
Modeling the Effect of Spatial Structure on Solid Tumor Evolution and Circulating Tumor DNA Composition

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
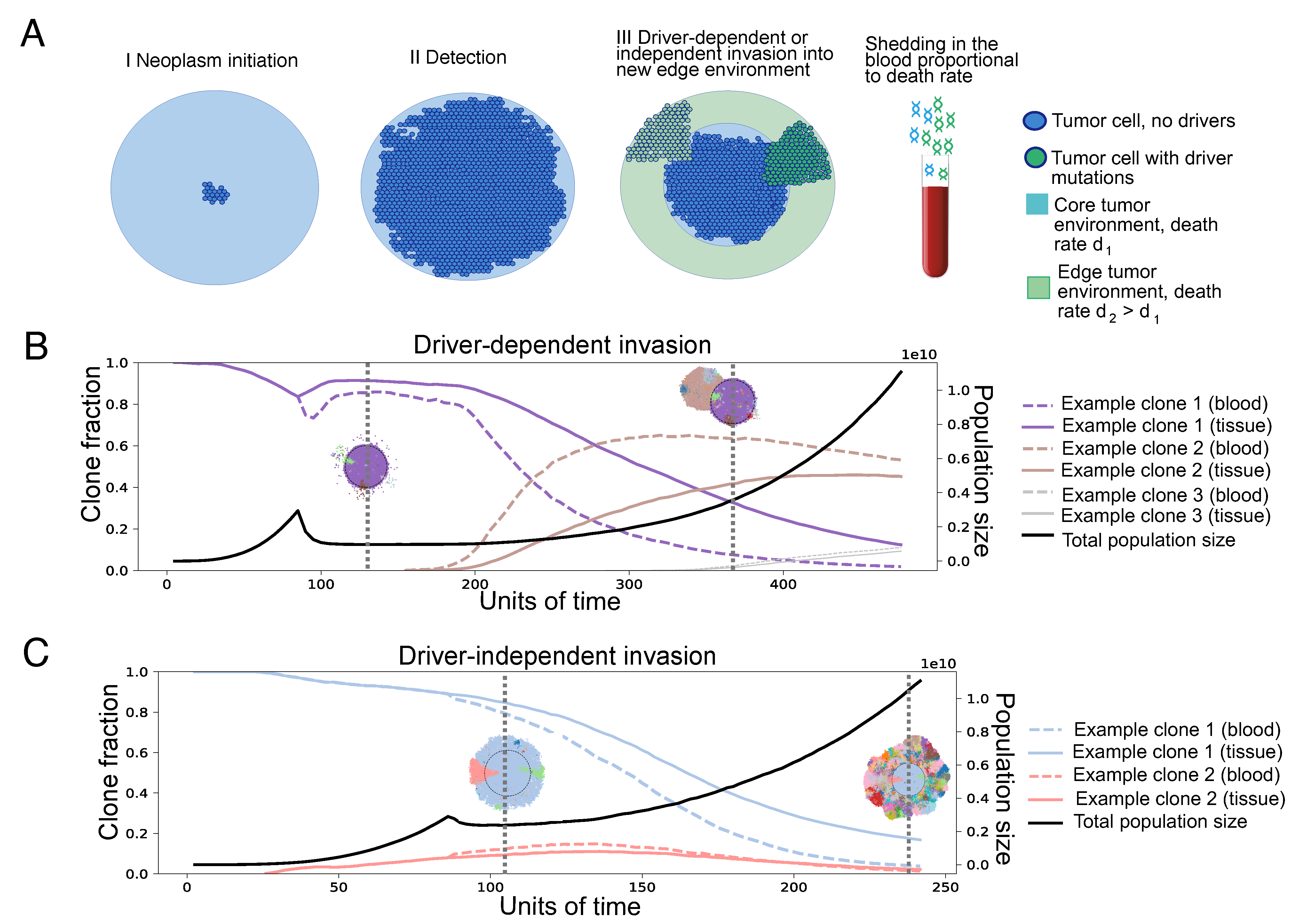
3. Results
3.1. Spatial Differences in Apoptosis and Shedding Can Bias Clone Fractions in ctDNA

3.2. Differential Shedding Leads to Overestimation of True Intratumor Heterogeneity
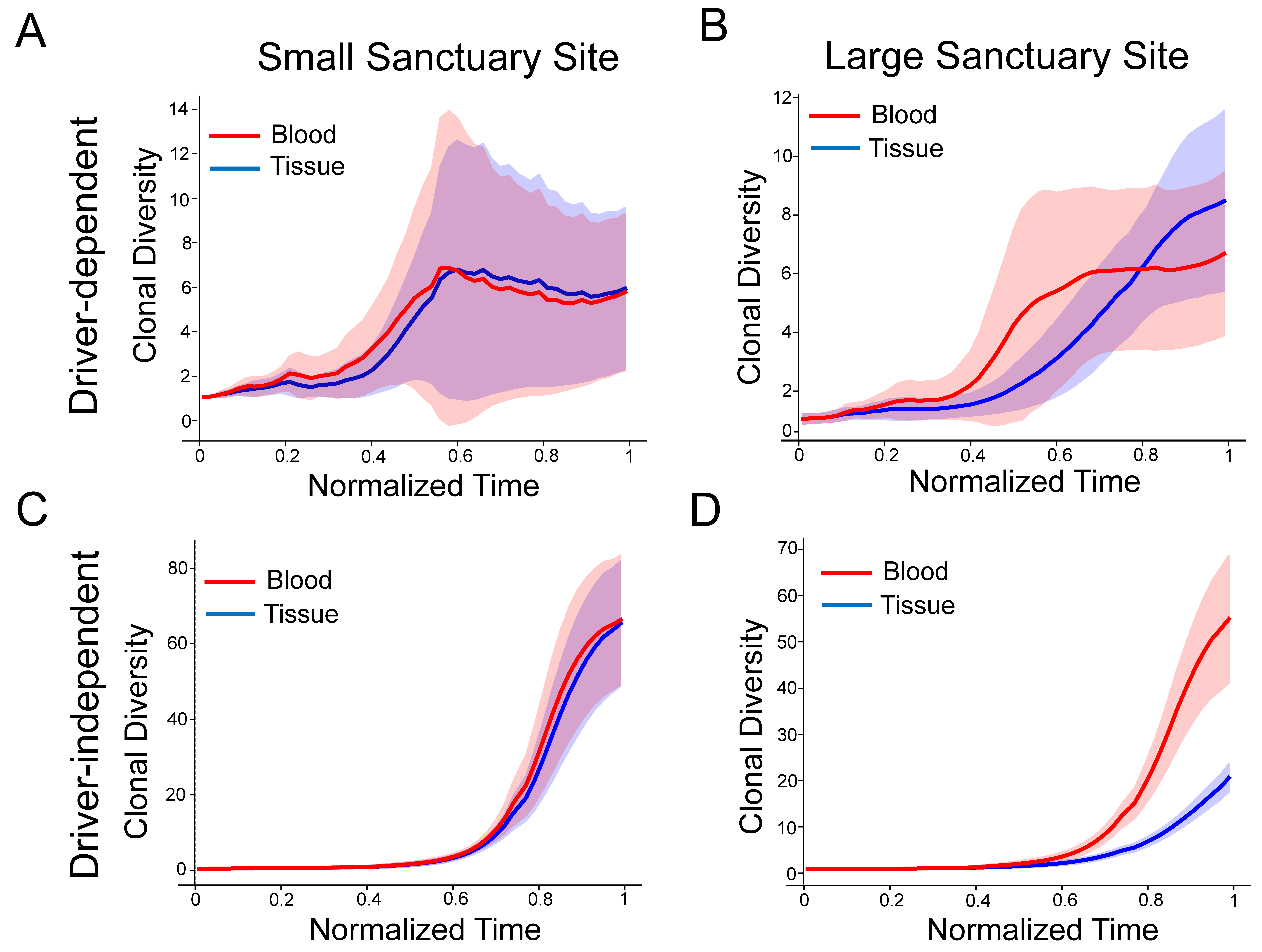
3.3. The Effect of Sequencing Detection Limits and Sanctuary Site Size on Observed VAFs in the Blood

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wan, J.C.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- De Rubis, G.; Krishnan, S.R.; Bebawy, M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, S.; Han, S.W. Utilizing Plasma Circulating Tumor DNA Sequencing for Precision Medicine in the Management of Solid Cancers. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2023, 55, 367–384. [Google Scholar] [CrossRef]
- Ulz, P.; Heitzer, E.; Geigl, J.B.; Speicher, M.R. Patient monitoring through liquid biopsies using circulating tumor DNA. Int. J. Cancer 2017, 141, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Kujala, J.; Hartikainen, J.M.; Tengström, M.; Sironen, R.; Auvinen, P.; Kosma, V.M.; Mannermaa, A. Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers 2022, 14, 1332. [Google Scholar] [CrossRef] [PubMed]
- Mattox, A.K.; Bettegowda, C.; Zhou, S.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Applications of liquid biopsies for cancer. Sci. Transl. Med. 2019, 11, eaay1984. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Davis, A.A.; Agte, S.; Pan, A.; Simon, N.I.; Iams, W.T.; Cruz, M.R.; Tamragouri, K.; Rhee, K.; Mohindra, N.; et al. Clinical implications of circulating tumor DNA tumor mutational burden (ctDNA TMB) in non-small cell lung cancer. Oncologist 2019, 24, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Zhao, E.; Bratman, S.V.; Siu, L.L. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: Current research, opportunities, and challenges. Sci. Adv. 2022, 8, eabi8618. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of genomic alterations by next-generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. Arch. Pathol. Lab. Med. 2018, 142, 1242–1253. [Google Scholar] [CrossRef]
