
Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989


Abstract
Simple Summary
Abstract

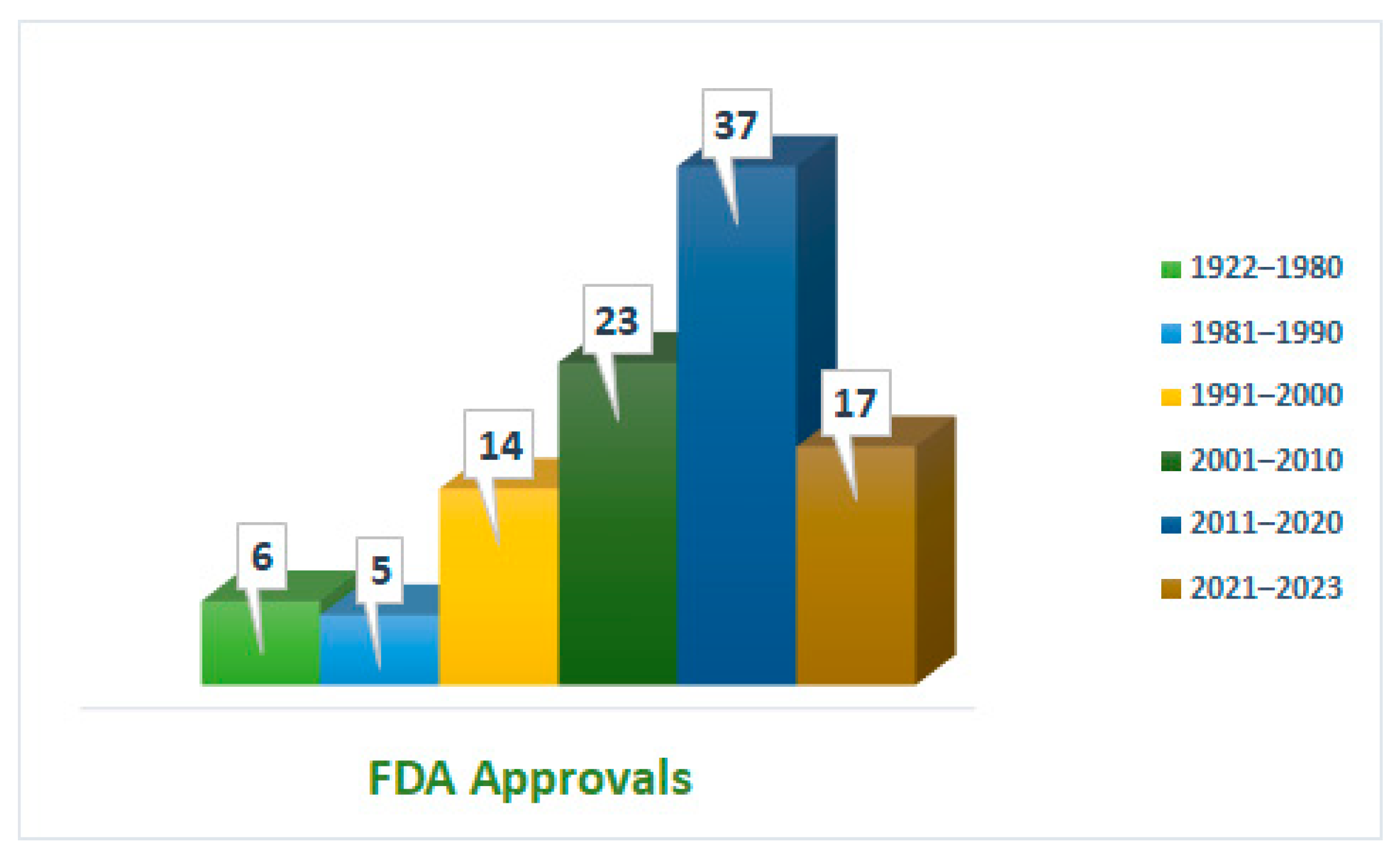
