
Molecular, Metabolic, and Subcellular Mapping of the Tumor Immune Microenvironment via 3D Targeted and Non-Targeted Multiplex Multi-Omics Analyses

 , , , , , , ,
, , , , , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
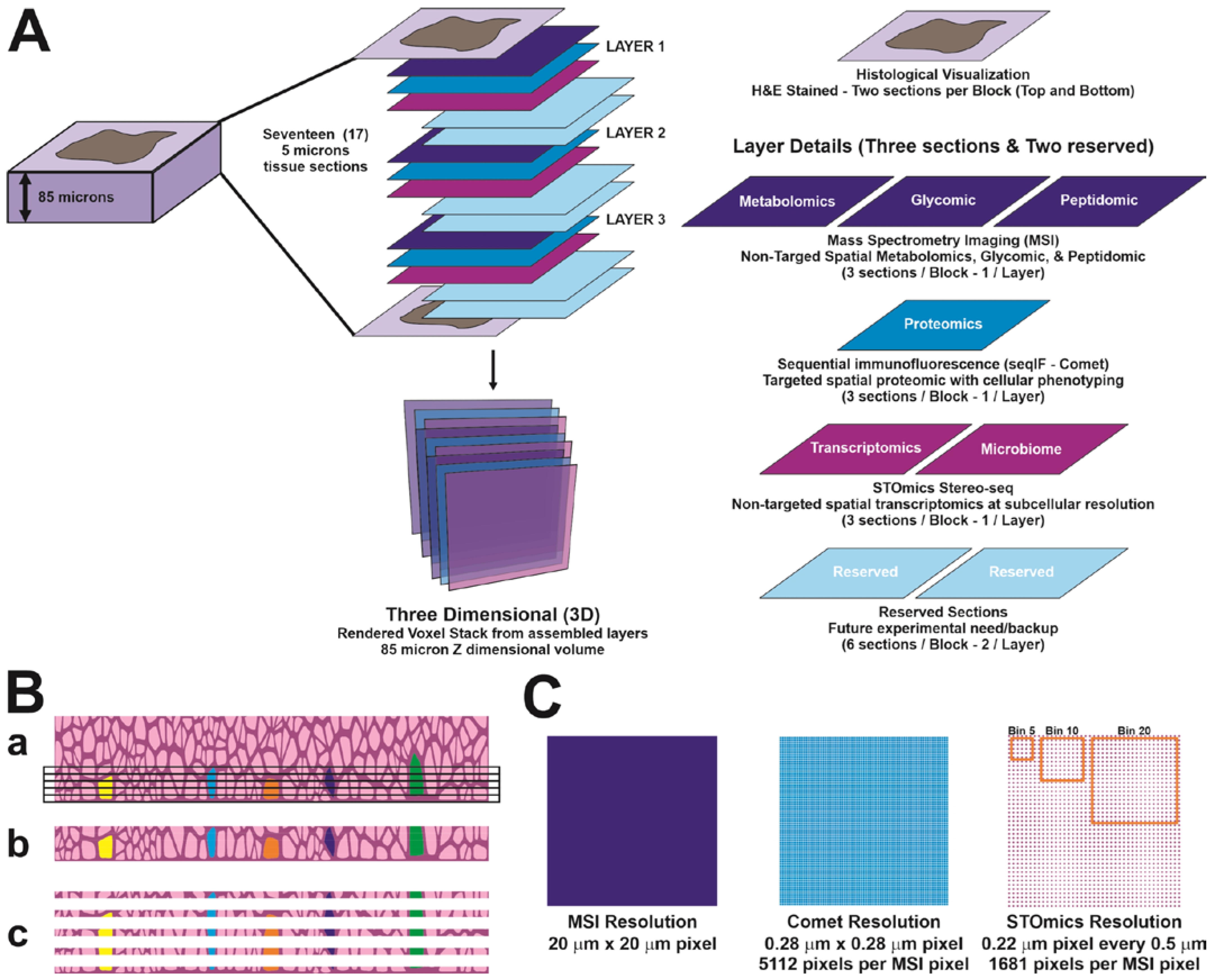
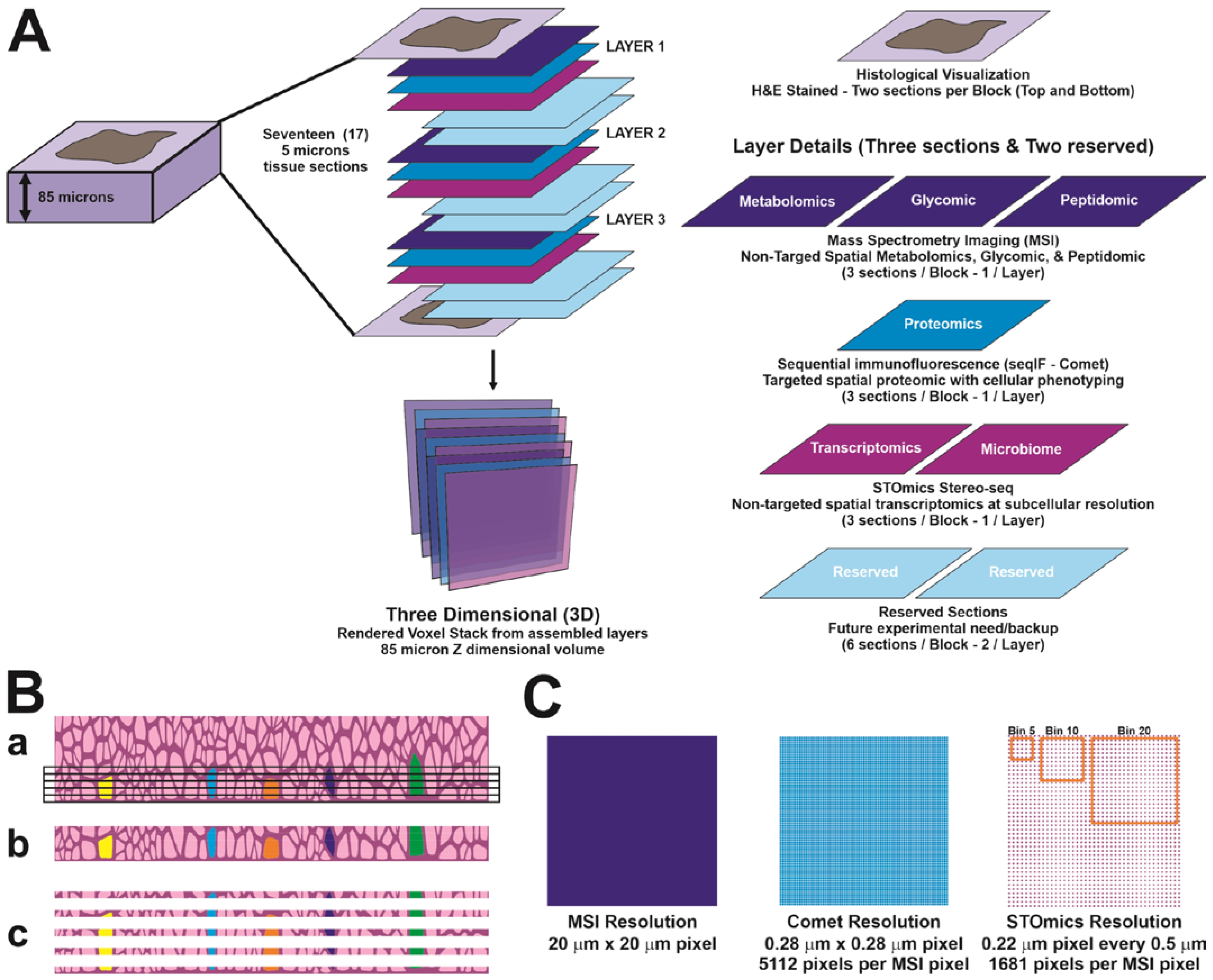
2.2. Sample Preparation for 3D Multi-Omics Analyses
2.3. Sequential Immunofluorescence Staining (seqIF)
2.3.1. Sequential Immunofluorescence Data Analysis
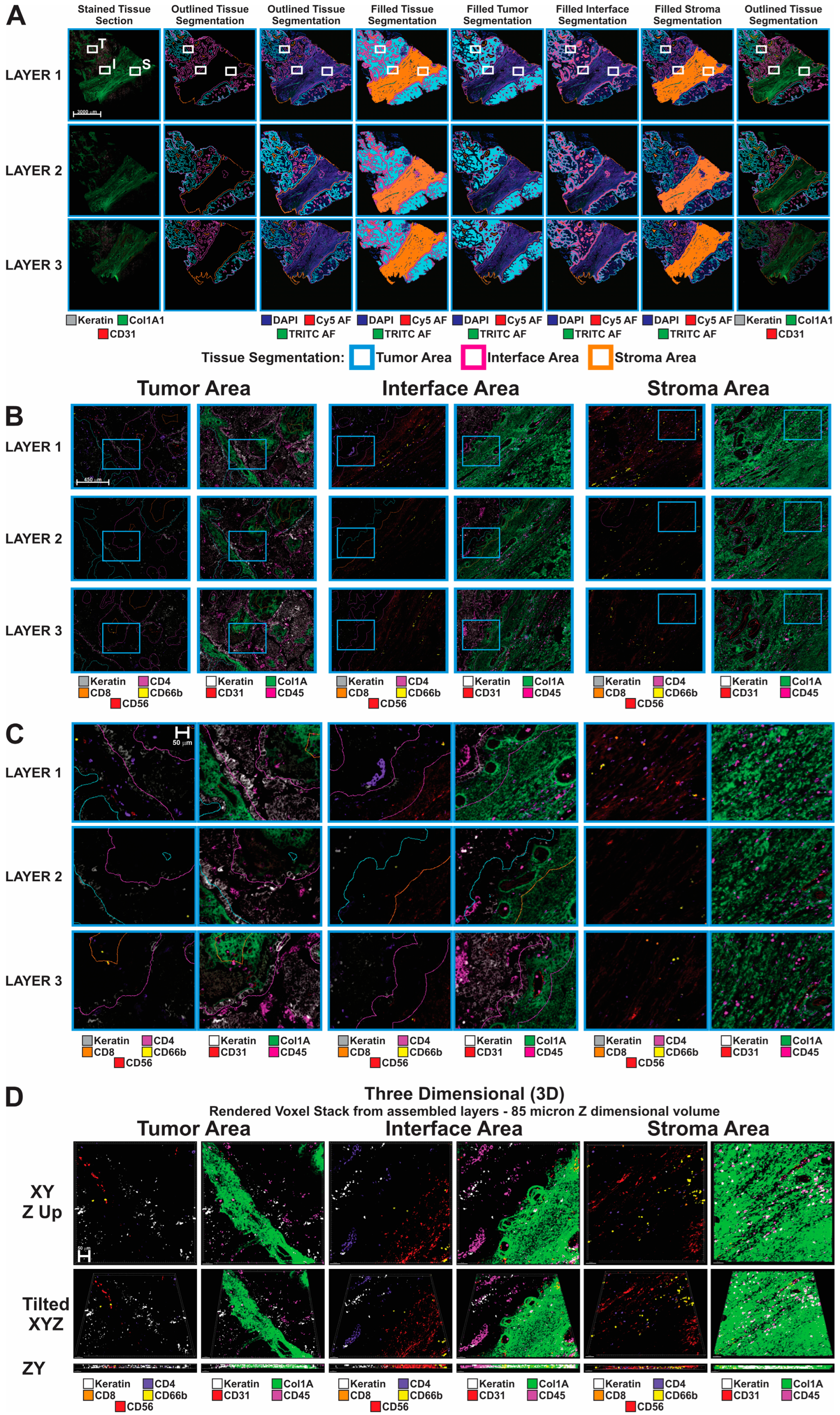
2.3.2. 3D Reconstruction of seqIF Images
2.4. Stereo-Seq Analysis
2.4.1. Stereo-Seq Raw Data Processing
2.4.2. Stereo-Seq Data Processing
2.5. MSI Analysis
2.5.1. MSI Sample Preparation
2.5.2. Mass Spectrometry Imaging
2.6. Integration of Multi-Omics Platforms
3. Results
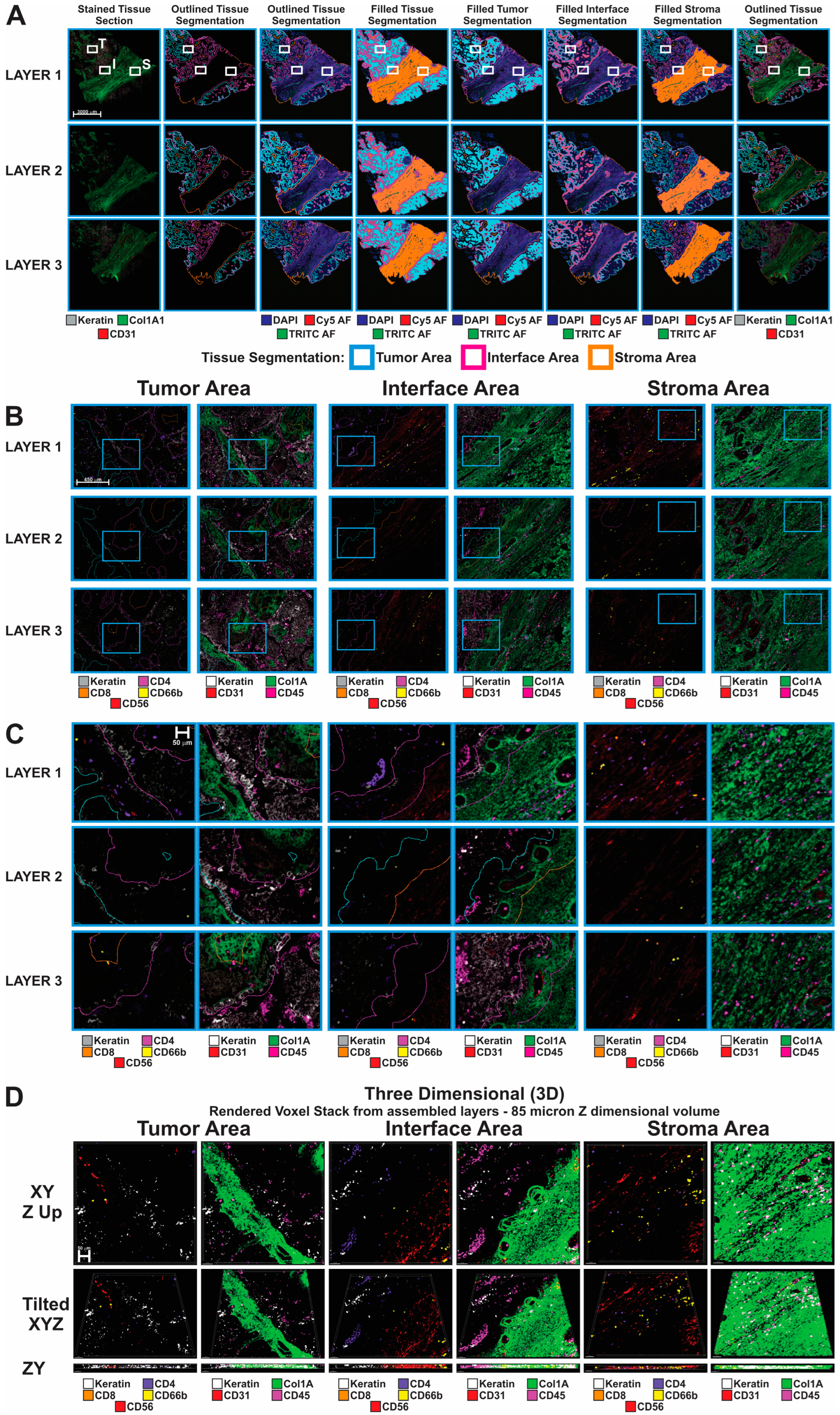
3.1. Sequential Immunofluorescence Analysis
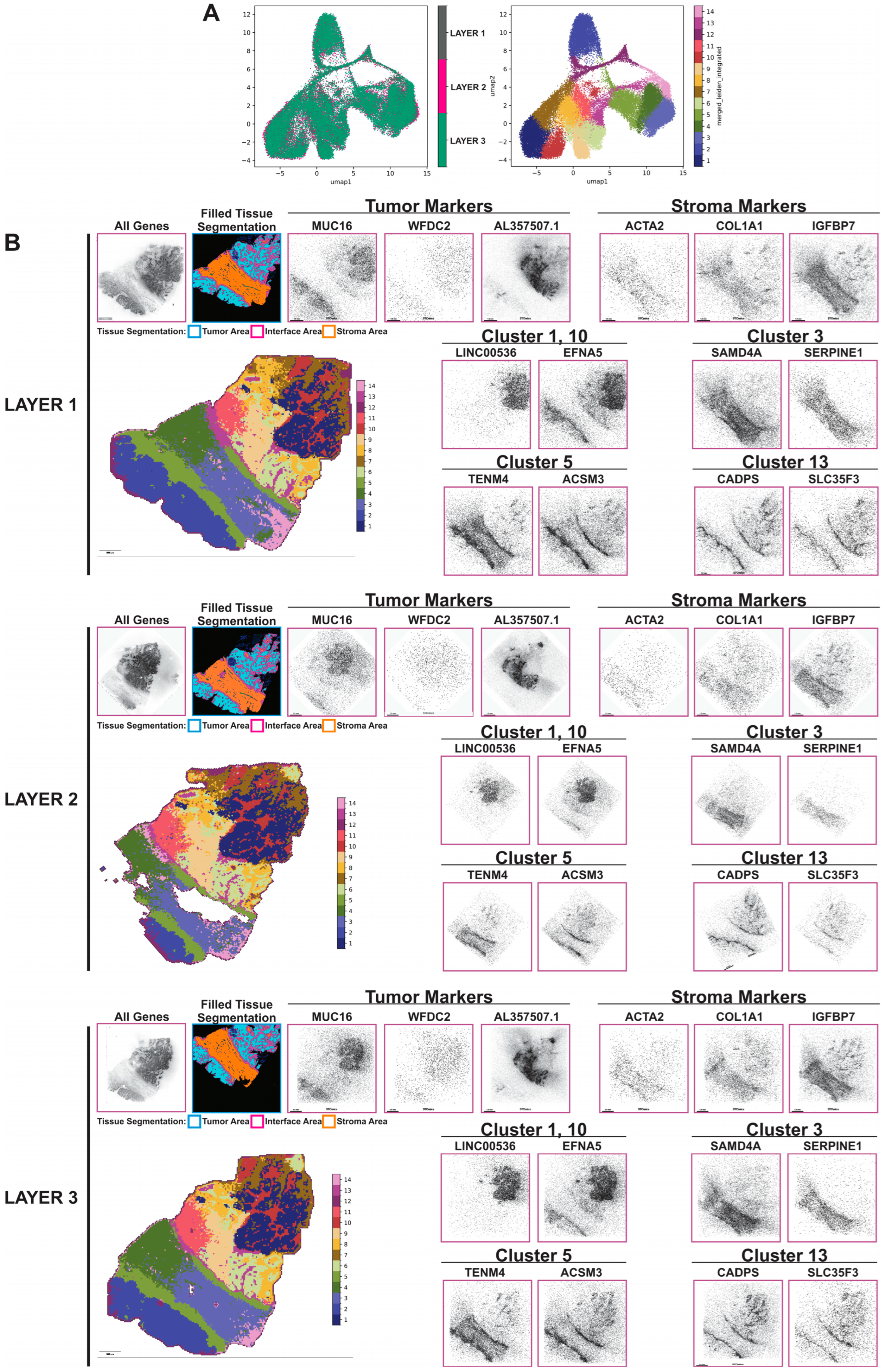
3.2. Generation of Robust Spatially Resolved Transcriptomic Profiles Using the Stereo-Seq Chip
High Levels of Tumor Heterogeneity Identified by Stereo-Seq Analysis and Leiden Clustering Analysis
3.3. Mass Spectrometry Imaging Analysis
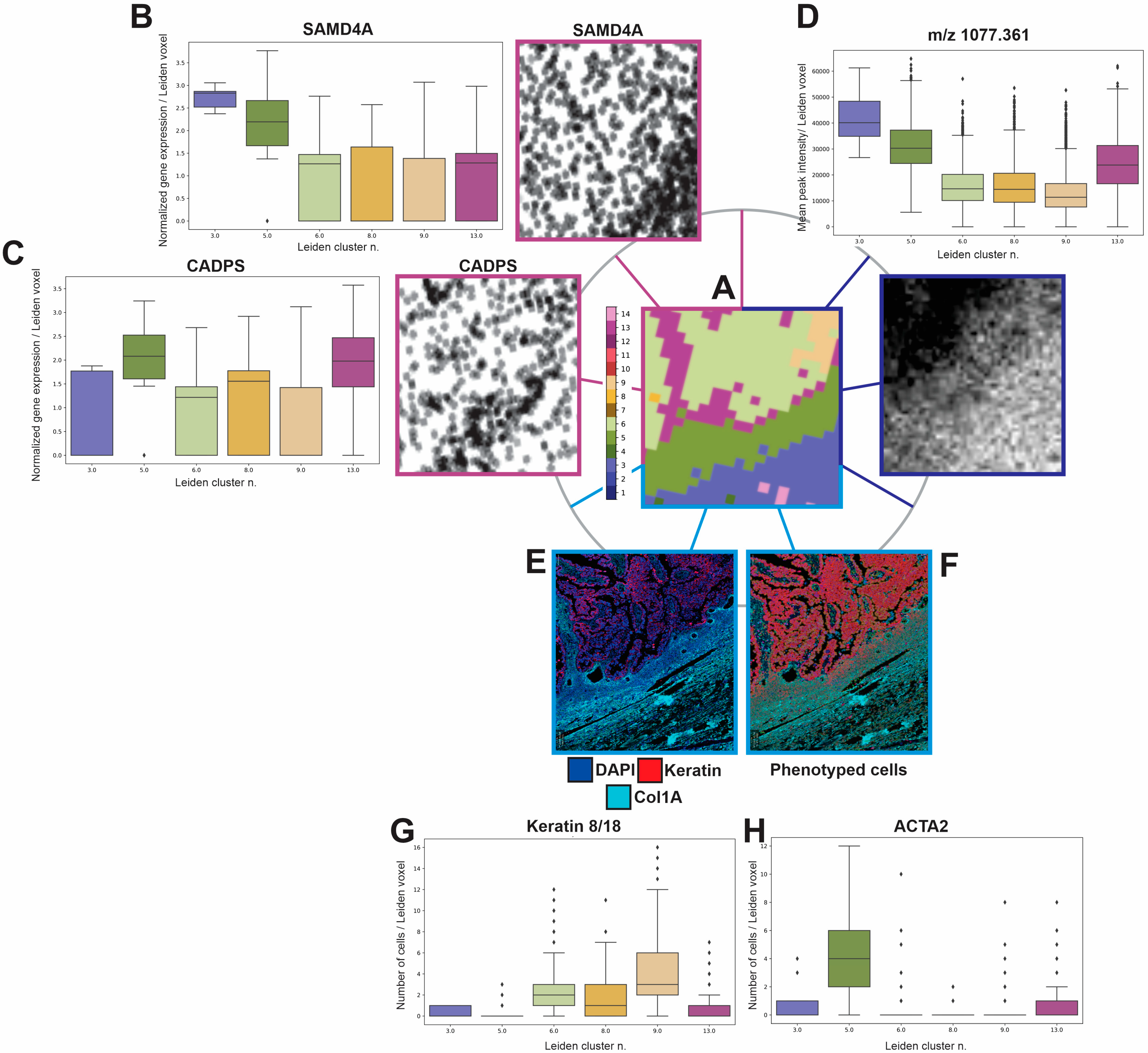
3.4. Multi-Modality Data Integration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdel-Salam, G.M.H.; Hellmuth, S.; Gradhand, E.; Kaseberg, S.; Winter, J.; Pabst, A.S.; Eid, M.M.; Thiele, H.; Nürnberg, P.; Budde, B.S.; et al. Biallelic MAD2L1BP (p31comet) mutation is associated with mosaic aneuploidy and juvenile granulosa cell tumors. JCI Insight 2023, 8, e170079. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Laung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Kaymak, I.; Williams, K.S.; Cantor, J.R.; Jones, R.G. Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell. 2021, 39, 28–37. [Google Scholar] [CrossRef]
- Bhai, P.; Turowec, J.; Santos, S.; Kerkhof, J.; Pickard, L.; Foroutan, A.; Breadner, D.; Cecchini, M.; Levy, M.A.; Stuart, A.; et al. Molecular profiling of solid tumors by next-generation sequencing: An experience from a clinical laboratory. Front. Oncol. 2023, 13, 1208244. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Schumacher, K.; Haensch, W.; Roefzaad, C.; Schlag, P.M. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001, 61, 3932–3936. [Google Scholar] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Kondratiev, S.; Sabo, E.; Yakirevich, E.; Lavie, O.; Resnick, M.B. Intratumoral CD8+ T lymphocytes as a prognostic factor of survival in endometrial carcinoma. Clin. Cancer Res. 2004, 10, 4450–4456. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Xia, S.; Chen, L.; Xu, W.; Huo, L.; Huang, D.; Shen, P.; Yang, C. Single-cell RNA sequencing reveals neurovascular-osteochondral network crosstalk during temporomandibular joint osteoarthritis: Pilot study in a human condylar cartilage. Heliyon 2023, 9, e20749. [Google Scholar] [CrossRef] [PubMed]