- Jahangiri, L.; Hurst, T. Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers 2019, 11, 1938. [Google Scholar] [CrossRef]
- Schwaederlé, M.C.; Patel, S.P.; Husain, H.; Ikeda, M.; Lanman, R.B.; Banks, K.C.; Talasaz, A.; Bazhenova, L.; Kurzrock, R. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 5101–5111. [Google Scholar] [CrossRef]
- Ma, F.; Zhu, W.; Guan, Y.; Yang, L.; Xia, X.; Chen, S.; Li, Q.; Guan, X.; Yi, Z.; Qian, H.; et al. ctDNA dynamics: A novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget 2016, 7, 66020. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.C.; Strohbehn, G.W.; Karrison, T.G.; Rouhani, S.J.; Segal, J.P.; Shergill, A.; Hoffman, P.C.; Patel, J.D.; Garassino, M.C.; Vokes, E.E.; et al. Brief Report: Discordance Between Liquid and Tissue Biopsy-Based Next-Generation Sequencing in Lung Adenocarcinoma at Disease Progression. Clin. Lung Cancer 2023, 24, e117–e121. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Pantel, K.; Müller, V.; Rack, B.; Kasimir-Bauer, S.; Janni, W.; Schwarzenbach, H. Apoptosis-related deregulation of proteolytic activities and high serum levels of circulating nucleosomes and DNA in blood correlate with breast cancer progression. BMC Cancer 2011, 11, 4. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, H.; Long, Y.; Li, P.; Gu, Y. The main sources of circulating cell-free DNA: Apoptosis, necrosis and active secretion. Crit. Rev. Oncol. 2021, 157, 103166. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.F.; Junqueira-Neto, S.; Pinheiro, J.; Machado, J.C.; Costa, J.L. Induction of apoptosis increases sensitivity to detect cancer mutations in plasma. Eur. J. Cancer 2020, 127, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of Apoptosis by Cancer Chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. JNCI J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Murthy, V.; Oshi, M.; Tokumaru, Y.; Endo, I.; Takabe, K. Increased apoptosis is associated with robust immune cell infiltration and cytolytic activity in breast cancer. Am. J. Cancer Res. 2021, 11, 3674–3687. [Google Scholar]