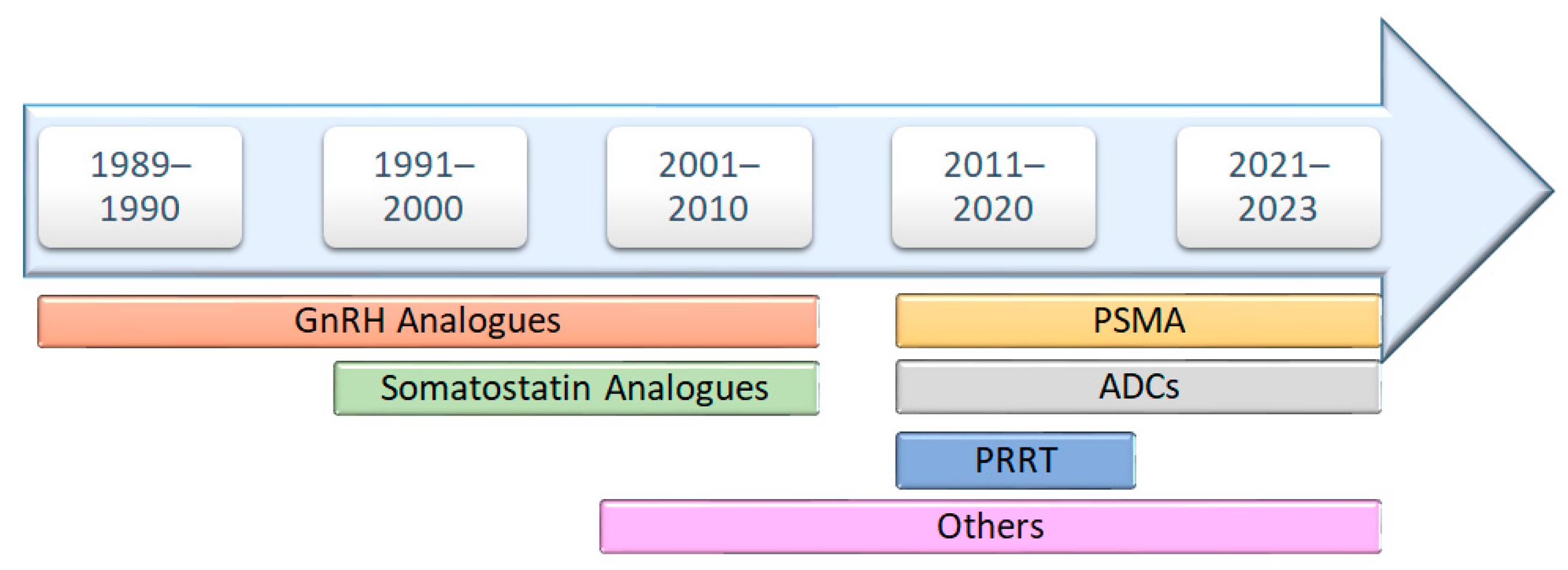
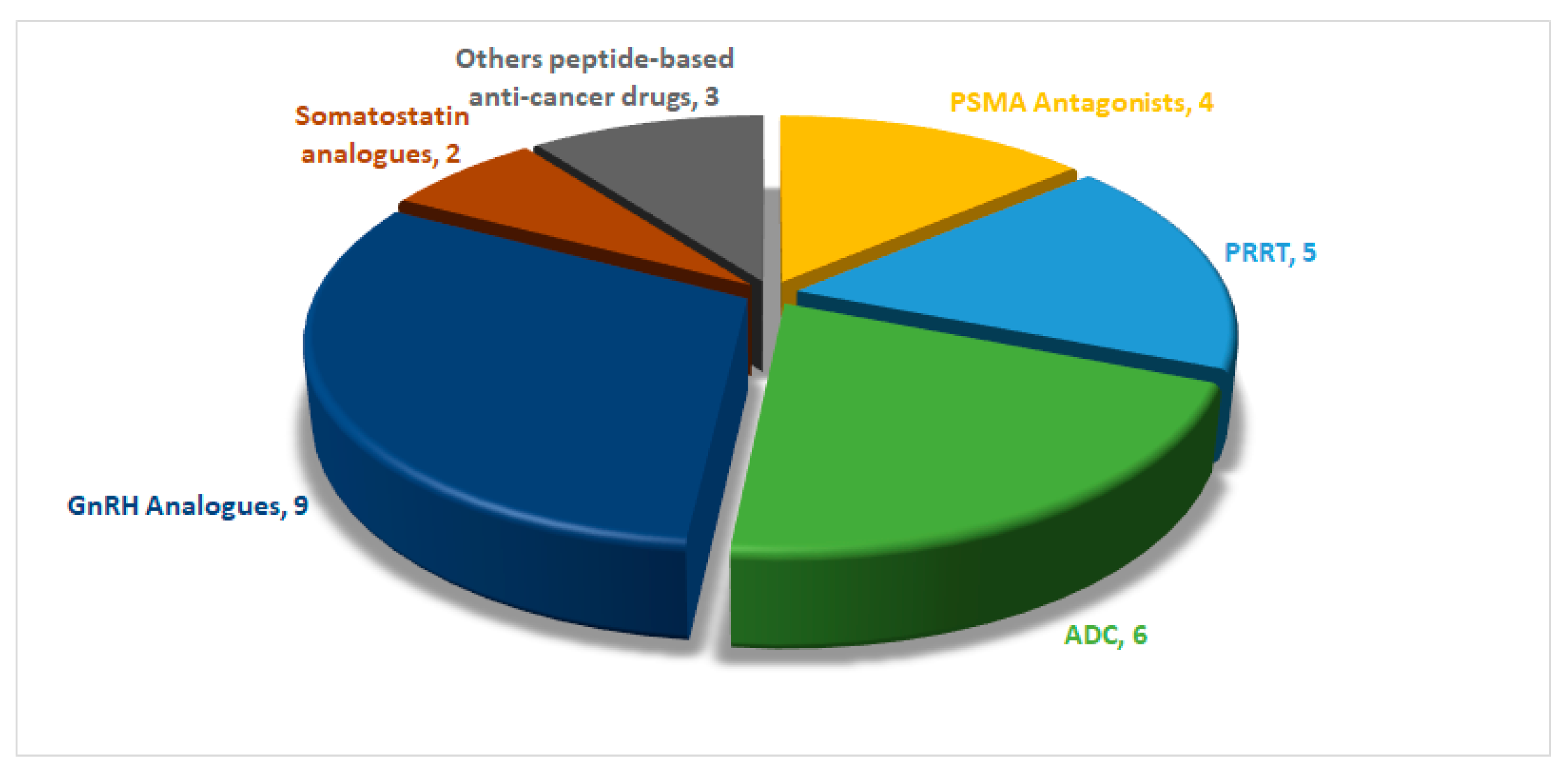
1. Introduction
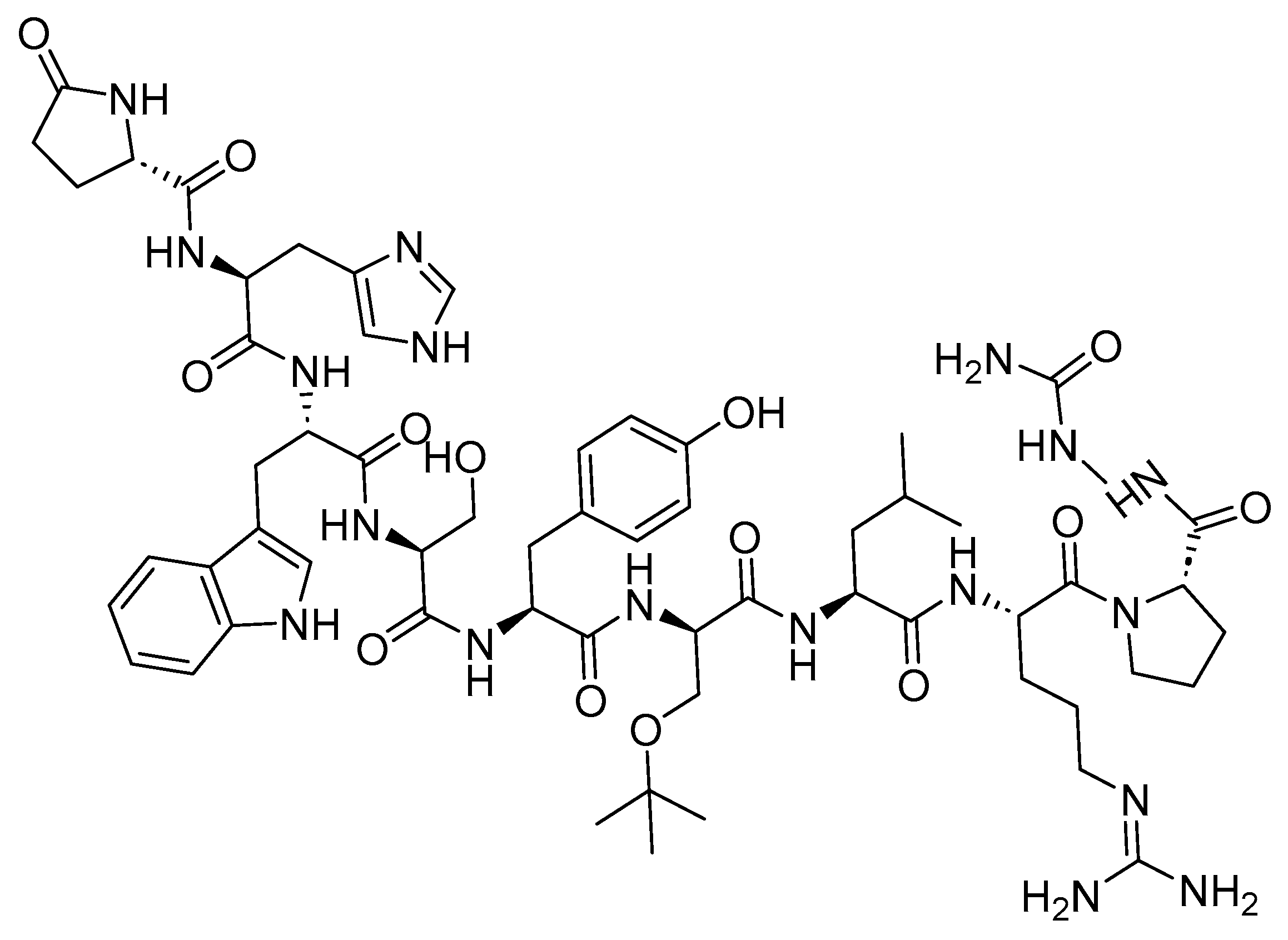
2. Prostate-Specific