- Nofech-Mozes, I.; Soave, D.; Awadalla, P.; Abelson, S. Pan-cancer classification of single cells in the tumour microenvironment. Nat. Commun. 2023, 14, 1615. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.O.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Seeley, E.H.; Liu, Z.; Yuan, S.; Stroope, C.; Cockerham, E.; Rashdan, N.A.; Delgadillo, L.F.; Finney, A.C.; Kumar, D.; Das, S.; et al. Spatially Resolved Metabolites in Stable and Unstable Human Atherosclerotic Plaques Identified by Mass Spectrometry Imaging. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1626–1635. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Clift, C.L.; Drake, R.R.; Mehta, A.; Angel, P.M. Multiplexed imaging mass spectrometry of the extracellular matrix using serial enzyme digests from formalin-fixed paraffin-embedded tissue sections. Anal. Bioanal. Chem. 2021, 413, 2709–2719. [Google Scholar] [CrossRef]
- Black, S.; Phillips, D.; Hickey, J.W.; Kennedy-Darling, J.; Venkataraaman, V.G.; Samusik, N.; Goltsev, Y.; Schürch, C.M.; Nolan, G.P. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 2021, 16, 3802–3835. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, Y.; Wang, X.; Fan, J.; Gao, Q. Spatial omics: Navigating to the golden era of cancer research. Clin. Transl. Med. 2022, 12, e696. [Google Scholar] [CrossRef]
- Zhu, Y.; Ferri-Borgogno, S.; Sheng, J.; Yeung, T.-L.; Burks, J.K.; Cappello, P.; Jazaeri, A.A.; Kim, J.-H.; Han, G.H.; Birrer, M.J.; et al. SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment. Cancers 2021, 13, 1777. [Google Scholar] [CrossRef] [PubMed]
