]. The primary limitation of previous studies lies in their broad approach to manipulating DNA methylation. DNA methylation inhibitors, due to their genome-wide action, cannot conclusively establish a locus-specific elevation in promoter methylation leading directly to an increase in gene expression. However, in recent years, there has been remarkable progress in developing epigenetic editing technologies, resulting in a diverse range of tools that enable comprehensive exploration of tumour biology [
25]. One particularly noteworthy advancement is the transformative impact of CRISPR-based methods on the realm of epigenetic editing. These techniques offer a revolutionary way to manipulate gene regulation precisely, creating opportunities to replicate these alterations in the context of cancer and metastasis [
25], with a level of precision that was previously unattainable.
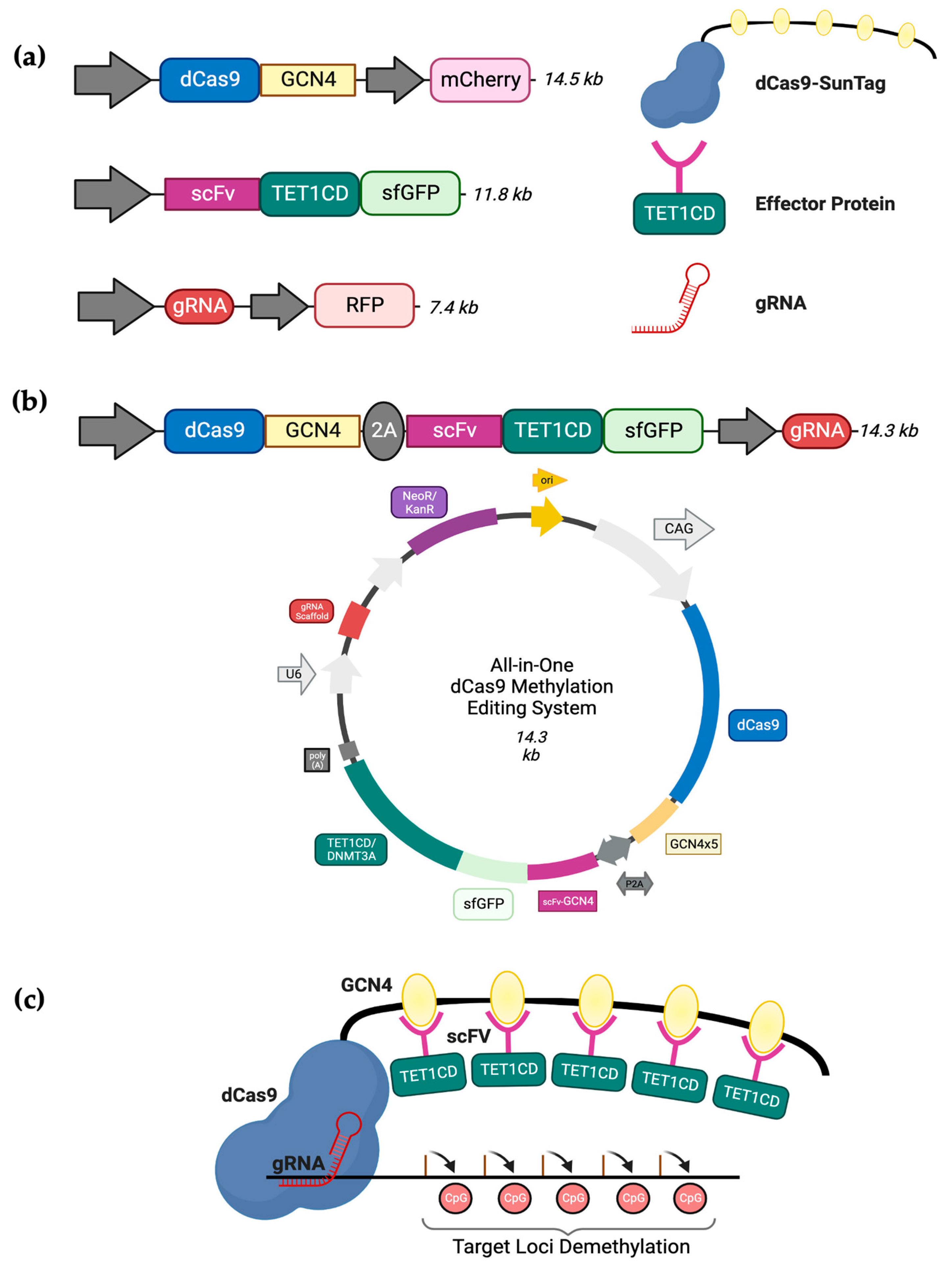
Based on the findings from our previous work [
26], attempting the concurrent transient transfection of three substantial plasmids is a challenging and time-consuming task. The primary objective of these transfection experiments was to generate enough cells for subsequent gene expression investigations and more extensive chromatin analysis. As a result, we suggest a novel approach: substituting the existing triple transfection method with a singular ‘All-in-one’ plasmid capable of expressing all three essential components of the dCas9-SunTag system. Thus, to enhance the efficiency of targeted DNA methylation editing, we obtained the All-in-one dCas9-CRISPR system described by Morita et al. [
27]. This plasmid includes the dCas9-SunTag, the TET1CD-scFv effector protein, a GFP tag, and a cloning site for gRNAs, all cloned into one single plasmid (
Figure 1). Similar to the three-component system [
28], the dCas9 enables the RNA-guided binding of our CRISPR All-in-one methylation editing system to a specific target site without altering any DNA sequence. Additionally, the SunTag component furnishes a repetitive epitope-based framework that can secure multiple instances of our effector construct using short-chain variable fragment (scFv) domains. This interaction of the dCas9-SunTag with a particular genomic locus is guided by a distinct gRNA construct. For this project, we utilised the construct pPlatTET-gRNA2 (Addgene #82559) for demethylation and cloned our unique gRNA_472-sequence. Similarly, for targeted methylation, we replaced the
TET1 catalytic domain with
DNMT3A using Gibson cloning, a reliable method described by Gibson et al. [
29] that utilises exonucleases to assemble DNA consistently and accurately in the correct sequence. Here, we demonstrate the successful delivery and efficiency of our All-in-one dCas9 CRISPR system to induce targeted DNA methylation changes in
EBF3 and establish paradoxical gene activation. Using subsequent gene expression analysis, we provide novel clues about the function of
EBF3 in cancer cells.
The results of this study suggest that specific modifications to the DNA methylome, especially in regions that govern gene expression, could have a significant impact on modifying the gene expression patterns crucial for metastasis. This hypothesis posits that the adaptable nature of epigenetic modifications, including DNA methylation, makes it easier for tumours to acquire the specific traits needed for successful metastasis [
8,
10].
2. Materials and Methods
2.1. Cell Culture
The CM150-post cell line is a patient-derived line, and it was cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen, Waltham, MA, USA) with the addition of 10% foetal calf serum (FCS) and 1% penicillin-streptomycin (Gibco, Grand Island, NY, USA). WM266-4 cell line, sourced from American Type Culture Collection (ATCC. CRL-1676
TM), was cultured in Minimum Essential Media Alpha (MEM a) (Invitrogen), supplemented with 1% penicillin-streptomycin (Gibco, NY, USA) and 10% foetal calf serum (FCS). These cell lines were grown in filter-capped cell culture flasks under standard conditions, maintained at 37 °C in a humidified atmosphere with 5% CO
2 and 21% O
2, as recommended. We have described the DNA methylomes of these cell lines in our previous works [
26,
30].
A standard protocol was employed to culture adherent human melanoma cell lines for all the cell lines used in this study. To initiate the culturing process, a frozen vial of 2 × 106 cells for each cell line was thawed from liquid nitrogen storage and seeded into a 75 cm2 filter-cap adherent tissue culture flask (Greiner Bio One, Monroe, NC, USA) with 14 mL of the corresponding culture medium. The cells were grown until they reached over 80% confluency (i.e., 5 × 104 cells/ cm2 before being trypsinised). The cells were then cultured in 175 cm2 filter-cap adherent tissue culture flasks (Greiner Bio One) with 23 mL of culture medium for experimental purposes. The time required to achieve 80% confluency in 175 cm2 flasks varied among cell lines and ranged from three to five days. When the cells achieved full confluence, they were prepared for transfection experiments, frozen for extended preservation, or collected for DNA/RNA extraction. The cells were monitored daily under a microscope to assess their morphology and condition and to ensure their viability at each stage of the cell culture process.
2.2. Plasmid DNA Isolation
In this study, we utilised the available construct pPlatTET-gRNA2 (Addgene #82559) for demethylation and cloned our respective gRNA_472-. The list of primer sequences used for inserting gRNAs and catalytic domain is provided in
Table S4. Similarly, for targeted methylation, we replaced the TET1 catalytic domain with DNMT3A. This was achieved using Gibson cloning, a reliable method described by Gibson et al. [
29] that utilises exonucleases to assemble DNA consistently and accurately in the correct sequence. The process was performed at a constant temperature with the aid of three enzymes: a 5′ exonuclease to create extensive overhangs, a polymerase to complete the gaps in the single-stranded regions, and a DNA ligase to join the ends of the annealed and filled gaps [
29].
Achieving optimal transfection efficiency and cell viability relies heavily on extracting high-quality plasmid DNA with minimal contamination of bacterial endotoxins. The plasmid DNA for this project was isolated from DH5α E. coli cells. To obtain bacterial colonies for plasmid DNA isolation, we streaked DH5α glycerol stocks onto LB agar plates supplemented with 100 μg/mL ampicillin and incubated at 37 °C overnight. The next day, individual colonies were selected and cultured in 5 mL of LB broth containing 100 μg/mL ampicillin, shaken at 200 rpm, and maintained at 37 °C for 6 h. Following this, the culture was introduced into a larger volume of 400 mL, supplemented with 100 μg/mL ampicillin, and incubated overnight at 37 °C with agitation at 200 rpm. The resulting 400 mL overnight culture was then subjected to purification using the GenCatchTM Plasmid DNA Maxi Prep Kit (Epoch Life Science, Missouri City, TX, USA) in accordance with the manufacturer’s guidelines. To determine the quantity of plasmid DNA, the Nanophotometer N120 was utilised.
2.3. Transient Delivery of the All-in-One System
This non-liposomal mode of transient transfection was performed using FuGENE HD (Promega, Madison, WI, USA) reagent [
31]. Around 5 × 10
6 cells were initially seeded for this transfection in each 10 cm plate. On the day of transfection, 600 μL of Opti-MEM Serum Free Medium (Invitrogen) was added to a 1.5 mL tube. Then, 1 μg of the pPlatTET/DNMTA plasmid DNA was added and vortexed. Following this, 18 μL of FuGENE HD was added to the tube and immediately vortexed. The mixture was left to incubate at ambient room temperature for 10 min. Subsequently, the reagent mixture was cautiously added drop by drop to the cell plate and placed in a 37 °C incubator with 5% CO
2 for a duration of 48 h. After the 48 h incubation period, the cell plates were examined using a microscope to observe GFP fluorescence and were then prepared for subsequent FACS analysis.
2.4. FACS Preparation and Sorting
The samples for FACS analysis were prepared based on previously published protocols from our lab [
26,
30]. Briefly, post-72 h of transfection, the cells were treated with trypsin and suspended in 250 μL of sterile auto MACS buffer containing 1 × DPBS, 1% FCS, and 2 mM EDTA. Before flow sorting, the cells were also treated with the LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit. BD FACS Aria Fusion was utilised to achieve isolation of the true cell population. For each transfection, the cells were sorted based on the presence of the transfected All-in-one system, which was identified using a specific fluorescent marker (GFP in this case).