Membrane Antigen (PSMA) Peptide Antagonists
2.1. 68Ga-PSMA-11 (68Ga Gozetotide)
2.2. Piflufolastat F 18 (Pylarify)
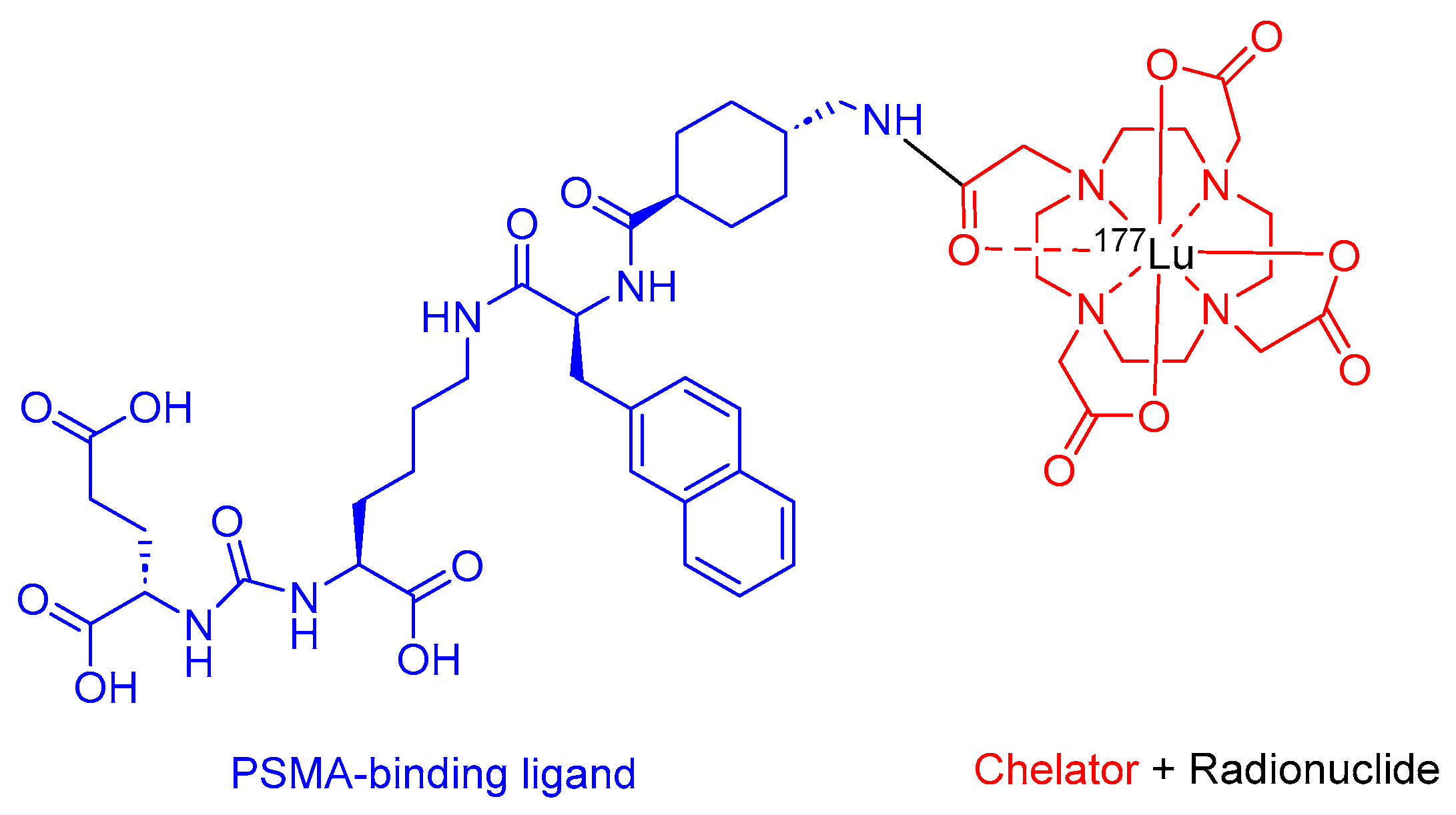