- Ferri-Borgogno, S.; Zhu, Y.; Sheng, J.; Burks, J.K.; Gomez, J.A.; Wong, K.K.; Wong, S.T.; Mok, S.C. Spatial Transcriptomics Depict Ligand-Receptor Cross-talk Heterogeneity at the Tumor-Stroma Interface in Long-Term Ovarian Cancer Survivors. Cancer Res. 2023, 83, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Heindl, A.; Lan, C.; Rodrigues, D.N.; Koelble, K.; Yuan, Y. Similarity and diversity of the tumor microenvironment in multiple metastases: Critical implications for overall and progression-free survival of high-grade serous ovarian cancer. Oncotarget 2016, 7, 71123–71135. [Google Scholar] [CrossRef]
- Arora, R.; Cao, C.; Kumar, M.; Sinha, S.; Chanda, A.; McNeil, R.; Samuel, D.; Arora, R.K.; Matthews, T.W.; Chandarana, S.; et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat. Commun. 2023, 14, 5029. [Google Scholar] [CrossRef]
- Hu, T.; Allam, M.; Cai, S.; Henderson, W.; Yueh, B.; Garipcan, A.; Ievlev, A.V.; Afkarian, M.; Beyaz, S.; Coskun, A.F. Single-cell spatial metabolomics with cell-type specific protein profiling for tissue systems biology. Nat. Commun. 2023, 14, 8260. [Google Scholar] [CrossRef] [PubMed]
- Kuett, L.; Catena, R.; Özcan, A.; Plüss, A.; Ali, H.R.; Al Sa’d, M.; Alon, S.; Aparicio, S.; Battistoni, G.; Balasubramanian, S.; et al. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat. Cancer 2022, 3, 122–133. [Google Scholar] [CrossRef]
- Rivest, F.; Eroglu, D.; Pelz, B.; Kowal, J.; Kehren, A.; Navikas, V.; Procopio, M.G.; Bordignon, P.; Pérès, E.; Ammann, M.; et al. Fully automated sequential immunofluorescence (seqIF) for hyperplex spatial proteomics. Sci. Rep. 2023, 13, 16994. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, N.; Burri, O.; Haub, P.; Guiet, R.; Seitz, A. An open-source whole slide image registration workflow at cellular precision using Fiji, QuPath and Elastix. Prontiers Comp. Sci. 2022, 3, 1–8. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef]
- Chen, A.; Sun, Y.; Lei, Y.; Li, C.; Liao, S.; Meng, J.; Bai, Y.; Liu, Z.; Liang, Z.; Zhu, Z.; et al. Single-cell spatial transcriptome reveals cell-type organization in the macaque cortex. Cell 2023, 186, 3726–3743.e24. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Powers, T.W.; Neely, B.A.; Shao, Y.; Tang, H.; Troyer, D.A.; Mehta, A.S.; Haab, B.B.; Drake, R.R. MALDI imaging mass spectrometry profiling of N-glycans in formalin-fixed paraffin embedded clinical tissue blocks and tissue microarrays. PLoS ONE 2014, 9, e106255. [Google Scholar] [CrossRef]
- Yang, F.; Liu, C.; Zhao, G.; Ge, L.; Song, Y.; Chen, Z.; Liu, Z.; Hong, K.; Ma, L. Long non-coding RNA LINC01234 regulates proliferation, migration and invasion via HIF-2alpha pathways in clear cell renal cell carcinoma cells. PeerJ 2020, 8, e10149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, G.; Lyu, X.; Rong, C.; Wang, Y.; Xu, Y.; Lyu, C. A Novel Predictive Model Associated with Osteosarcoma Metastasis. Cancer Manag. Res. 2021, 13, 8411–8423. [Google Scholar] [CrossRef]
- Rupp, C.; Scherzer, M.; Rudisch, A.; Unger, C.; Haslinger, C.; Schweifer, N.; Artaker, M.; Nivarthi, H.; Moriggl, R.; Hengstschläger, M.; et al. IGFBP7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor-stroma interaction. Oncogene 2015, 34, 815–825. [Google Scholar] [CrossRef]