For each cell line and plasmid transfection, both negative and positive control samples were incorporated. Following that, we employed live-dead stain gating in the experiments (utilising FSC-A and APC-Cy7-A) to separate the live cell population in the negative control sample. Initially, cells were gated based on their side scatter (SSC) and forward scatter (FSC) to exclude debris, if any. The FSC measurement allows for discrimination of cells based on size. As such, the dead or apoptotic cells, which are smaller in size, can be easily eliminated. Conversely, the SSC allows for distinguishing cells based on their complexity (i.e., granularity). Following that, gating the live-dead stain used in the experiments (FSC-A and APC-Cy7-A) was utilised to gate the cells for the negative control sample and isolate the live cell population. Subsequently, the live cell population was gated based on the presence of each of the three fluorophores in their respective channels. Following this, we further isolated the true cell population based on the presence of sfGFP in the FITC channel. A total number of 27,500 and 78,190 cells were acquired using plasmids pPlatDNMT3A_gRNA_472- and pPlatTET_gRNA_472-.
FACS data analysis was conducted utilising FlowJo 10.4.2 software (FlowJo, LLC, Vancouver, BC, USA). The data, saved as .fcs files, were uploaded to the FlowJo platform. Consistent gating and compensation settings were applied to all melanoma cell lines during the analysis in FlowJo, allowing for the evaluation of cell viability and efficiency of transfection in each FACS result.
2.5. Assessment of Targeted DNA Methylation Editing
Elaborate methods for next-generation and specific DNA methylation analysis techniques have already been documented in our prior publications [
26,
32]. Consequently, this publication will not delve into further specifics regarding these methodologies.
2.5.1. Sample Preparation for Illumina Mini-Sequencing
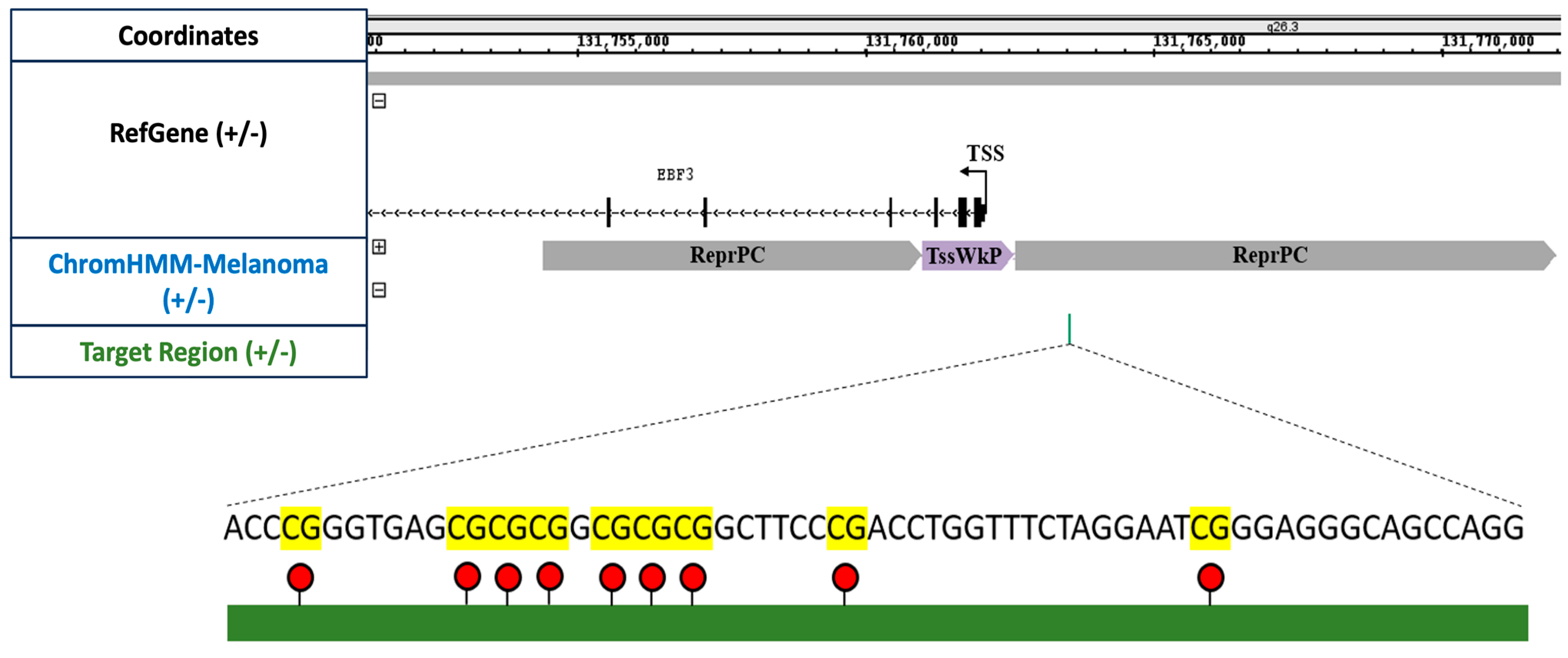
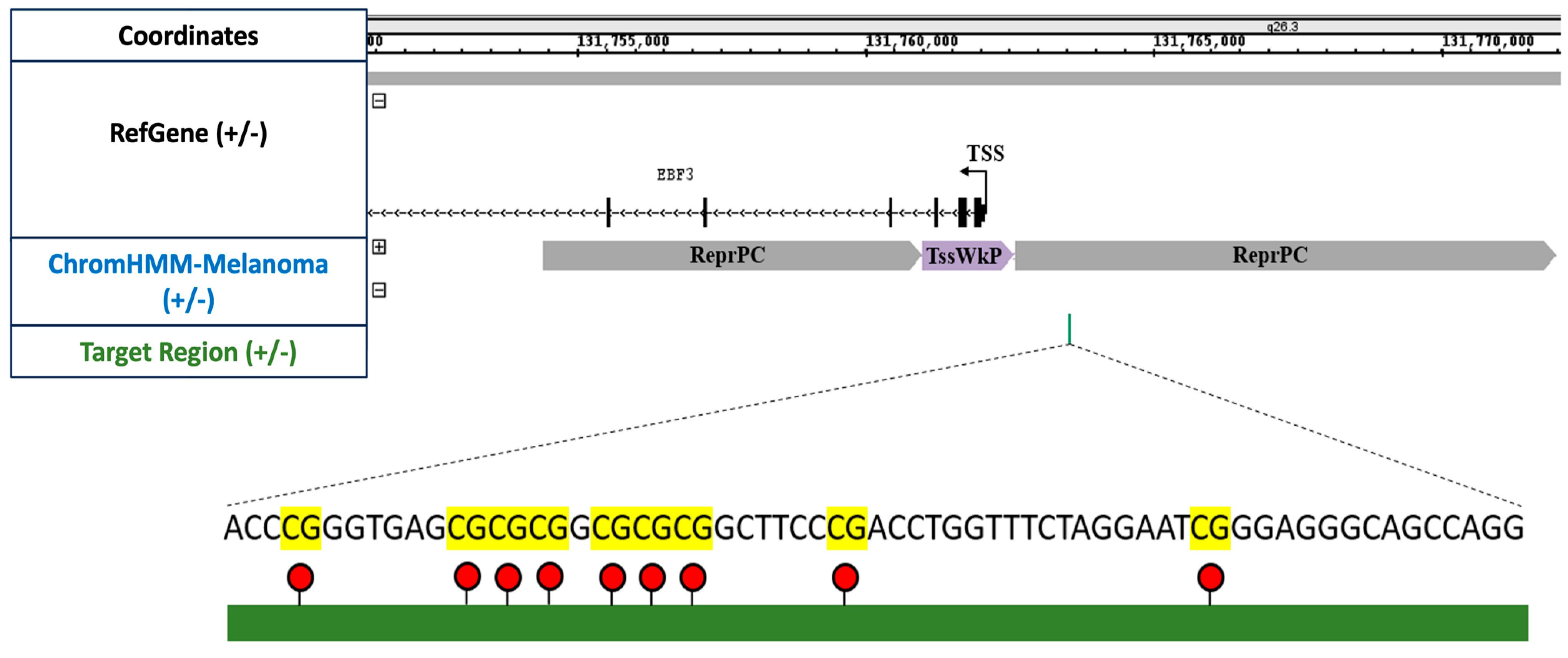
We employed the EZ DNA Methylation-Direct Kit from (Zymo Research, Orange, CA, USA) for DNA extraction and bisulfite conversion. This column-based kit employs cell lysis and bisulfite conversion, eliminating the necessity for DNA isolation, and is specifically designed for processing small cell quantities, thereby enhancing yield from a low cell input. Further, for amplicon-specific PCR, we amplified a 285 bp segment of the EBF3 promoter region using KAPA HiFi DNA Polymerase (2X Ready Mix) in a 10 μL reaction volume. The primer pairs EBF3_993_F and EBF3_993_R were utilised in a touch-down PCR approach. After PCR amplification, the presence of the product was validated by visualising it on a 2% agarose gel.
The amplified product from each sample was subsequently subjected to purification through the use of AMPure XP magnetic beads. Following the purification process, the concentration of each sample was determined using the Qubit High Sensitivity 1X dsDNA Kit (Invitrogen). Based on the quantification results, each sample was subsequently diluted to achieve a concentration of 1 ng/μL.
To enable multiplexing and demultiplexing of many samples, it is necessary to identify sequencing reads that pertain to each individual sample. A second round of PCR was therefore performed to index each sample with a unique combination of Illumina forward and reverse adaptor index sequences. Following indexing, the samples can be grouped together (multiplexed) into a unified library for sequencing. To create this library, 3 μL of each index PCR product was combined and subsequently purified again using the Qubit High Sensitivity 1X dsDNA Kit from Invitrogen. The library’s quality, including the presence of primers or non-specific products, was assessed using the Agilent BioAnalyser system and the BioAnalyser 2100 High Sensitivity DNA Assay.
2.5.2. Illumina Sequencing and Data Analysis
For sequencing, a total of four samples were used. These samples included unedited and edited cells for each cell line in triplicate. Approximately 2500 cells were initially used for bisulfite conversion, which was subsequently amplified for library preparation. Mean DNA methylation levels for both the target region (nine CpG sites, 58 bp long) and the complete EBF3 amplicon (34 CpG sites, 285 bp long) were analysed. The results were also generated using three replicates for each of the samples. The relative DNA methylation change was calculated using the formula mentioned in our previous publication [
26].
To analyse the raw Mini-Seq sequencing data for DNA methylation analysis, multiple bioinformatic programmes are required. The method explained in this section relies on an existing UNIX-based bioinformatic framework described previously [
32]. Briefly, for each identified sample, two read files are produced, R1 and R2. These pair-ended sequencing reads are joined using PEAR (Paired-End reAd mergeR) to create full-length reads [
33]. The PEAR programme uses quality scores and statistical analysis to merge reads [
33]. The combined sequencing reads are subjected to a quality control check using FastQC (Babraham Bioinformatics, Babraham, UK) to identify and eliminate sequencing data with low-quality output from further analysis. To eliminate Illumina adaptor index sequences, Trim Galore! (Version 0.5.0, Babraham Bioinformatics) was employed. Each read’s alignment and methylation data were subsequently processed using the BiQ Analyser HT package [
34]. The results were analysed and presented graphically with GraphPad Prism 9 (v.9.0.1). These data were also statistically analysed using an unpaired
t-test to determine the mean difference in relation to the standard error between the two groups.