2.3. Lutetium 177Lu Vipivotide Tetraxetan (Pluvicto)
2.4. Flotufolastat F 18 (Posluma)
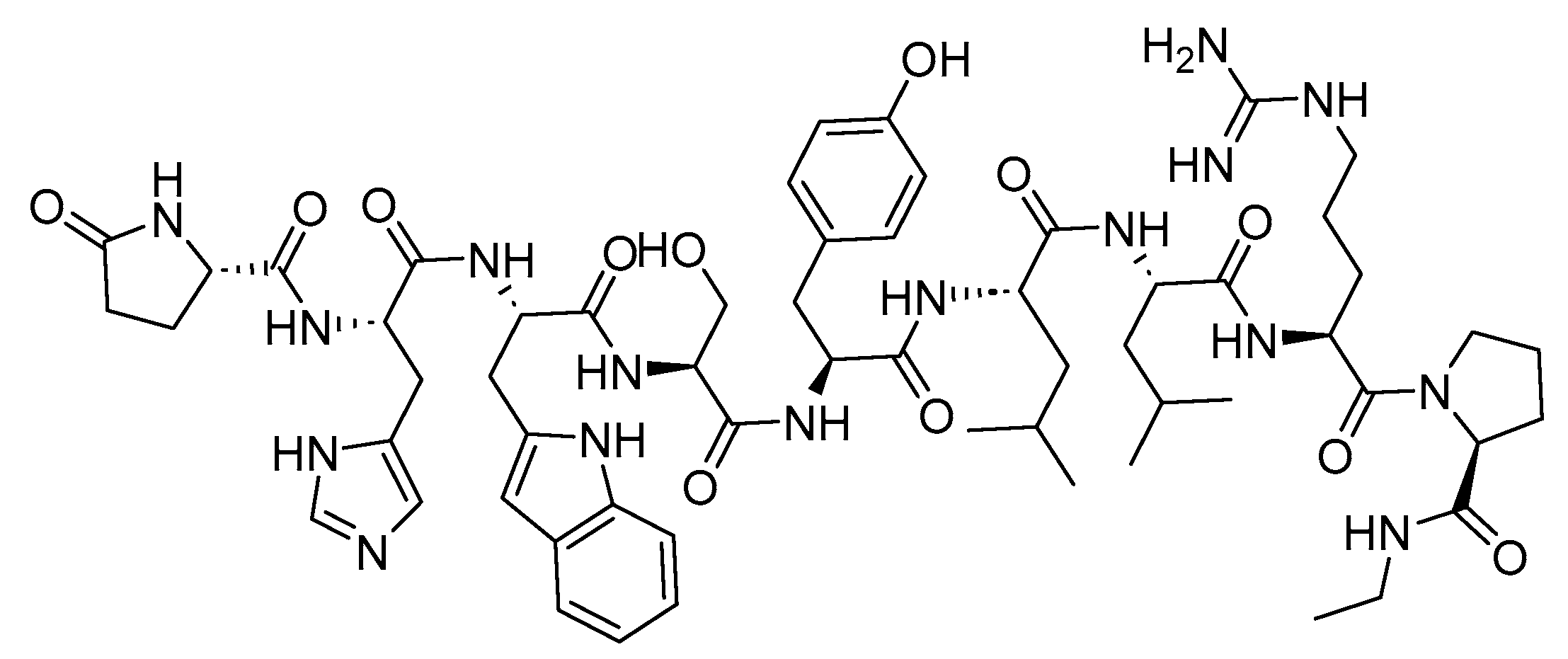