- Walent, J.H.; Porter, B.W.; Martin, T.F. A novel 145 kd brain cytosolic protein reconstitutes Ca(2+)-regulated secretion in permeable neuroendocrine cells. Cell 1992, 70, 765–775. [Google Scholar] [CrossRef]
- Nestvogel, D.B.; Merino, R.M.; Leon-Pinzon, C.; Schottdorf, M.; Lee, C.; Imig, C.; Brose, N.; Rhee, J.-S. The Synaptic Vesicle Priming Protein CAPS-1 Shapes the Adaptation of Sensory Evoked Responses in Mouse Visual Cortex. Cell Rep. 2020, 30, 3261–3269.e4. [Google Scholar] [CrossRef]
- Artyomov, M.N.; Van den Bossche, J. Immunometabolism in the Single-Cell Era. Cell Metab. 2020, 32, 710–725. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | PNGaseF | Glycan Matrix | Trypsin | Peptide Matrix | |
|---|---|---|---|---|---|
| Matrix/Enzyme | DAN | PNGaseF | CHCA | Trypsin | CHCA |
| Concentration (mg/mL) | 10 | 0.1 | 10 | 0.1 | 10 |
| Solvent | 50% ACN | Water | 70% ACN, 0.1% TFA, 10 mM AmPhos | 9% ACN, 100 mM AmBic | 70% ACN, 0.1% TFA, 10 mM AmPhos |
| Flow Rate (mL/min) | 0.1 | 0.025 | 0.12 | 0.01 | 0.12 |
| Number of Passes | 4 | 15 | 3 | 12 | 4 |
| Nozzle Temperature (°C) | 60 | 45 | 75 | 30 | 75 |
| Track Speed (mm/min) | 1200 | 1200 | 1200 | 750 | 1200 |
| Track Spacing (mm) | 3 | 3 | 3 | 3 | 3 |
| Track Pattern | CC | CC | HH | HH | HH |
| Nozzle Height (mm) | 40 | 40 | 40 | 40 | 40 |
| Metabolites | Glycans | Peptides | |
|---|---|---|---|
| Polarity | Negative | Positive | Positive |
| m/z range | 50–600 | 700–3500 | 600–4500 |
| Number of laser shots | 200 | 300 | 300 |