2.6. Sample Preparation for Gene Expression Assessment
2.6.1. RNA Isolation
RNA was extracted from approximately 1000 cells following FACS sorting, utilising the RNAeasy Mini Kit from Qiagen. Initially, the cells were lysed in 350 μL of RLT lysis buffer, and then, 350 μL of 70% ethanol was added. The mixture of RLT and ethanol was thoroughly mixed before being transferred to an RNA spin column and subjected to centrifugation for 30 s at 8000× g. Subsequently, the eluate was discarded, and 700 μL of RW1 buffer was introduced to the RNA spin column, followed by a 15 s centrifugation at 8000× g. The column then underwent two additional washes, each with 500 μL of RPE buffer, at 8000× g. Afterwards, the column was centrifuged at maximum speed for two minutes to ensure complete membrane drying. Finally, 30 μL of RNAse-free water was applied to the column for RNA extraction, and the extracted RNA was collected in a fresh 1.5 mL Eppendorf tube by centrifugation at 8000× g for one minute. To determine the RNA concentration, the QubitTM RNA High Sensitivity Assay Kit (Invitrogen) was utilised with the QubitTM Fluorometer 4 (Invitrogen).
2.6.2. qRT-PCR
Approximately 10 ng of RNA was converted to cDNA in a 30 μL reaction using qScript XLT cDNA Supermix Kit (Quantabio, Beverly, MA, USA) in a thermocycler. Single-stranded cDNA of 1 ng/μL was used for qRT-PCR. The SYBR Premix Ex (TaKaRa, Shiga, Japan, SYBR Premix Ex Taq) was used to prepare reactions in 20 μL volumes, and dispensed into LightCycler 480 Multiwell Plate 96-well plates. Each sample was run in triplicate as technical replicates for every experiment. The reference genes chosen for this study were RPL32 and SRP14. These reference genes were selected based on previous data from our lab group studying EBF3 expression in melanoma cells [
10]. Real-time PCR reactions were run on the LightCycler 480 (Roche, Vienna, Austria). After acquiring the C
t values for each replicate, the values were normalised to the reference genes, RPL32 and SRP14, using the 2
−ΔΔCt method [
35]. Subsequently, the results were analysed and presented graphically with GraphPad Prism 9 (v.9.0.1). This data was analysed using an unpaired
t-test to determine the mean difference in relation to the standard error between the two groups.
2.7. RNA Sequencing Analysis
2.7.1. RNA-Seq Alignment and Differential Expression Analyses
Approximately 150 ng/μL of unedited RNA and 10 ng/μL of edited RNA samples were sequenced on Illumina NextSeq 500 (AgResearch, Aotearoa, New Zealand) with 150 bp paired ends. Given the limited number of cells obtained from FACS analysis, RNA-Seq was performed without any replicates (n = 1). We have followed our previously published protocols and pipelines for RNA extraction, library preparation, and data analysis [
36,
37,
38,
39]. After sequencing, raw reads were assessed using FASTQC and MultiQC. The reads were then aligned to the human genome (GRCh37) with STAR [