3. Peptide Receptor Radionuclide Therapy (PRRT)
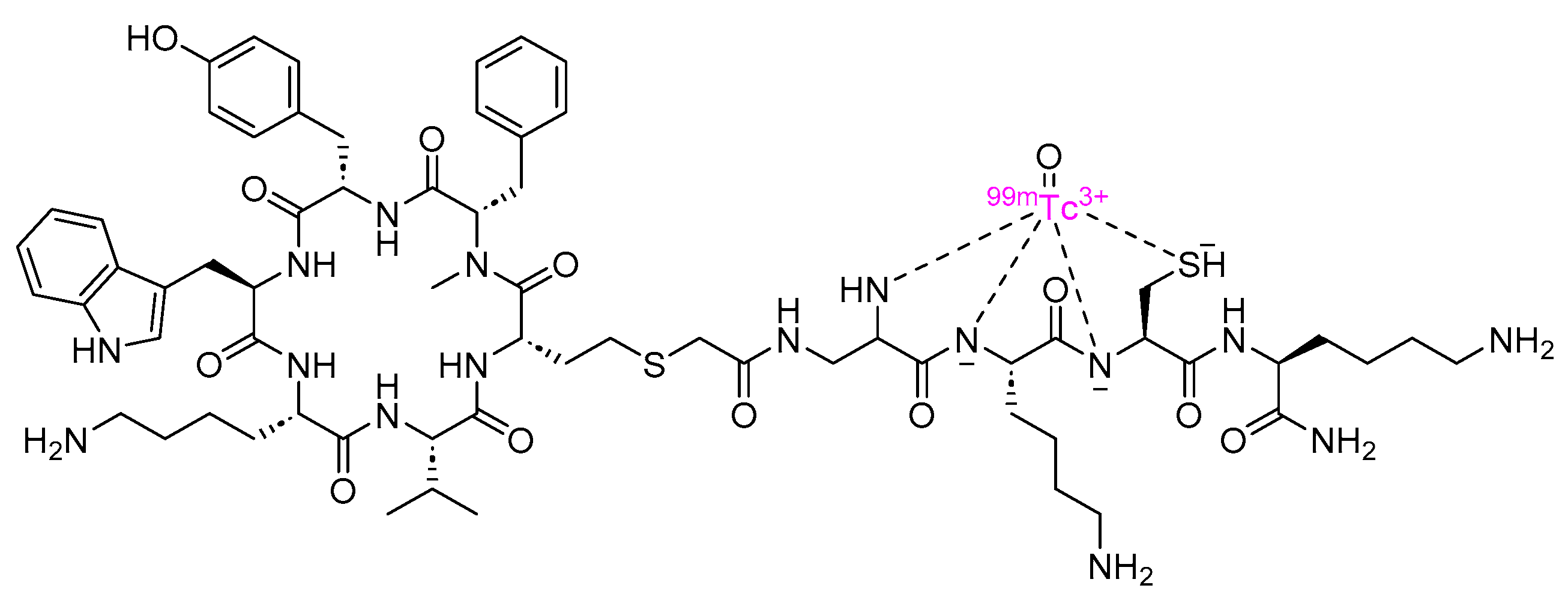
3.1. Depreotide (Neotect)
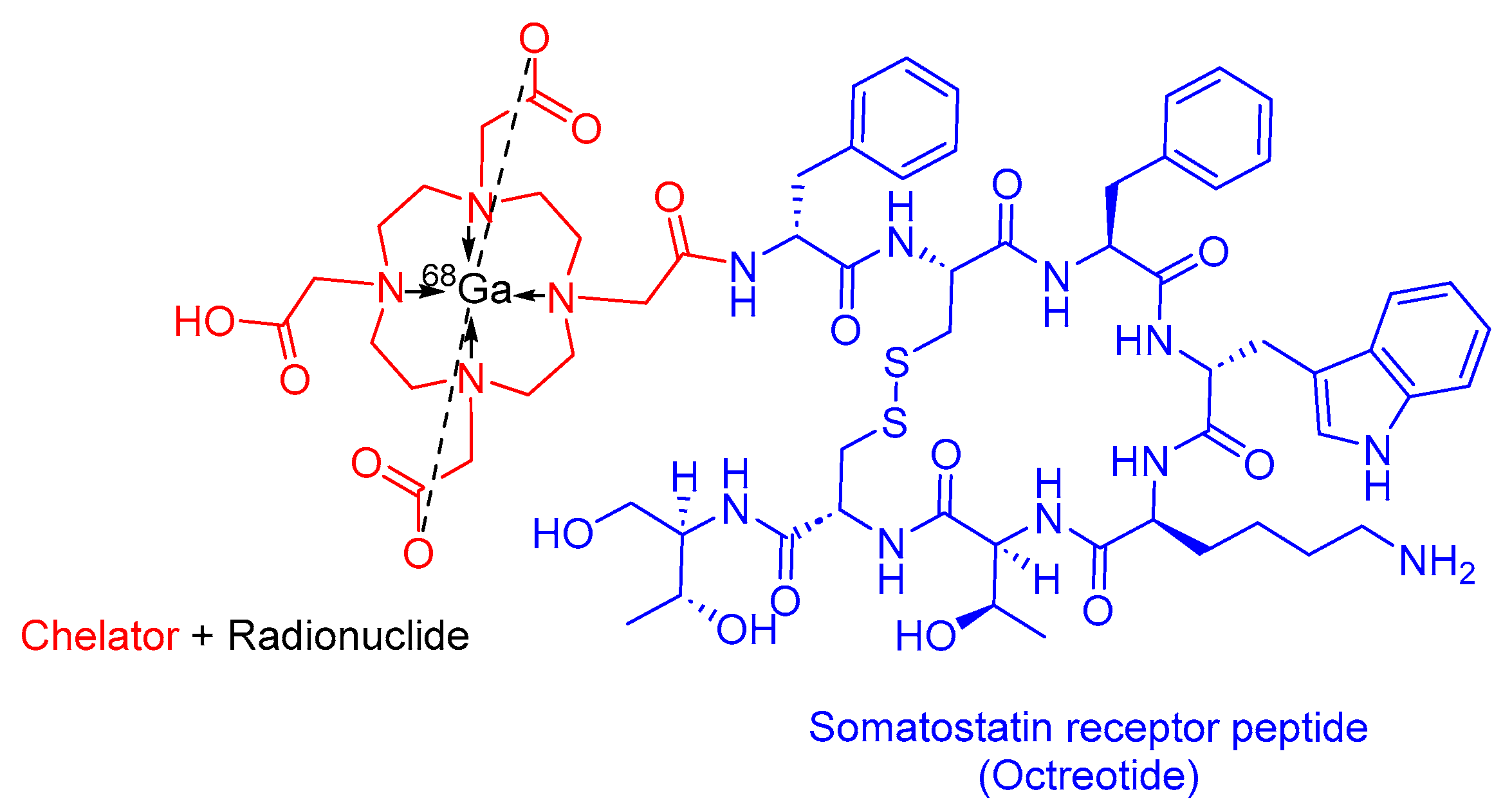