- Giordano, S.; Morosi, L.; Veglianese, P.; Licandro, S.A.; Frapolli, R.; Zucchetti, M.; Cappelletti, G.; Falciola, L.; Pifferi, V.; Visentin, S.; et al. 3D Mass Spectrometry Imaging Reveals a Very Heterogeneous Drug Distribution in Tumors. Sci. Rep. 2016, 6, 37027. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Schnitzer, S.; Brüne, B. Tumor hypoxia and cancer progression. Cancer Lett. 2006, 237, 10–21. [Google Scholar] [CrossRef]
- Kato, K.; Uchida, J.; Kukita, Y.; Kumagai, T.; Nishino, K.; Inoue, T.; Kimura, M.; Imamura, F. Transient appearance of circulating tumor DNA associated with de novo treatment. Sci. Rep. 2016, 6, 38639. [Google Scholar] [CrossRef]
- Rostami, A.; Lambie, M.; Caberry, W.Y.; Stambolic, V.; Waldron, J.N.; Bratman, S.V. Senescence, necrosis, and apoptosis govern circulating cell-free DNA release kinetics. Cell Rep. 2020, 31, 107830. [Google Scholar] [CrossRef]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef]
- Abouali, H.; Hosseini, S.A.; Purcell, E.; Nagrath, S.; Poudineh, M. Recent advances in device engineering and computational analysis for characterization of cell-released cancer biomarkers. Cancers 2022, 14, 288. [Google Scholar] [CrossRef]
- Stetson, D.; Ahmed, A.; Xu, X.; Nuttall, B.R.; Lubinski, T.J.; Johnson, J.H.; Barrett, J.C.; Dougherty, B.A. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis. Oncol. 2019, 3, 3. [Google Scholar] [CrossRef]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Rhrissorrakrai, K.; Utro, F.; Levovitz, C.; Parida, L. Lesion Shedding Model: Unraveling site-specific contributions to ctDNA. Briefings Bioinform. 2023, 24, bbad059. [Google Scholar] [CrossRef]
- Fu, F.; Nowak, M.A.; Bonhoeffer, S. Spatial Heterogeneity in Drug Concentrations Can Facilitate the Emergence of Resistance to Cancer Therapy. PLOS Comput. Biol. 2015, 11, e1004142. [Google Scholar] [CrossRef]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef]
- Bozic, I.; Paterson, C.; Waclaw, B. On measuring selection in cancer from subclonal mutation frequencies. PLoS Comput. Biol. 2019, 15, e1007368. [Google Scholar] [CrossRef]