40]. Next, the read counting was performed using the package Subread (version 2.0.3-GCC-10.3.0 D) and function feature count. The data were subsequently analysed for differentially expressed genes (DEGs) with Edge R (version 3.40.2) [
41] in R studio (version.2022.07.1+554), with a False Discovery Rate (FDR) < 0.05.
2.7.2. Pathway Analyses
Differentially expressed genes common between the two melanoma cell lines were further analysed for pathway enrichment using Metascape [
42]. Additionally, the ENRICHR [
43] online platform was used to identify the histone and transcription factor enrichment of the common genes with several databases: Epigenomics roadmap histone modification ChIP-Seq, TRANSFAC/JASPAR transcription factor binding profiles, and ENCODE/ChEA consensus target genes. This analysis aimed to identify and elucidate the significance of enriched histone modifications and transcription factors associated with the differentially expressed common genes in our study.
2.8. CUT&RUN Experiments
About 2500 cells were counted for CM150-post and WM266-4 from FACS-sorted cells for each antibody (IgG, HEK27Me3 and H3K27Ac), with 500,000 cells counted for the baseline. CUT&RUN experiments were conducted following the manufacturer’s protocol with slight modifications (Epicypher, Durham, NC, USA) [
44]. Eluted DNA libraries were prepared using the NEBNext UltraTM II DNA Library Prep Kit for Illumina (NEB #E7645S/L). Replicated sequencing libraries (n = 2) were sent to Genohub (Austin, TX, USA) for sequencing. A total of 35 uniquely barcoded libraries from the CUT and RUN protocol were pooled, and 150 bp paired-end sequencing was performed on the Novaseq X Plus sequencer.
4. Discussion
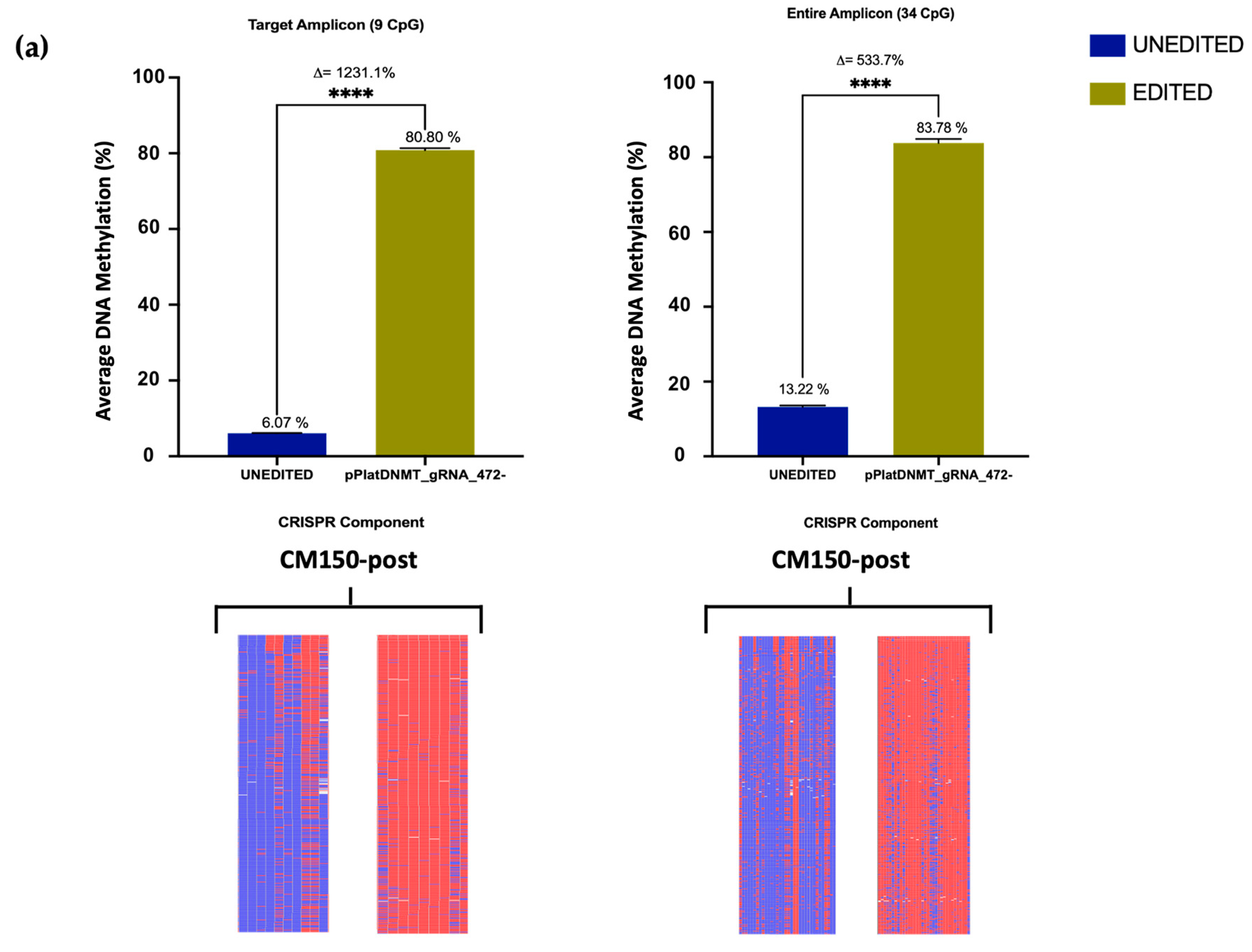
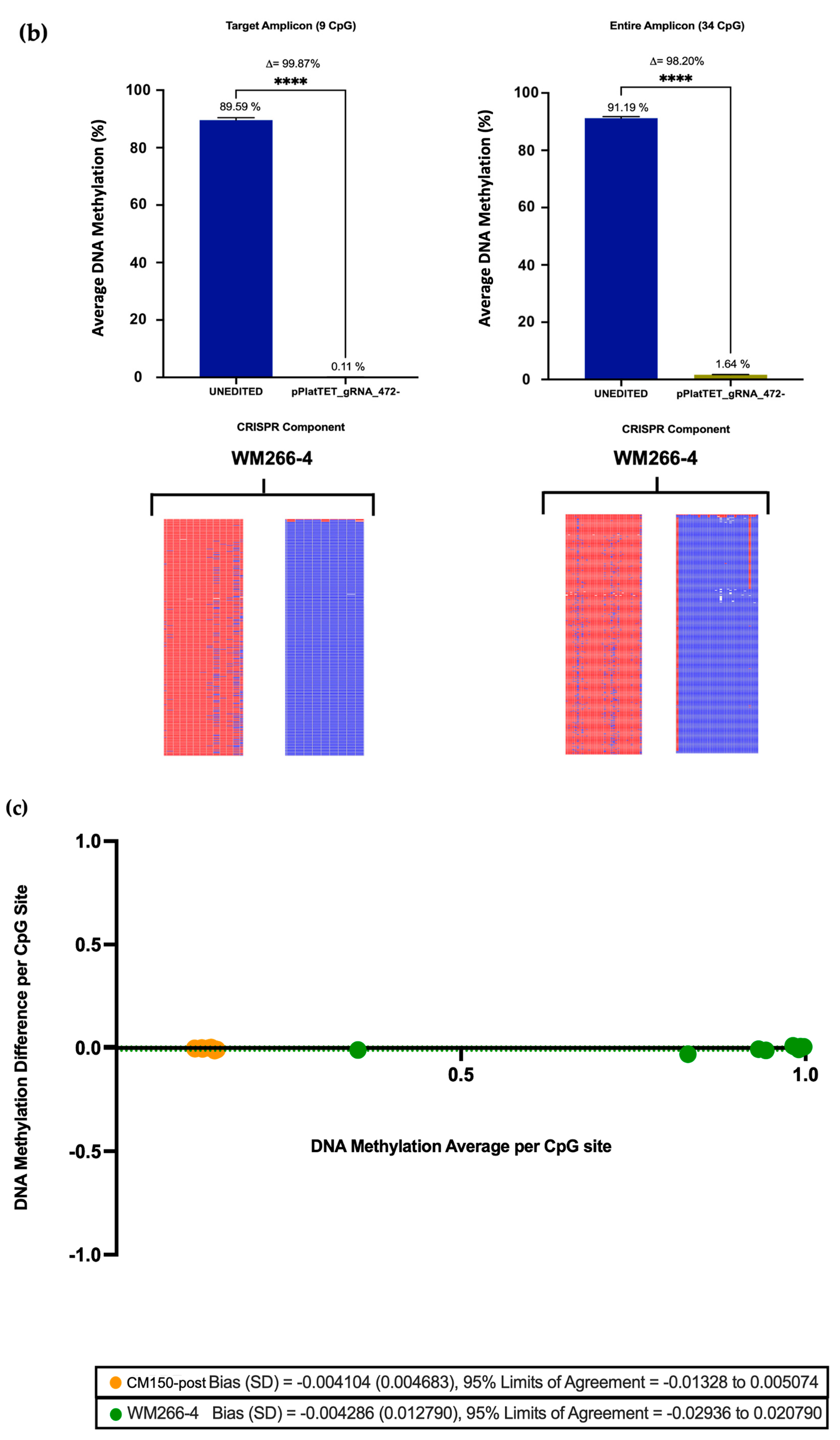
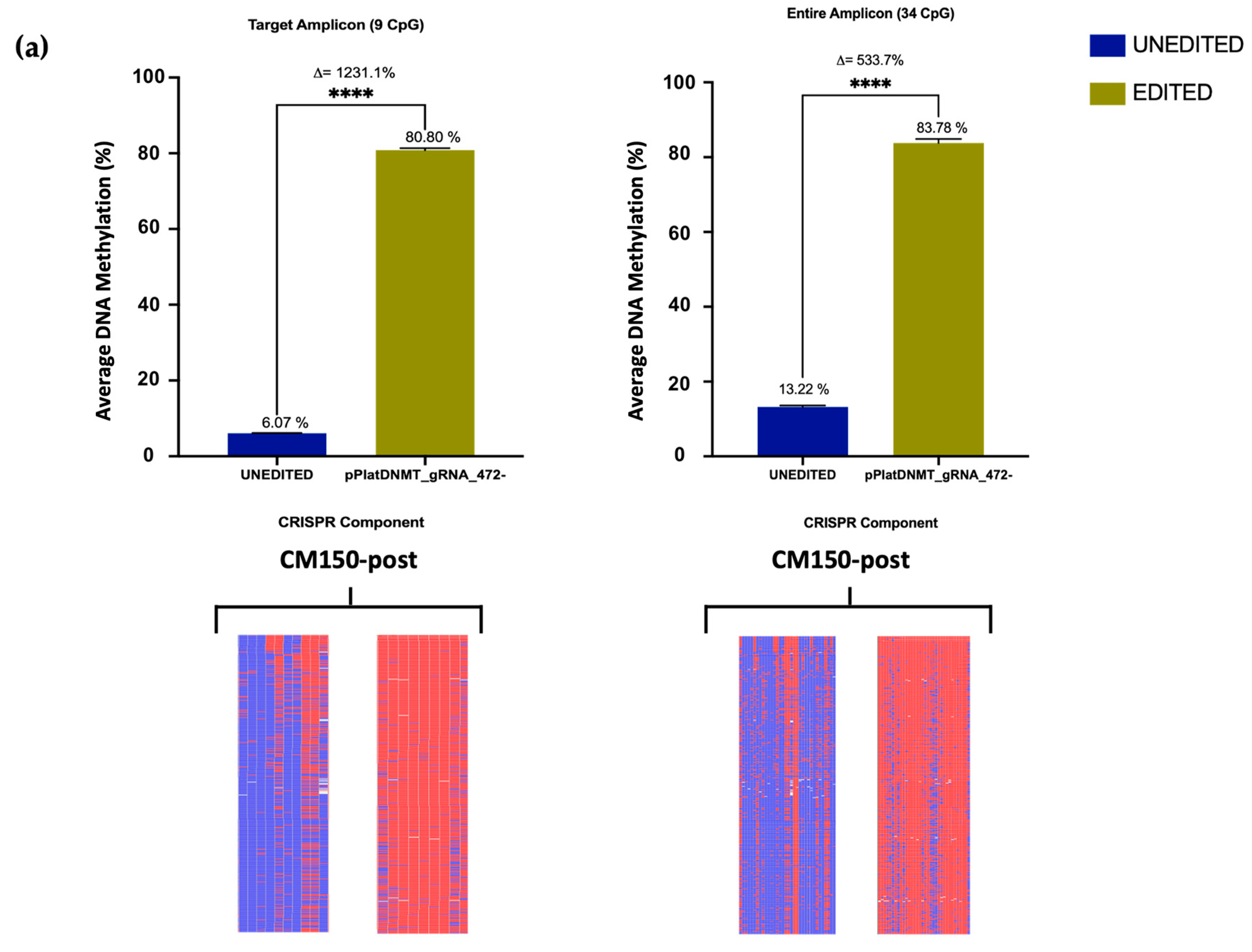
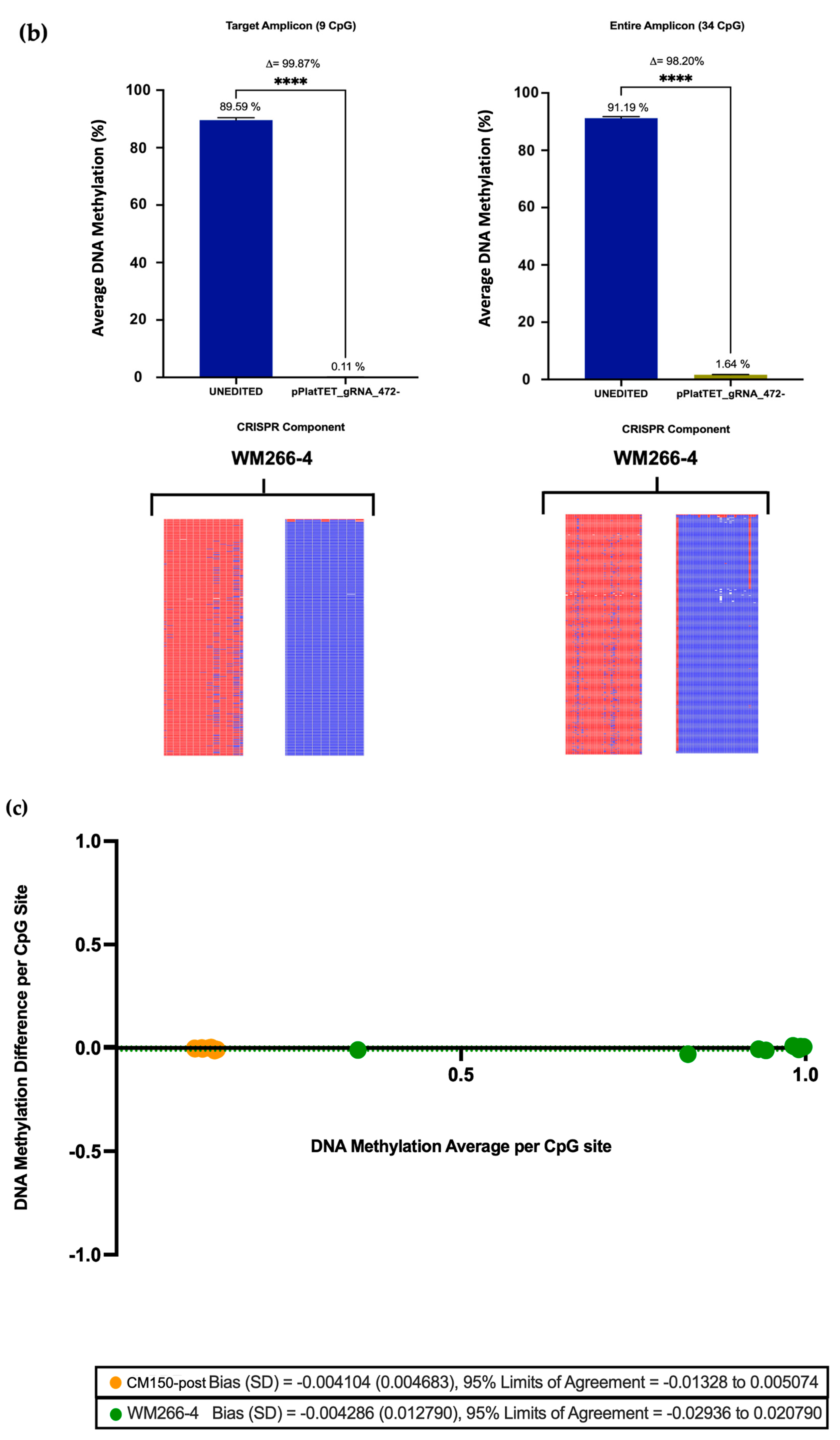
Previous editing of our target
EBF3 locus using a dCas9-SunTag-scFvDNMT3A/TET1CD system was able to induce notable methylation changes in three melanoma cell lines [
26]. However, using the All-in-one system here, editing efficiency was significantly augmented, with an average methylation increase of approximately 80% in the CM150-post cell line (
Section 3.3), rising from the initial 13.22%. Conversely, nearly complete demethylation was achieved in the WM266-4 cell line (
Section 3.3), reducing the methylation level to a mere 0.11% from the initial 91.2%, respectively. A previous study utilised the dCas9-SunTag and scFv-TET1CD and indicated successful induction of targeted DNA demethylation of the
Fgf21 promoter in hepatoma both in vitro and in vivo [
48]. Moreover, Josipović et al. utilised a TET1-dCas9 construct similar to ours and demonstrated precise DNA demethylation at specific target sites in ovarian cancer cell lines [
49]. Taken together, the significant methylation changes observed confirm the efficacy of the CRISPR All-in-one editing system.