3.2. 68Ga-DOTATATE(Netspot)
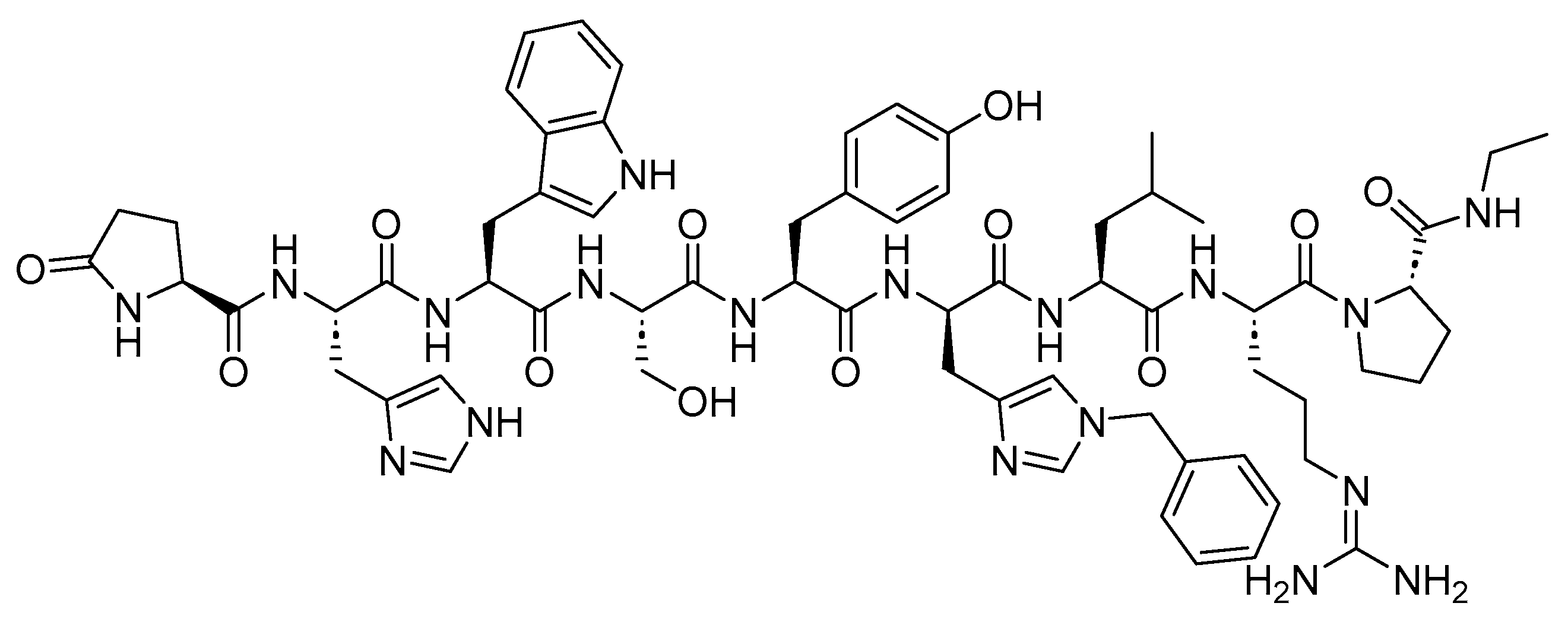
3.3. [177Lutetium]Lu-DOTA-TATE (Lutathera)
3.4. 68Ga-DOTATOC
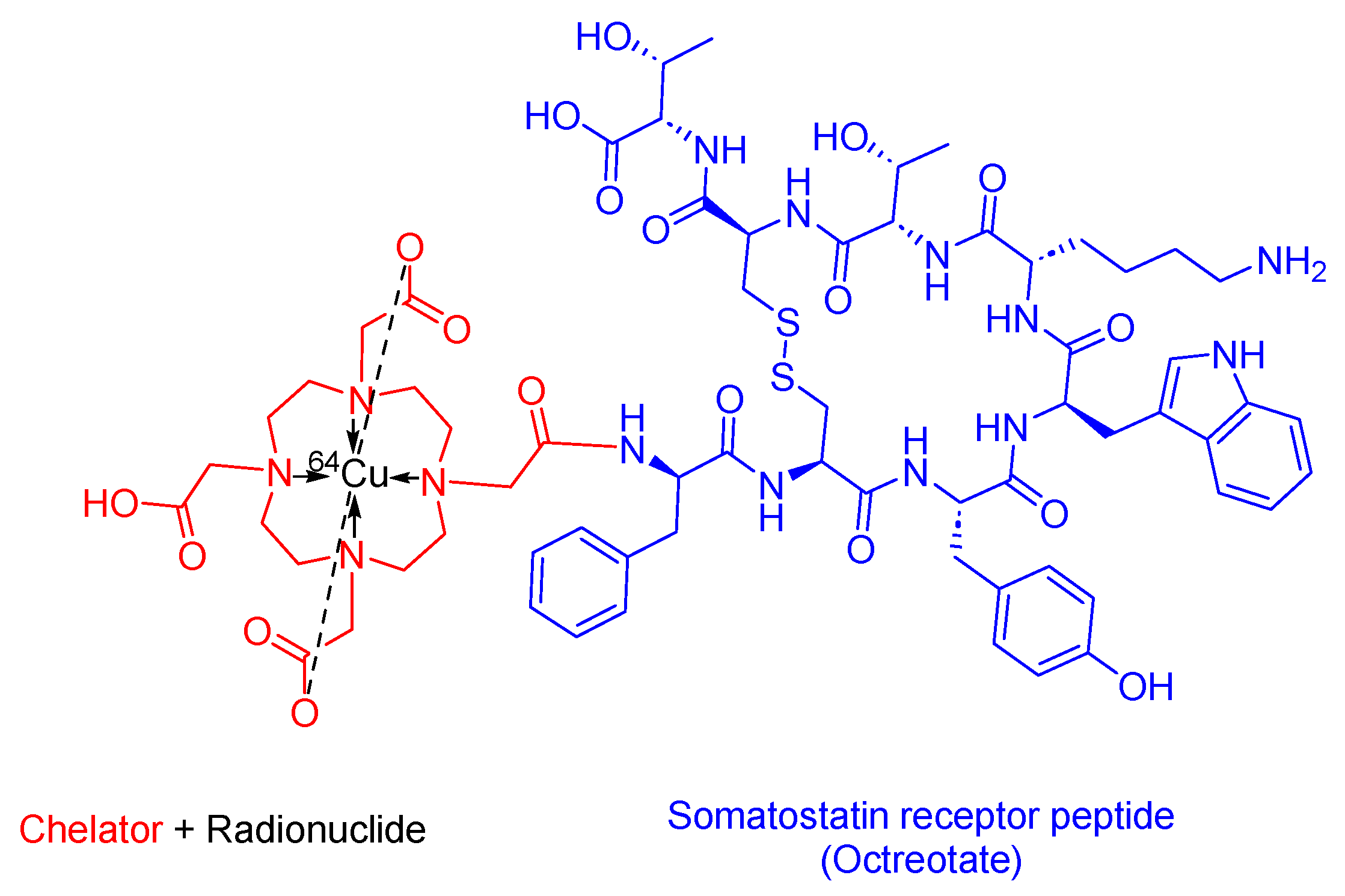