- Chkhaidze, K.; Heide, T.; Werner, B.; Williams, M.J.; Huang, W.; Caravagna, G.; Graham, T.A.; Sottoriva, A. Spatially constrained tumour growth affects the patterns of clonal selection and neutral drift in cancer genomic data. PLoS Comput. Biol. 2019, 15, e1007243. [Google Scholar] [CrossRef]
- Noble, R.; Burri, D.; Le Sueur, C.; Lemant, J.; Viossat, Y.; Kather, J.N.; Beerenwinkel, N. Spatial structure governs the mode of tumour evolution. Nat. Ecol. Evol. 2022, 6, 207–217. [Google Scholar] [CrossRef]
- Lewinsohn, M.A.; Bedford, T.; Müller, N.F.; Feder, A.F. State-dependent evolutionary models reveal modes of solid tumour growth. Nat. Ecol. Evol. 2023, 7, 581–596. [Google Scholar] [CrossRef]
- Del Monte, U. Does the cell number 109 still really fit one gram of tumor tissue? Cell Cycle 2009, 8, 505–506. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Buckland, S.; Magurran, A.; Green, R.; Fewster, R. Monitoring change in biodiversity through composite indices. Philosophical Trans. R. Soc. B Biol. Sci. 2005, 360, 243–254. [Google Scholar] [CrossRef]
- Chin, R.I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef]
- Fridland, S.; Choi, J.; Nam, M.; Schellenberg, S.J.; Kim, E.; Lee, G.; Yoon, N.; Chae, Y.K. Assessing tumor heterogeneity: Integrating tissue and circulating tumor DNA (ctDNA) analysis in the era of immuno-oncology-blood TMB is not the same as tissue TMB. J. Immunother. Cancer 2021, 9, e002551. [Google Scholar] [CrossRef]
- Bredno, J.; Lipson, J.; Venn, O.; Gross, S.; Fields, A.P.; Beausang, J.F.; Liu, Q.; Brooks, J.D.; Chen, X.; Lopatin, R.; et al. Tumor area and microscopic extent of invasion to determine circulating tumor DNA fraction in plasma and detectability of colorectal cancer (CRC). J. Clin. Oncol. 2020, 38, 243. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; McNamara, K.; Reiter, J.G.; Sun, R.; Hu, Z.; Ma, Z.; Ding, J.; Suarez, C.J.; Tilk, S.; Raghavendra, A.; et al. Clonal replacement and heterogeneity in breast tumors treated with neoadjuvant HER2-targeted therapy. Nat. Commun. 2019, 10, 657. [Google Scholar] [CrossRef]