Recent studies, including our own, have demonstrated a paradoxical role of DNA methylation in transcriptional regulation [
10,
19]. In certain scenarios, promoter methylation appears to directly activate gene expression. This is supported by instances of significant gene expression levels, even when there is noticeable promoter hypermethylation. A prior study involving melanoma cell lines demonstrated a decrease in the expression of the
EBF3 gene [
10] upon decitabine (DNA methylating inhibitor) treatment. However, decitabine induces global changes in methylation patterns, making it non-specific and incapable of establishing a direct cause-and-effect relationship. To offer concrete proof of gene activation driven by methylation, we opted for a precise methylation editing approach using the All-in-one system. Several early publications provide evidence for gene activation induced by hypermethylation. Early observations in the scientific literature focused on the hTERT gene, which has been widely believed to have a significant impact on telomerase activation in various cancer types [
50]. Numerous studies, including those conducted on colorectal cancer, HPV-induced carcinogenesis, and brain tumours, have established a positive association between promoter hypermethylation of hTERT and enhanced transcriptional activity [
50,
51,
52]. A recent study on prostate cancer also identified a positive corelation between hypermethylation and enhanced gene expression when evaluating the downstream regulatory impact of DNA methylation [
53]. Additionally, gene body CGI hypermethylation was shown to serve as a predictive indicator for increased levels of oncogenes in hepatocellular carcinoma (HCC) mouse models [
54]. Furthermore, Unoki and Nakamura [
55] showed that increased methylation in the intron 1 of the tumour suppressor
EGR2, as opposed to the promoter region, led to gene activation resembling that of an enhancer. Additionally, De Larco et al. [
56] discovered a pair of hypermethylated CpG sites located upstream of the
IL8 promoter, serving as a transcriptional activator linked to the shift toward metastasis in breast carcinoma cell lines. Similarly, findings from a recent study demonstrated that intragenic hypermethylation of MMP9 is linked to the overexpression of MMP-9, thereby contributing to the development and progression of cancer [
57]. Alongside these examples, our findings challenge the long-standing notion of DNA methylation as an exclusively gene-silencing mechanism. In alignment with our hypothesis, we noted enhanced
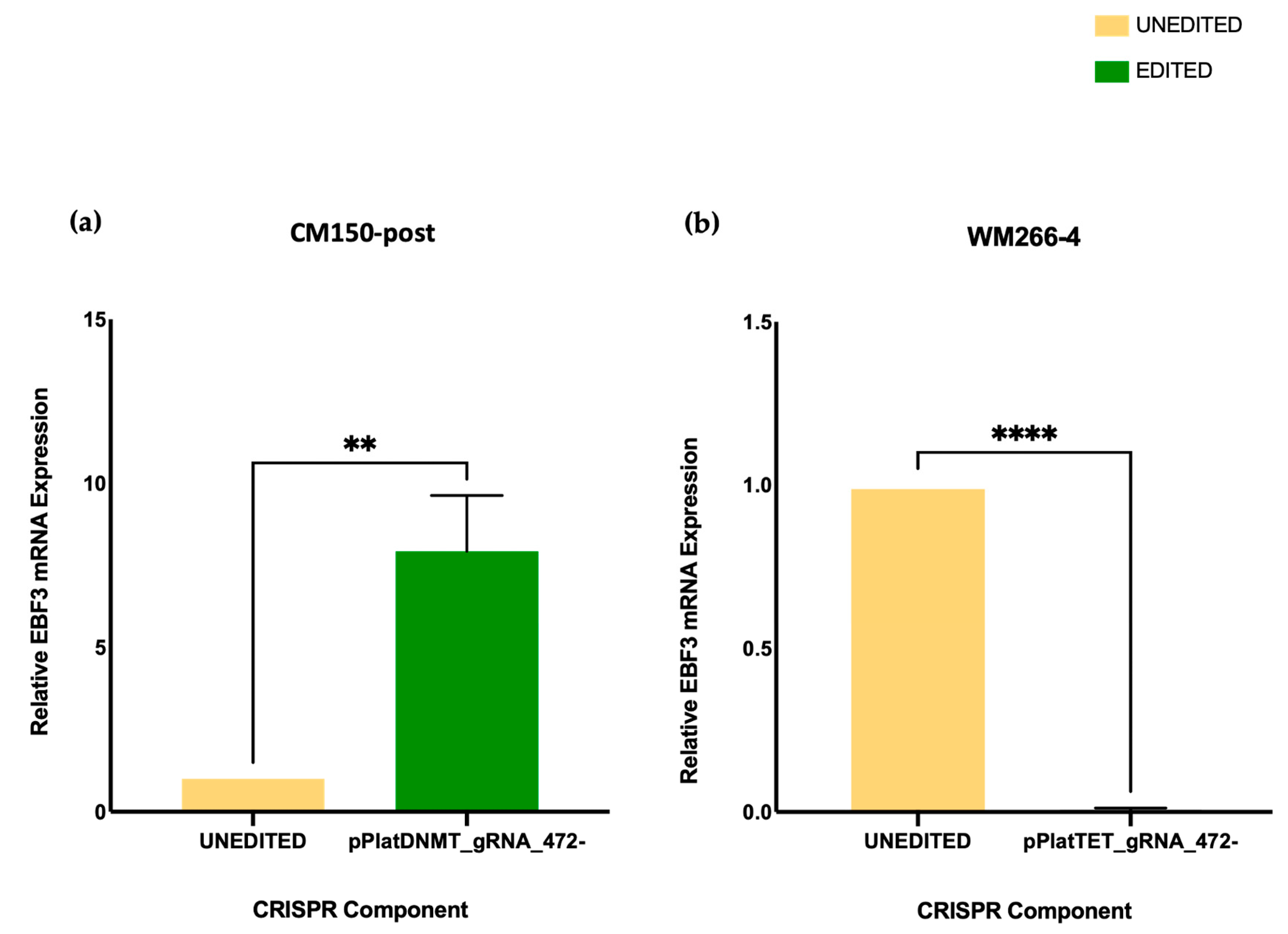
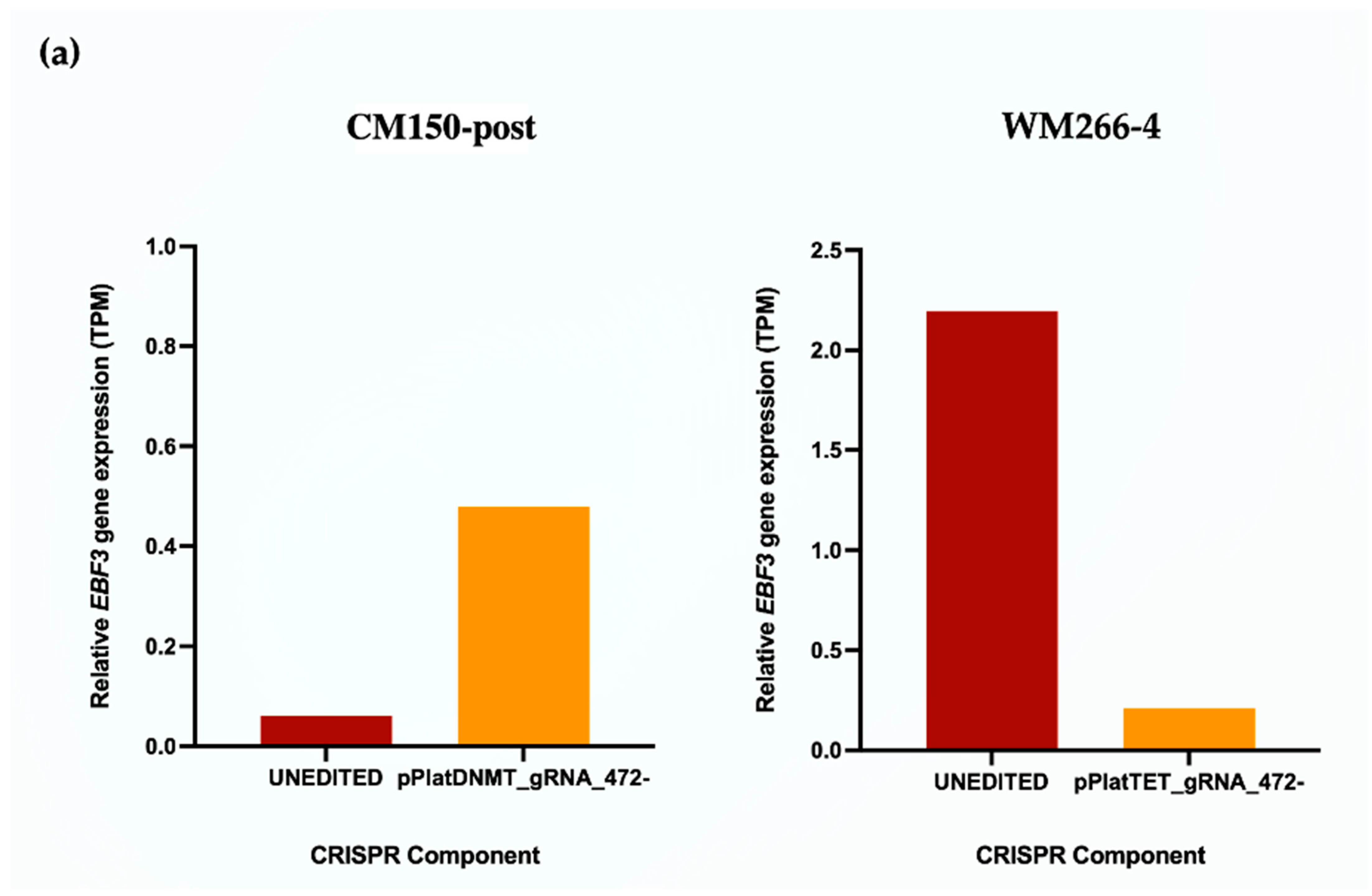
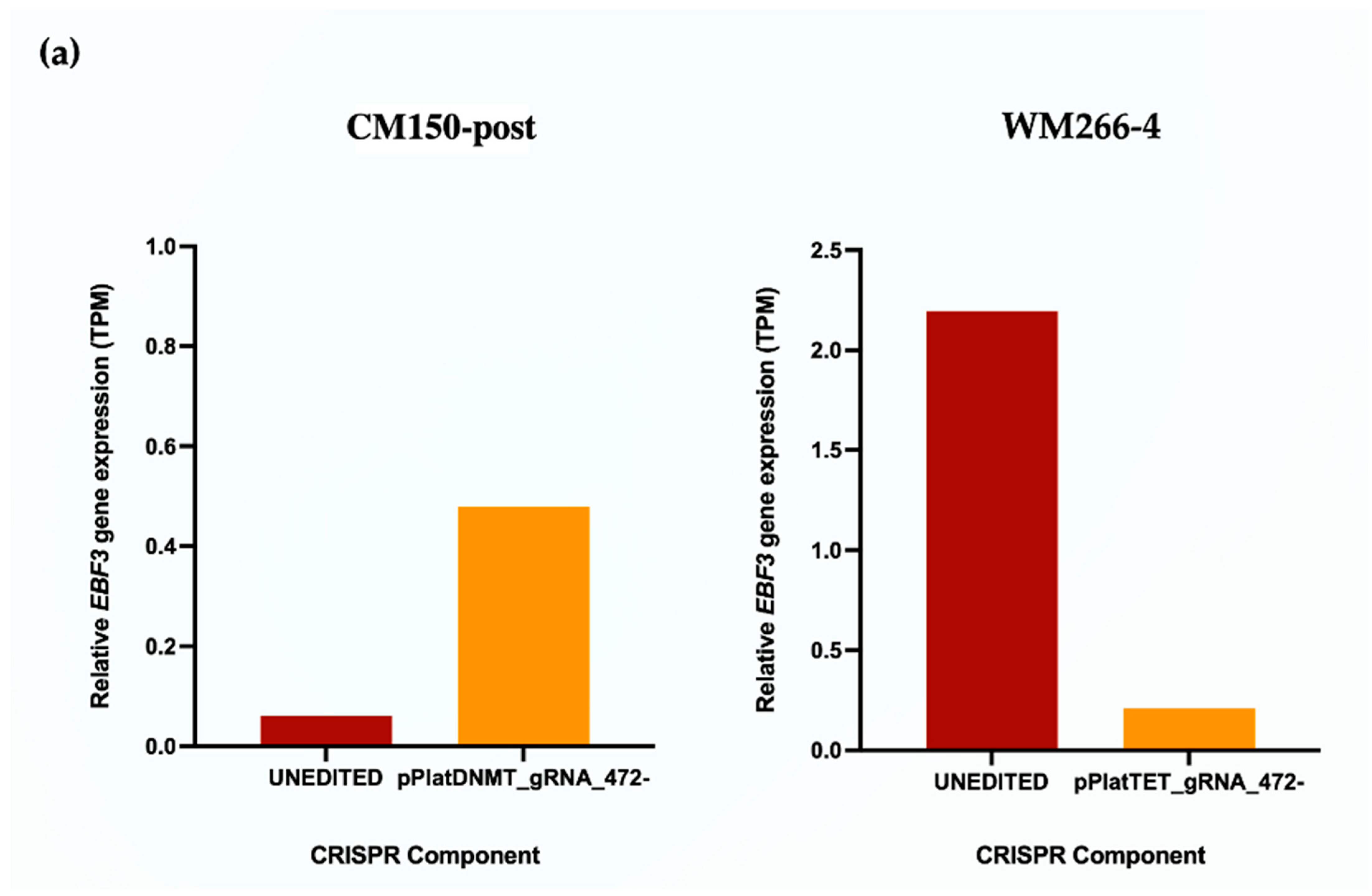
EBF3 gene expression upon promoter methylation induction in CM150-post, while this gene expression decreased upon promoter demethylation in WM266-4 cells with targeted DNA methylation editing. Whether or not this change in gene expression can subsequently alter cell phenotype is a notable aspect but was beyond the scope of this study.