3.5. 64Cu-DOTATATE (Detectnet)
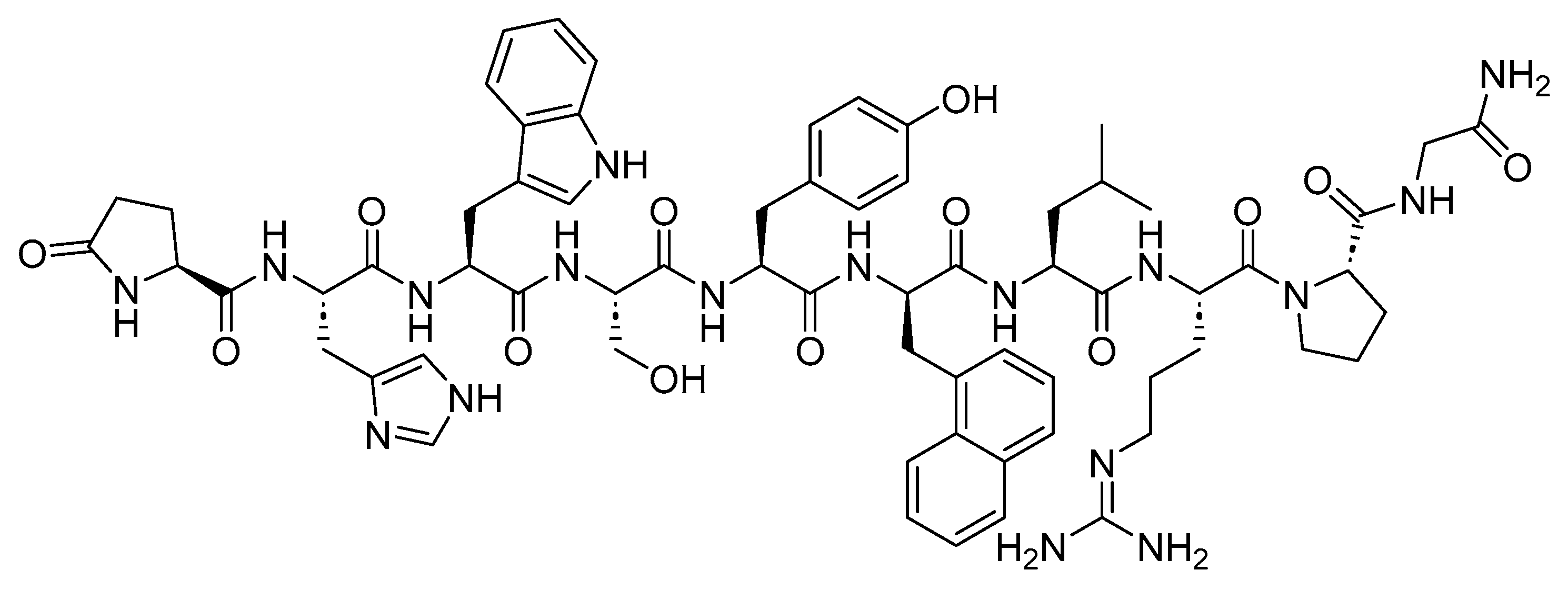
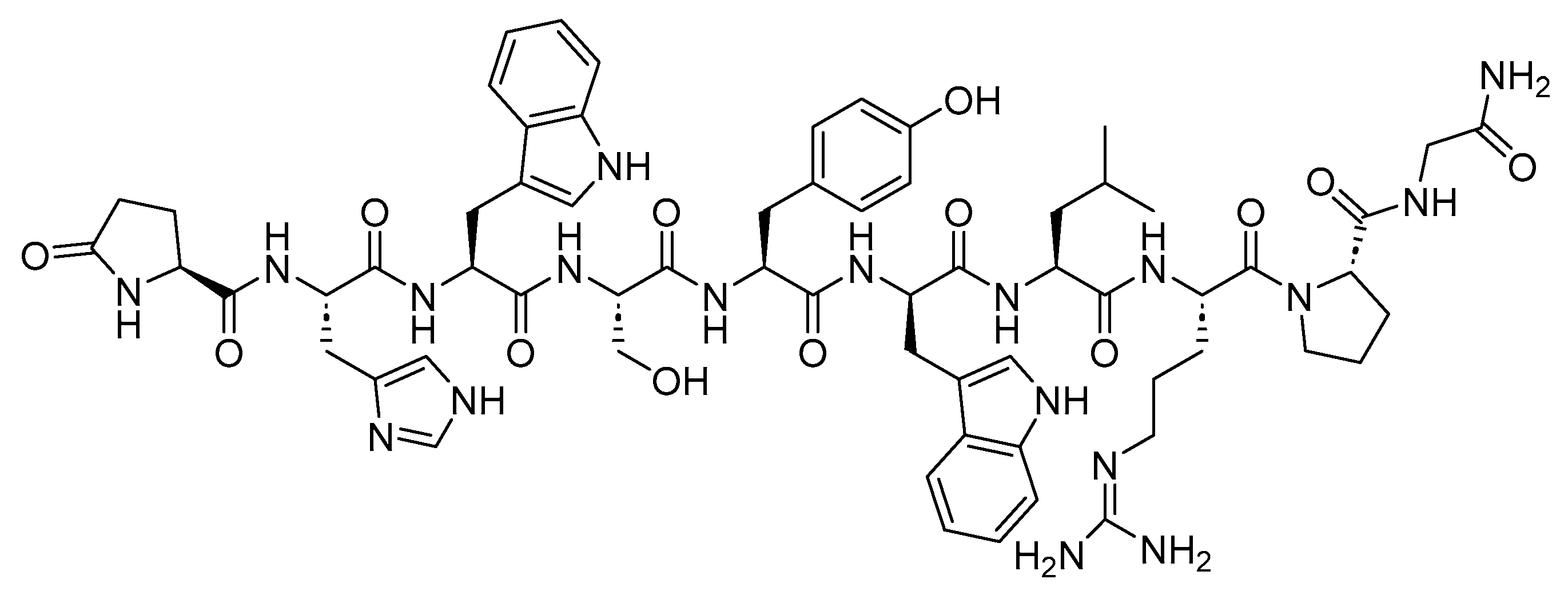
4. Somatostatin Analogs