- Barry, P.; Vatsiou, A.; Spiteri, I.; Nichol, D.; Cresswell, G.D.; Acar, A.; Trahearn, N.; Hrebien, S.; Garcia-Murillas, I.; Chkhaidze, K.; et al. The spatiotemporal evolution of lymph node spread in early breast cancer. Clin. Cancer Res. 2018, 24, 4763–4770. [Google Scholar] [CrossRef]
- Kammesheidt, A.; Tonozzi, T.R.; Lim, S.W.; Braunstein, G.D. Mutation detection using plasma circulating tumor DNA (ctDNA) in a cohort of asymptomatic adults at increased risk for cancer. Int. J. Mol. Epidemiol. Genet. 2018, 9, 1. [Google Scholar]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.K. Clonal hematopoiesis in liquid biopsy: From biological noise to valuable clinical implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef]
- Friedman, R. Drug resistance in cancer: Molecular evolution and compensatory proliferation. Oncotarget 2016, 7, 11746. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Final tumor size |
|---|---|
| R | Core / sanctuary site radius |
| b | Initial cell birth rate |
| Cell death rate in the tumor core | |
| Cell death rate in the tumor edge | |
| s | Driver mutation fitness advantage |
| Poisson-distributed driver mutation rate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachman, T.; Bartlett, D.; LaFramboise, W.; Wagner, P.; Schwartz, R.; Carja, O. Modeling the Effect of Spatial Structure on Solid Tumor Evolution and Circulating Tumor DNA Composition. Cancers 2024, 16, 844. https://doi.org/10.3390/cancers16050844
Rachman T, Bartlett D, LaFramboise W, Wagner P, Schwartz R, Carja O. Modeling the Effect of Spatial Structure on Solid Tumor Evolution and Circulating Tumor DNA Composition. Cancers. 2024; 16(5):844. https://doi.org/10.3390/cancers16050844
Chicago/Turabian StyleRachman, Thomas, David Bartlett, William LaFramboise, Patrick Wagner, Russell Schwartz, and Oana Carja. 2024. "Modeling the Effect of Spatial Structure on Solid Tumor Evolution and Circulating Tumor DNA Composition" Cancers 16, no. 5: 844. https://doi.org/10.3390/cancers16050844
APA StyleRachman, T., Bartlett, D., LaFramboise, W., Wagner, P., Schwartz, R., & Carja, O. (2024). Modeling the Effect of Spatial Structure on Solid Tumor Evolution and Circulating Tumor DNA Composition. Cancers, 16(5), 844. https://doi.org/10.3390/cancers16050844