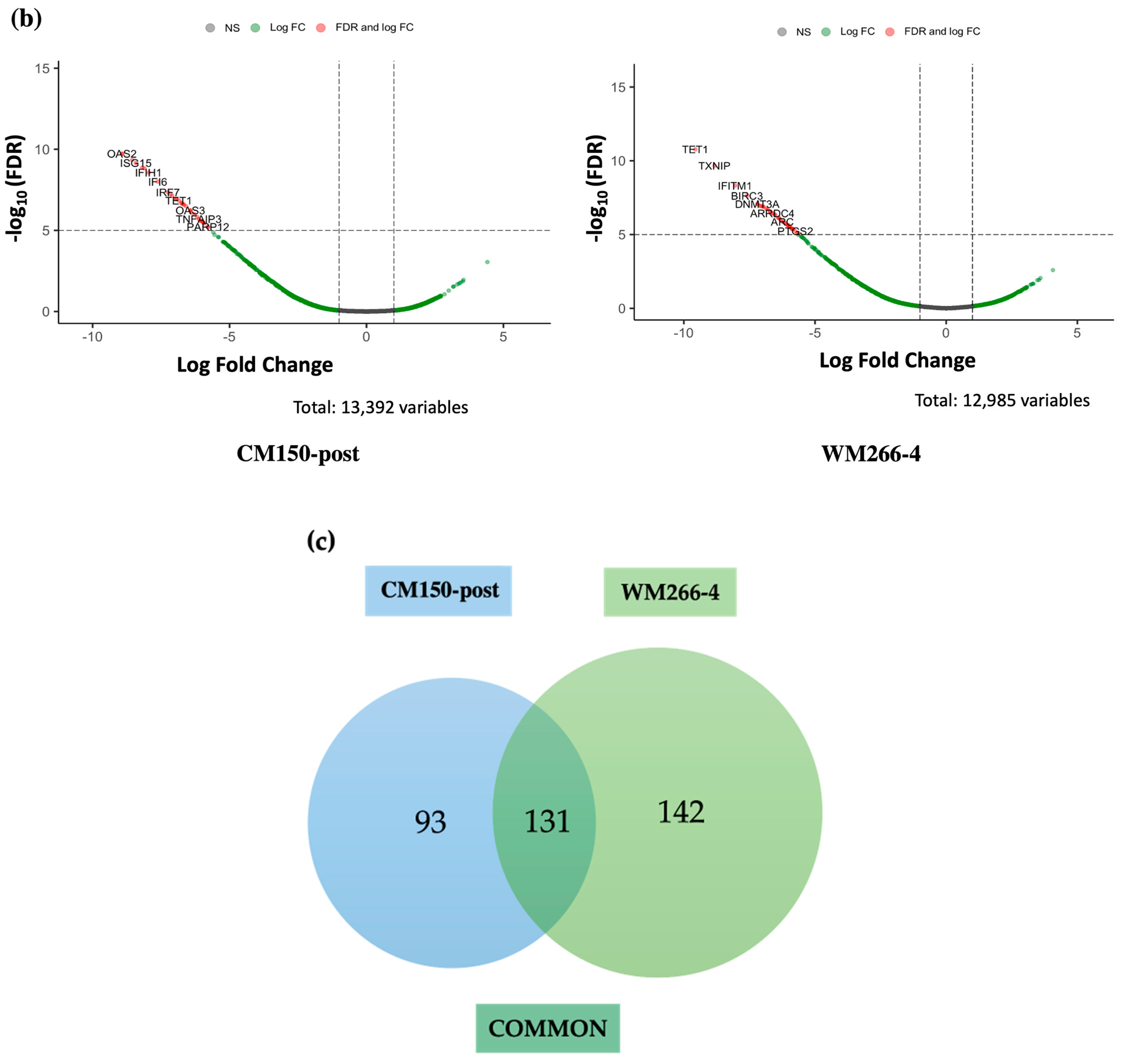
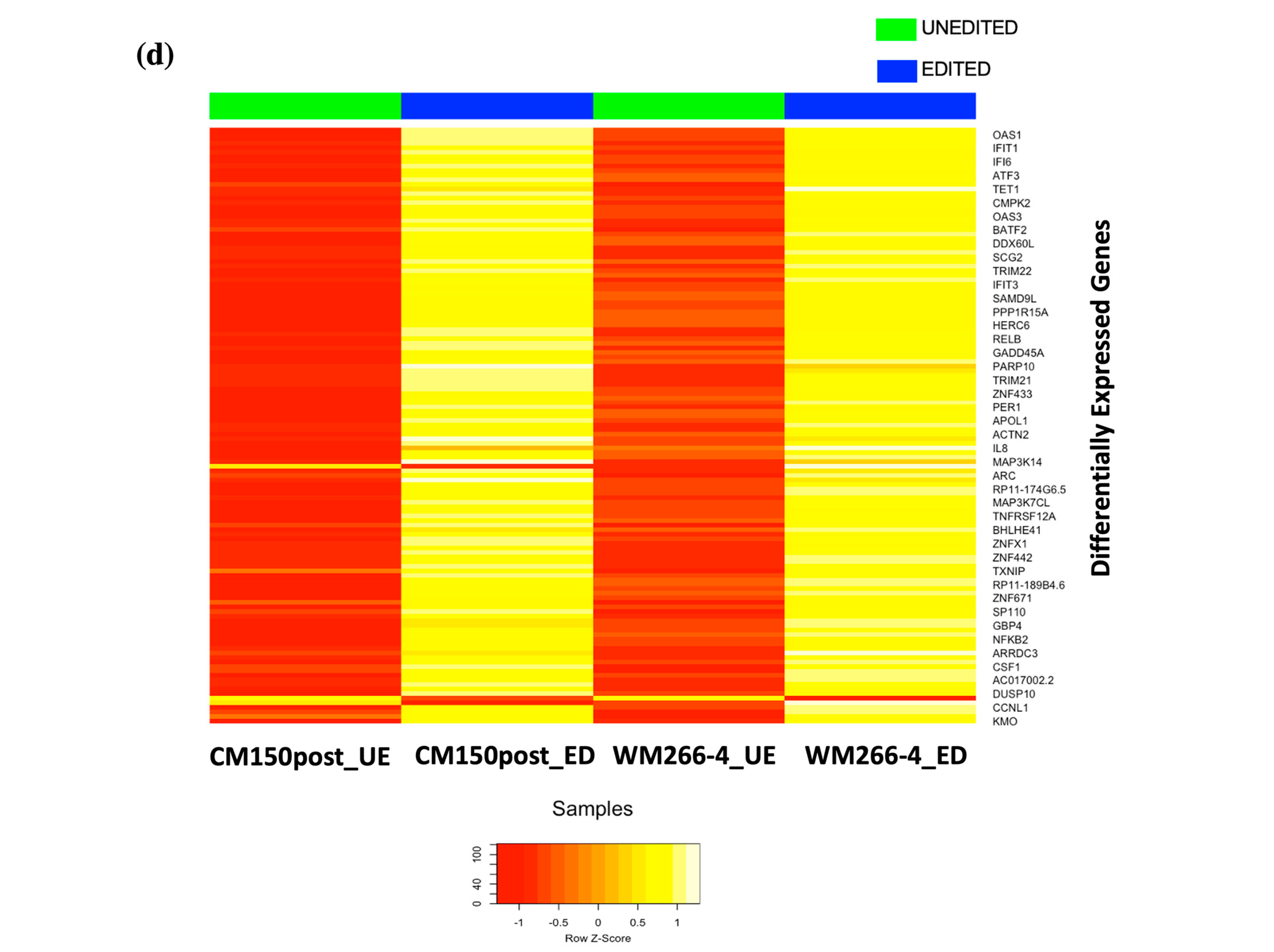
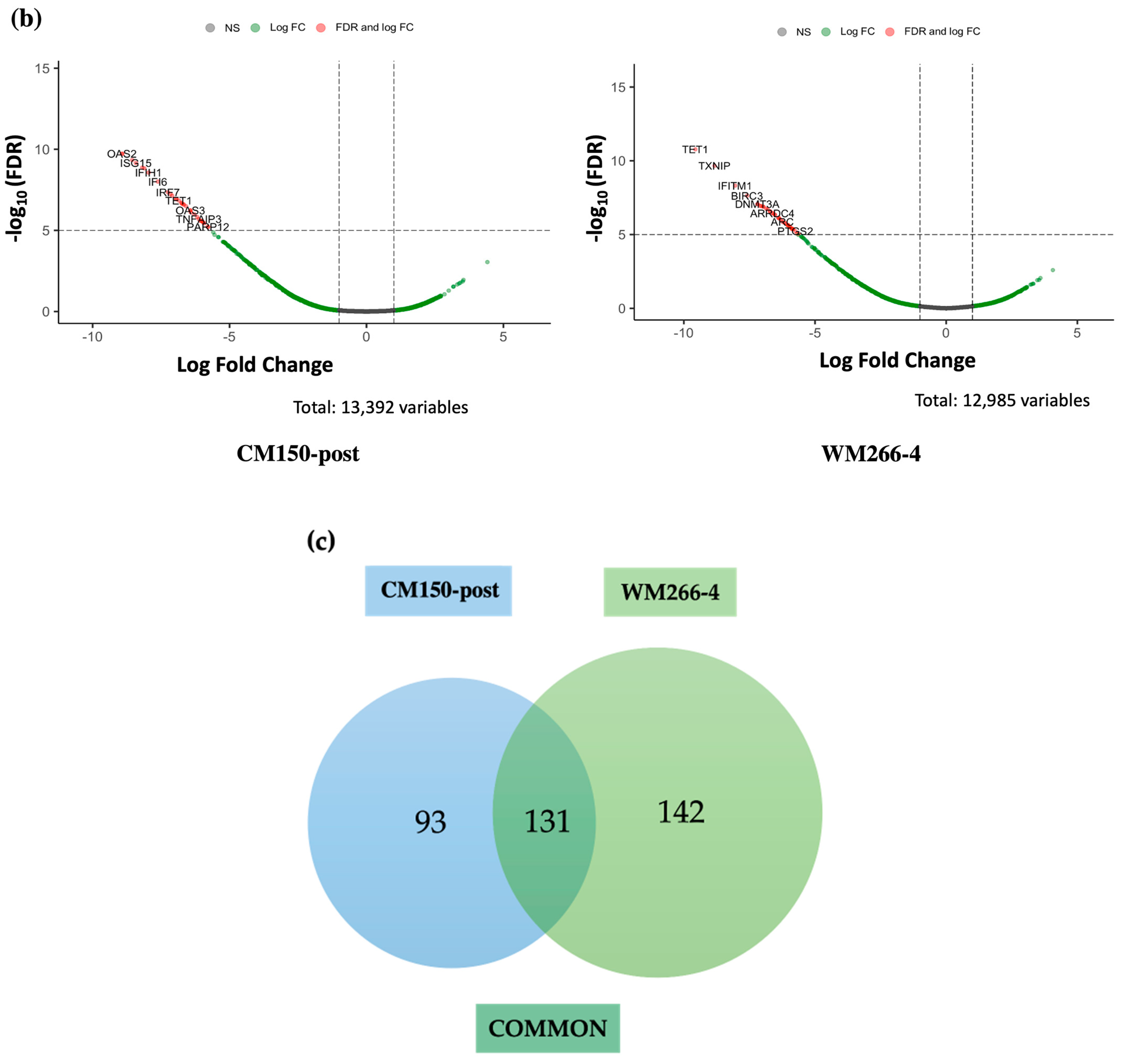
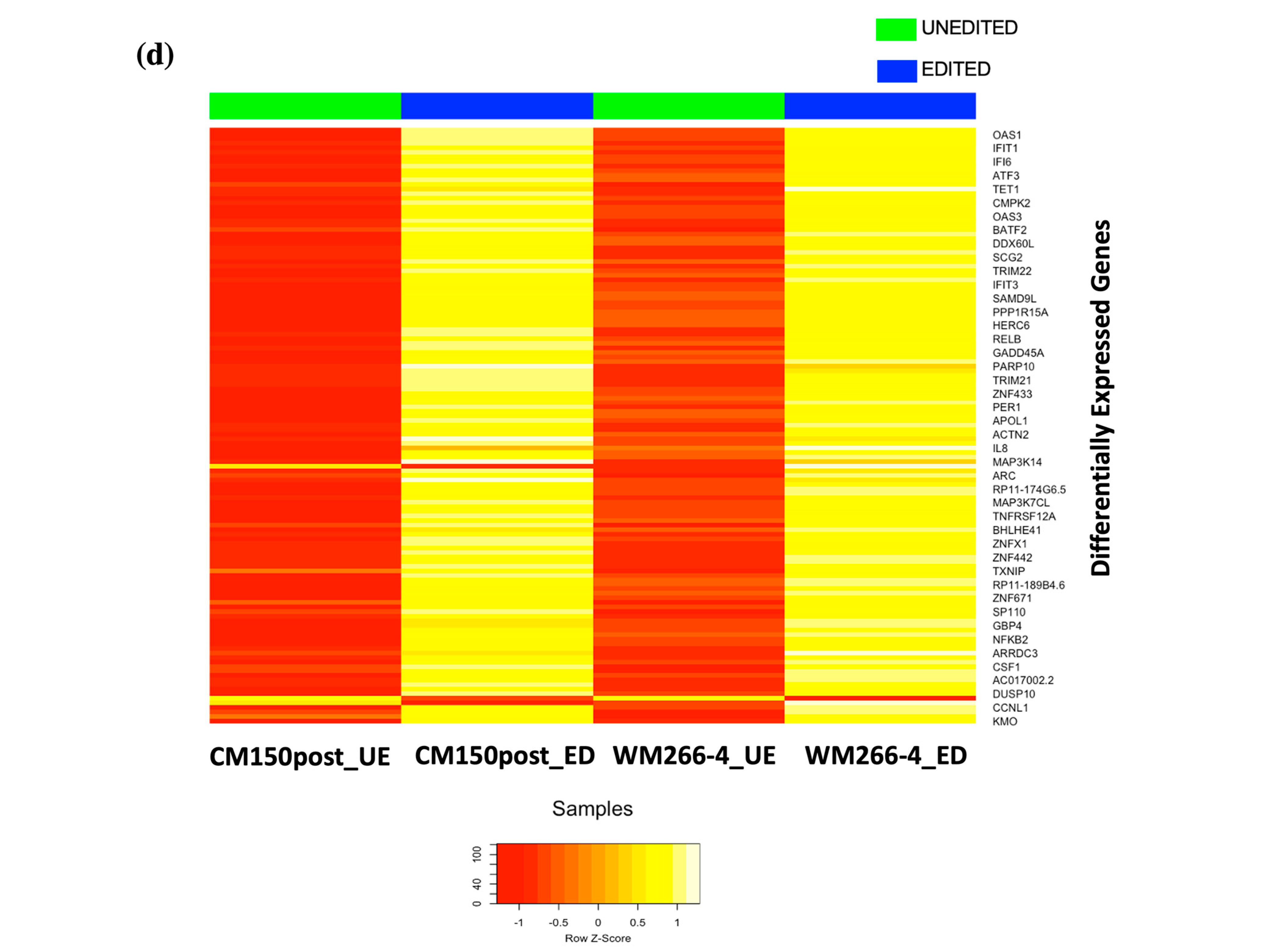
RNA-seq expression analysis of
EBF3 confirmed a decrease in gene expression when demethylating the promoter region and an increase in gene expression when methylating the promoter region. These results successfully validated the paradoxical gene expression observed in qRT-PCR in the context of
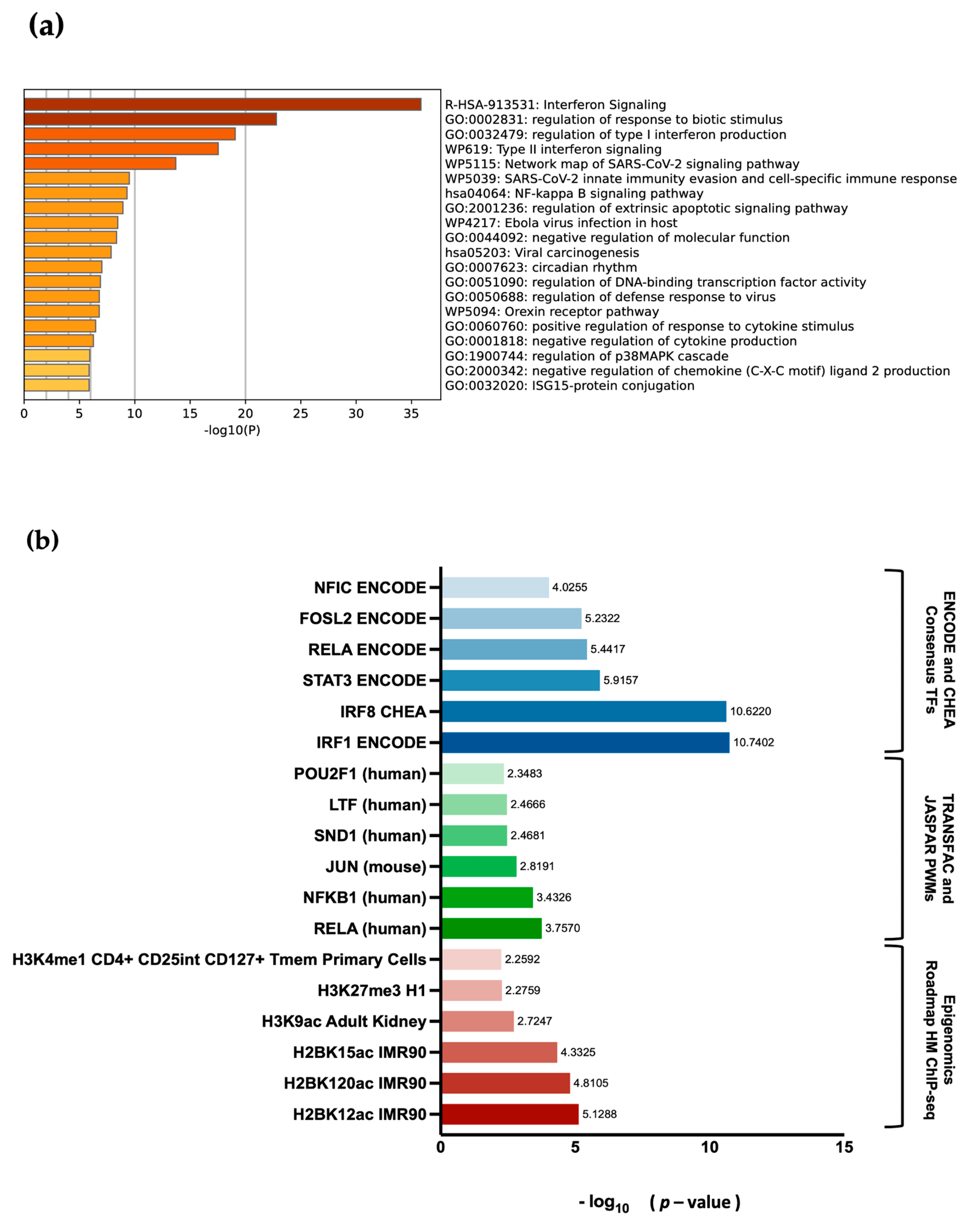
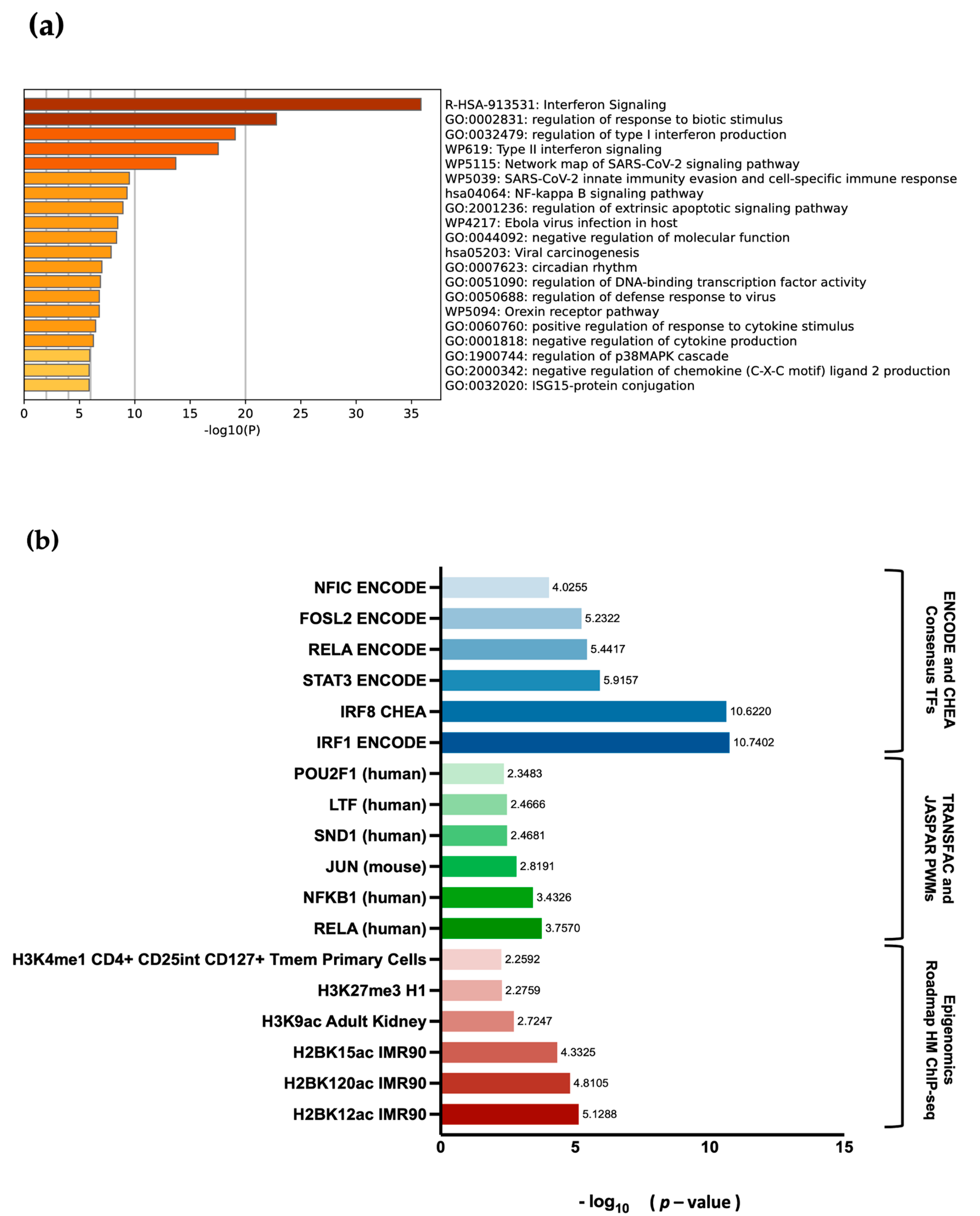
EBF3. Differential gene expression and pathway enrichment analysis revealed a strong association with immune regulatory pathways. The significant enrichment of the common genes in the interferon signalling pathway is an indication that they might play a crucial role in the body’s antiviral defense mechanisms. Interferons are signalling proteins released in response to viral infections, and they induce the expression of numerous genes that are involved in antiviral defense [
58]. In the context of
EBF3 editing, our results suggests that
EBF3 may indirectly influence these genes, potentially by affecting B cell development and function, which can impact the immune response [
21,
59]. Similarly, the enrichment of genes related to the regulation of immune responses further underscores the importance of the common genes identified in maintaining a balanced immune system. It is likely that
EBF3 editing or dysregulation could lead to changes in the expression of these genes, impacting the body’s ability to respond to infections or regulate immune responses effectively. This is highly consistent with the fact that the common DEGs also display high enrichment for transcription factors of the RELA/NF-κB family,
IRF1 and
IRF8, which are involved in the regulation of immune responses [
47]. It is possible that
EBF3, as a transcription factor itself, could interact or crosstalk with NF-κB family members to influence immune-related gene expression. Additionally, the enrichment of H2BK12ac indicates that these genes are actively transcribed, indicating their involvement in the ongoing and changing activities of cells related to the immune response. This could include processes such as immune cell activation, gene expression regulation, or other mechanisms crucial for the immune system’s function [
60].