| Funnel 1 RF (Vpp) | 75 | 450 | 450 |
| Funnel 2 RF (Vpp) | 100 | 500 | 500 |
| Multipole RF (Vpp) | 150 | 500 | 600 |
| Collision Energy (eV) | 10 | 10 | 10 |
| Collision RF (Vpp) | 500 | 2700 | 3400 |
| Quadrupole Ion Energy (eV) | 5 | 5 | 5 |
| Transfer time (μs) | 35 | 140 | 180 |
| Pre Pulse Storage (μs) | 2 | 14 | 18 |
| Sample Type | Sample Name | MID under Tissue Area | Median Reads (per bin200) | Median MID (per bin200) | Median Gene types (per bin200) | Mitochondria Transcripts | Microbiome Transcripts |
|---|---|---|---|---|---|---|---|
| HGSOC | HGSOC_4 | 82.40% | 349,016 | 8442 | 4102 | <2% | 104,870 |
| HGSOC_9 | 66.80% | 489,331 | 6193 | 3122 | <2% | 52,250 | |
| HGSOC_14 | 77.70% | 504,198 | 9964 | 4396 | <2% | 42,360 | |
| AEH | AEH_4 | 59.68% | 253,798 | 9401 | 4169 | <2% | 35,650 |
| AEH_9 | 66.70% | 145,040 | 9055 | 4000 | <2% | 24,980 | |
| AEH_14 | 74.18% | 159,337 | 10,224 | 4345 | <2% | 40,300 |
| Mean Gene Type Number | Mean Gene Type Number | ||||||
|---|---|---|---|---|---|---|---|
| Bin | RNA Capture Area | HGSOC_4 | HGSOC_9 | HGSOC_14 | AEH_4 | AEH_9 | AEH_14 |
| 1 | 0.5 μm × 0.5 μm | 1.2 | 1.16 | 1.25 | 1.2 | 1.21 | 1.21 |
| 20 | 10 μm × 10 μm | 82.62 | 59.44 | 97.85 | 91.64 | 90.87 | 102 |
| 50 | 25 μm × 25 μm | 461 | 333 | 536 | 505 | 502 | 561 |
| 100 | 50 μm × 50 μm | 1501 | 1101 | 1706 | 1592 | 1586 | 1752 |
| 150 | 75 μm × 75 μm | 2741 | 2045 | 3050 | 2816 | 2810 | 3078 |