Our findings on the paradoxical activation of
EBF3 in melanoma, influenced by methylation changes, intersect with observations by Li et al. on the IFN pathway in cholangiocarcinoma [
61]. This comparison suggests a potentially broader role for the IFN pathway in cancer, beyond its known immune functions. Further research is needed to explore how the activation of
EBF3 affects the IFN pathway in melanoma, potentially offering new insights into immune-mediated mechanisms in cancer progression and treatment strategies. However, the limited number of replicates and low expression of
EBF3 hindered our ability to accurately establish the global effect of targeted methylation editing on RNA-Seq data and its role in the enriched pathways.
Furthermore,
EBF3, as a transcription factor, interacts with histones and chromatins to regulate gene expression and chromatin structure. As such,
EBF3 influences the accessibility of target genes and plays a role in shaping the transcriptional landscape in various cellular processes, including development, differentiation, and disease, such as cancer [
62]. H3K27Me3 and H3K27Ac, two of the important histone modification marks, could be associated with the mechanism of chromatin remodelling and paradoxical gene activation. H3K27Me3 acts as a mark for the recruitment of proteins that promote chromatin compaction and transcriptional repression, leading to the silencing of gene expression alongside maintaining cellular identity [
63]. Therefore, H3K27Me3 might be involved in preserving the undifferentiated state of cancer stem cells (CSCs) through the repression of differentiation-related genes like
EBF3. This, in turn, enhances the self-renewal ability of CSCs and facilitates tumour progression [
64]. In contrast, as H3K27Ac is a key mark associated with active enhancers, as such, its aberrant deposition or redistribution can result in the activation of genes that promote tumourigenesis [
65]. Alterations in H3K27Ac levels and distribution are part of broader epigenetic remodelling events observed in cancer cells. These changes in H3K27Ac can occur due to dysregulation of histone acetyltransferases (HATs) and histone deacetylases (HDACs), which regulate the acetylation status of histones [
65,
66]. Overall, we hypothesised that editing DNA methylation at the EBF3 promoter and the associated change in gene expression could alter chromatin-bound H3K27Me3 and H3K27Ac around this region. Thus, to assess whether targeted methylation editing at the
EBF3 locus impacts the local chromatin-associated histone modifications, we performed Cleavage Under Targets and Release Using Nuclease (CUT and RUN) assays on the baseline, unedited, and edited melanoma cell lines [
67]. Data visualisation using Integrated Genome Browser (IGB) (
Figure S1) revealed pronounced peak enrichment within the enhancer regions that emphasised the functional relevance of H3K27Ac as a reliable indicator of enhancer activity, providing evidence that these regions are plausibly active and engaged in regulatory processes. Additionally, the robust peak enrichment observed in polycomb-repressed (ReprPC) states for H3K27Me3 baseline conditions strongly suggested that the region of interest is polycomb-repressed, as expected. Unfortunately, the absence of peak enrichment in the unedited and edited condition presented a limitation in elucidating the underlying chromatin state and interactions that could potentially contribute to a comprehension of the paradoxical gene expression phenomenon observed in
EBF3.
In summary, the findings suggest that EBF3, through its putative role in B cell development and function, may have a significant impact on immune responses and immune regulation. Alterations in EBF3 expression or activity, such as through epigenetic editing, could potentially disrupt the finely tuned immune system by influencing the expression of genes involved in interferon signalling, immune response regulation, and immune-related transcription factors. Understanding these connections could have potential implications for therapies associated with immune-related diseases such as cancer. However, further research is needed to establish the precise mechanisms and implications of EBF3 editing on global transcriptomic regulation.
Overall, these results demonstrated the successful and efficient DNA methylation editing using an All-in-one system and also provided the first direct and causal evidence of promoter methylation causing activation of gene transcription in the context of EBF3.
4.1. Limitations and Troubleshooting
4.1.1. Transfection Efficiency Improvisation
In the context of this study, we have demonstrated the methylation editing efficiency of the three-component system and the All-in-one construct. As outlined in the results presented in
Section 3.2, the use of the All-in-one construct in conjunction with FuGENE HD demonstrated a substantial enhancement in transfection efficiency and the number of positively sorted cells via FACS. This improvement was particularly pronounced when compared to the utilisation of the three-component system [
26] and lipofectamine 3000, confirming a significant advancement over previous methodologies. The FuGENE HD reagent is characterised by its cationic polymer properties, allowing it to create complexes with nucleic acids referred to as polyplexes or micelles when in aqueous solutions. These structures are typically less harmful to the cell membrane [
68]. This unique capability to traverse the cell membrane effectively, facilitating the delivery of exogenous DNA and promoting protein expression, while simultaneously preserving the integrity and stability of the membrane, is crucial for the successful application of this reagent. Furthermore, these enhanced outcomes were accompanied by improved cell health and a reduction in cellular debris. The findings from this study were similar to a recent study that compared different transfection methods in the context of high throughput cellular assays [
69]. The study revealed that FuGENE-mediated transfection was the most efficient method when compared to the effectiveness of Lipofectamine 3000 and calcium phosphate for transiently expressing two vital voltage-gated ion channels involved in pain signalling pathways [
69].
4.1.2. Effect of Plasmid Size and Quality in Transfection Efficiency
Our previous work using the three-component system [
26] revealed another crucial insight concerning the impact of plasmid size on transfection efficiency. In this study, we found that the size of a plasmid can significantly impact transfection and editing efficiency. Plasmids that are larger in size can often face challenges in entering cells and navigating through the cellular environment, which can result in reduced transfection rates. Furthermore, within the cell, larger plasmids may be less stable, replicate less efficiently, and provide limited access to target sites for editing processes. These factors collectively contribute to lower editing efficiency when working with larger plasmids. This was seen with the limited number of cells acquired from FACS using the three-component system (33.76 kb). In contrast, plasmids with comparatively smaller sizes, such as the All-in-one construct (14.28 kb), are generally preferred as they are more efficiently taken up by cells, move more easily within the cell, and provide better access to target DNA sites for editing, leading to higher overall success rates in targeted methylation editing. As such, we employed cells acquired from transfection using the All-in-one plasmid construct for further gene expression and downstream analysis. With the All-in-one plasmids, we achieved significantly better transfection efficiency and a higher number of FACS-sorted cells. Although this cohort of cells was subsequently employed for downstream analyses, it is worth noting that the need for further amplification of cell numbers remained.
Cell toxicity during transfection can also frequently be attributed to the plasmid DNA itself [
70]. The presence of residual substances such as endotoxins, salts, and lipopolysaccharides post-DNA extraction may lead to cell death and hinder the molecular outcome of lipofectamine’s cationic lipid molecules [
71]. Thus, ensuring high quality and purity of DNA preparation was a significant aim of this study. The early plasmid isolation kit, Zyppy Plasmid Miniprep Kit (Zymo Research), produced low-quantity DNA leading to poor transfection efficiency. As such, we improved plasmid isolation experiments from commercially available GenCatch
TM Plasmid DNA columns. Using this approach, we were able to achieve a high quantity and concentration of each plasmid and generate sufficient DNA to eliminate further variations due to disparate batch preparations.
4.1.3. Effect of Cell Type in Efficient Editing
In spite of the extensive application of gene transfer approaches in cellular and molecular biology research, the ability to achieve safe, efficient, and reproducible transfection of melanoma cells has remained challenging [
72]. This assertion gains support from the fact that there is relatively little research conducted on the specific topic of targeted methylation editing in melanoma cell lines. This scarcity of research in the field makes it challenging to gather comprehensive insights or draw robust conclusions regarding the effects of methylation editing in melanoma cells. Furthermore, to address this knowledge gap and to understand the effect of cell type in transfection, we performed transfection in Hek293FT cells. As seen from the results (
Figure 3), there was a clear increase in the sfGFP-positive cells in Hek293FT cells compared to CM150-post and WM266-4. Since melanoma cells originate from melanocytes (skin cells), they have robust defence mechanisms to protect themselves from damage, including DNA repair mechanisms and innate immune responses [
73]. These defence mechanisms can act as barriers to transfection. They can rapidly degrade or eliminate foreign genetic material before it can integrate into the cell’s genome or express the desired genes [
73]. Genetic heterogeneity and complex cellular properties could also contribute to poor transfection efficiency [
74]. These results highlight that different cell lines may exhibit significantly varying transfection efficiencies. For instance, Choudhury et al. [
75] reported up to 84.6% transfection efficiencies in HeLa cells and 56.2% in MCF7 cells using a dCas9-TET1CD single construct system, indicating substantial variability in transfection efficiency among different cell lines. However, further exploration and investigation are required to provide a more complete comprehension of the impact of cell type on transfection and editing efficiencies.
GraphPad Prism 9 (v.9.0.1). These data were also statistically analysed using an unpaired t-test to determine the mean difference in relation to the standard error between the two groups.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}