| 200 | 100 μm × 100 μm | 3985 | 3010 | 4362 | 3989 | 4000 | 4339 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferri-Borgogno, S.; Burks, J.K.; Seeley, E.H.; McKee, T.D.; Stolley, D.L.; Basi, A.V.; Gomez, J.A.; Gamal, B.T.; Ayyadhury, S.; Lawson, B.C.; et al. Molecular, Metabolic, and Subcellular Mapping of the Tumor Immune Microenvironment via 3D Targeted and Non-Targeted Multiplex Multi-Omics Analyses. Cancers 2024, 16, 846. https://doi.org/10.3390/cancers16050846
Ferri-Borgogno S, Burks JK, Seeley EH, McKee TD, Stolley DL, Basi AV, Gomez JA, Gamal BT, Ayyadhury S, Lawson BC, et al. Molecular, Metabolic, and Subcellular Mapping of the Tumor Immune Microenvironment via 3D Targeted and Non-Targeted Multiplex Multi-Omics Analyses. Cancers. 2024; 16(5):846. https://doi.org/10.3390/cancers16050846
Chicago/Turabian StyleFerri-Borgogno, Sammy, Jared K. Burks, Erin H. Seeley, Trevor D. McKee, Danielle L. Stolley, Akshay V. Basi, Javier A. Gomez, Basant T. Gamal, Shamini Ayyadhury, Barrett C. Lawson, and et al. 2024. "Molecular, Metabolic, and Subcellular Mapping of the Tumor Immune Microenvironment via 3D Targeted and Non-Targeted Multiplex Multi-Omics Analyses" Cancers 16, no. 5: 846. https://doi.org/10.3390/cancers16050846
APA StyleFerri-Borgogno, S., Burks, J. K., Seeley, E. H., McKee, T. D., Stolley, D. L., Basi, A. V., Gomez, J. A., Gamal, B. T., Ayyadhury, S., Lawson, B. C., Yates, M. S., Birrer, M. J., Lu, K. H., & Mok, S. C. (2024). Molecular, Metabolic, and Subcellular Mapping of the Tumor Immune Microenvironment via 3D Targeted and Non-Targeted Multiplex Multi-Omics Analyses. Cancers, 16(5), 846. https://doi.org/10.3390/cancers16050846