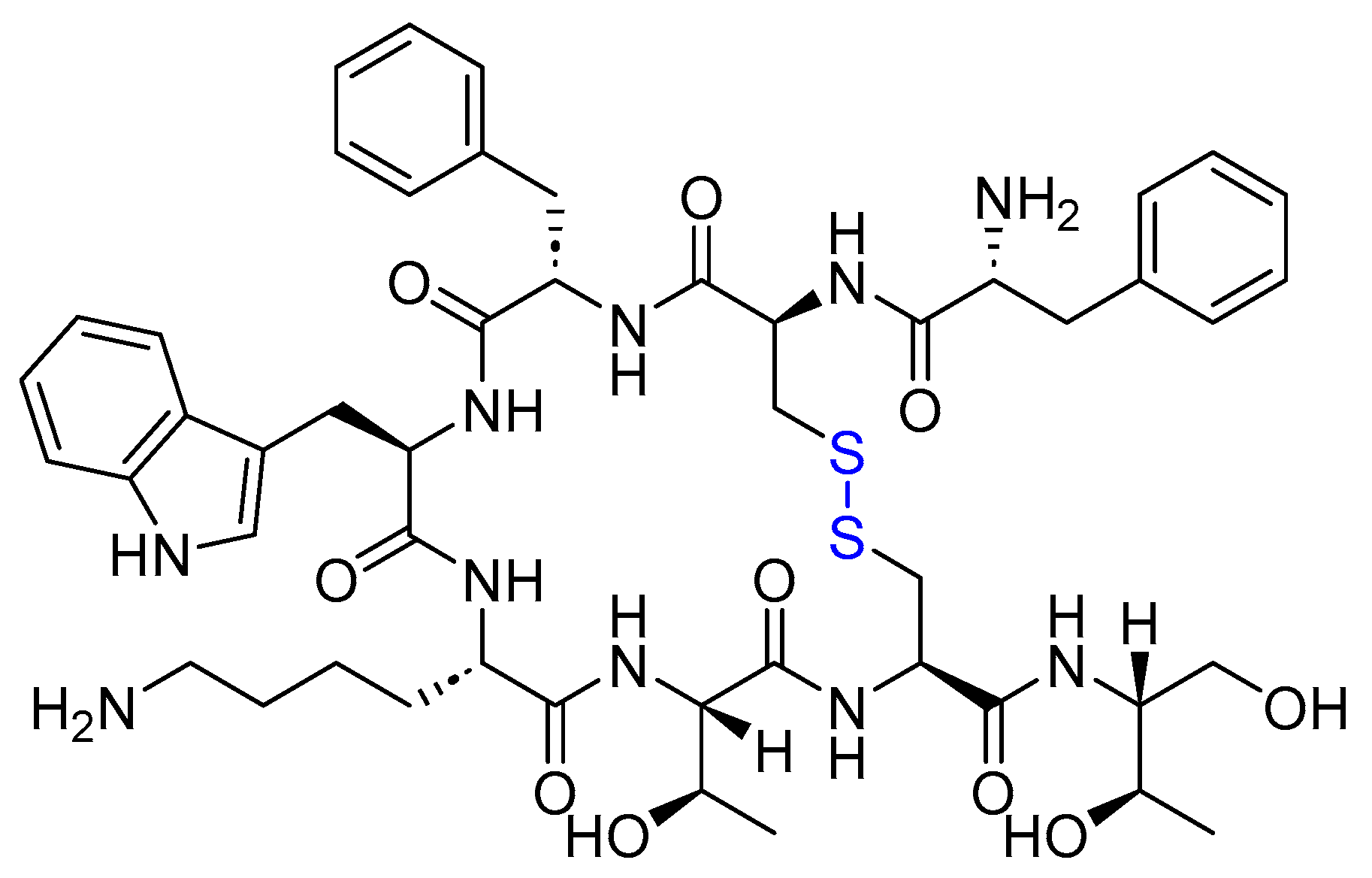
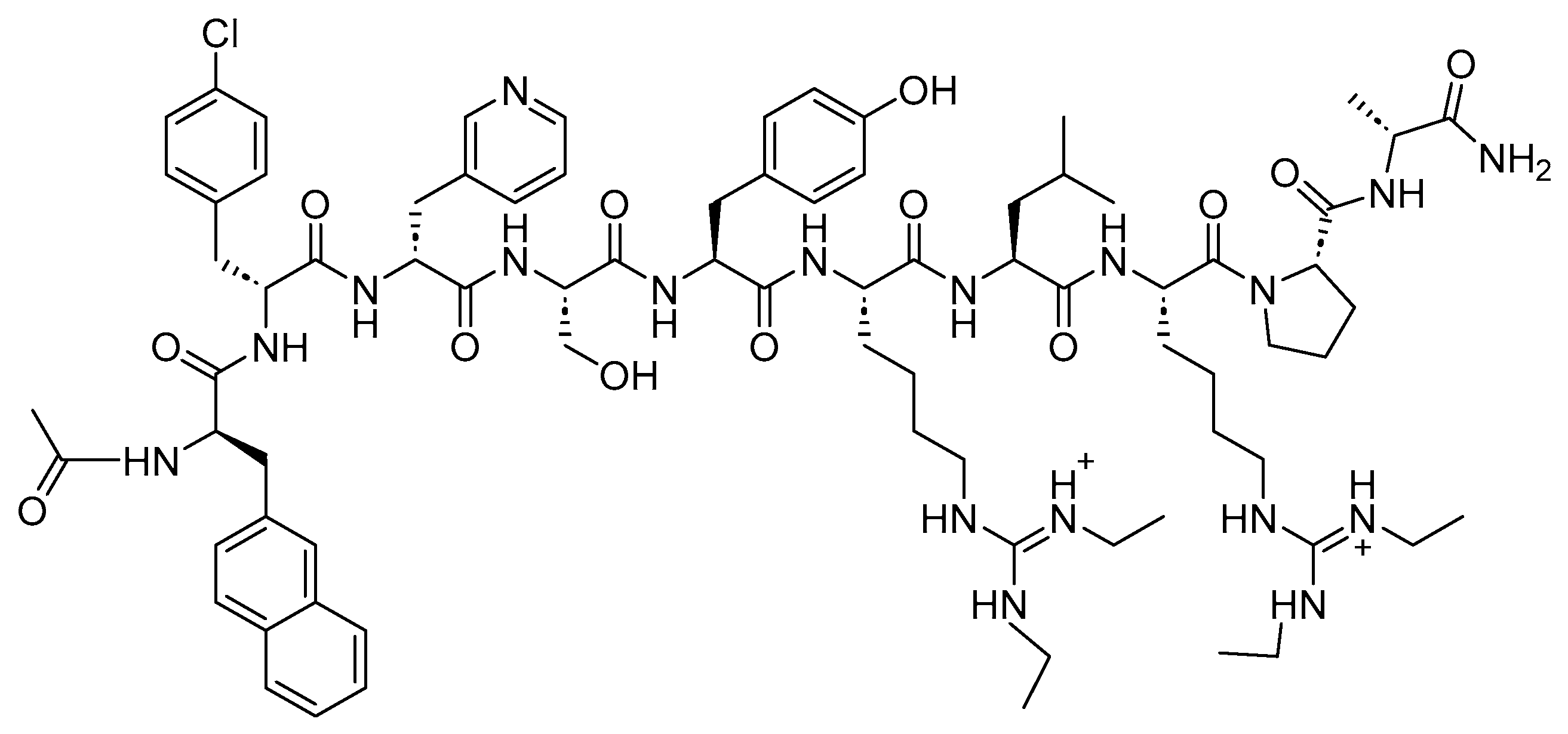
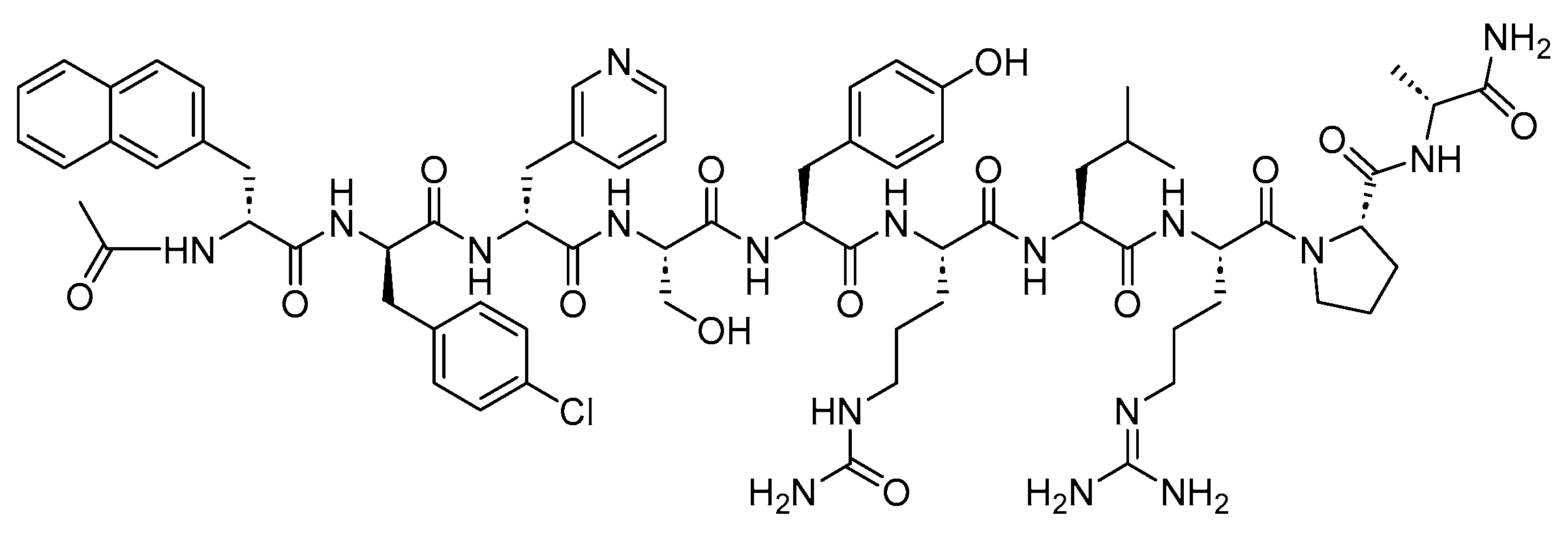
4.1. Octreotide (Sandostatin)
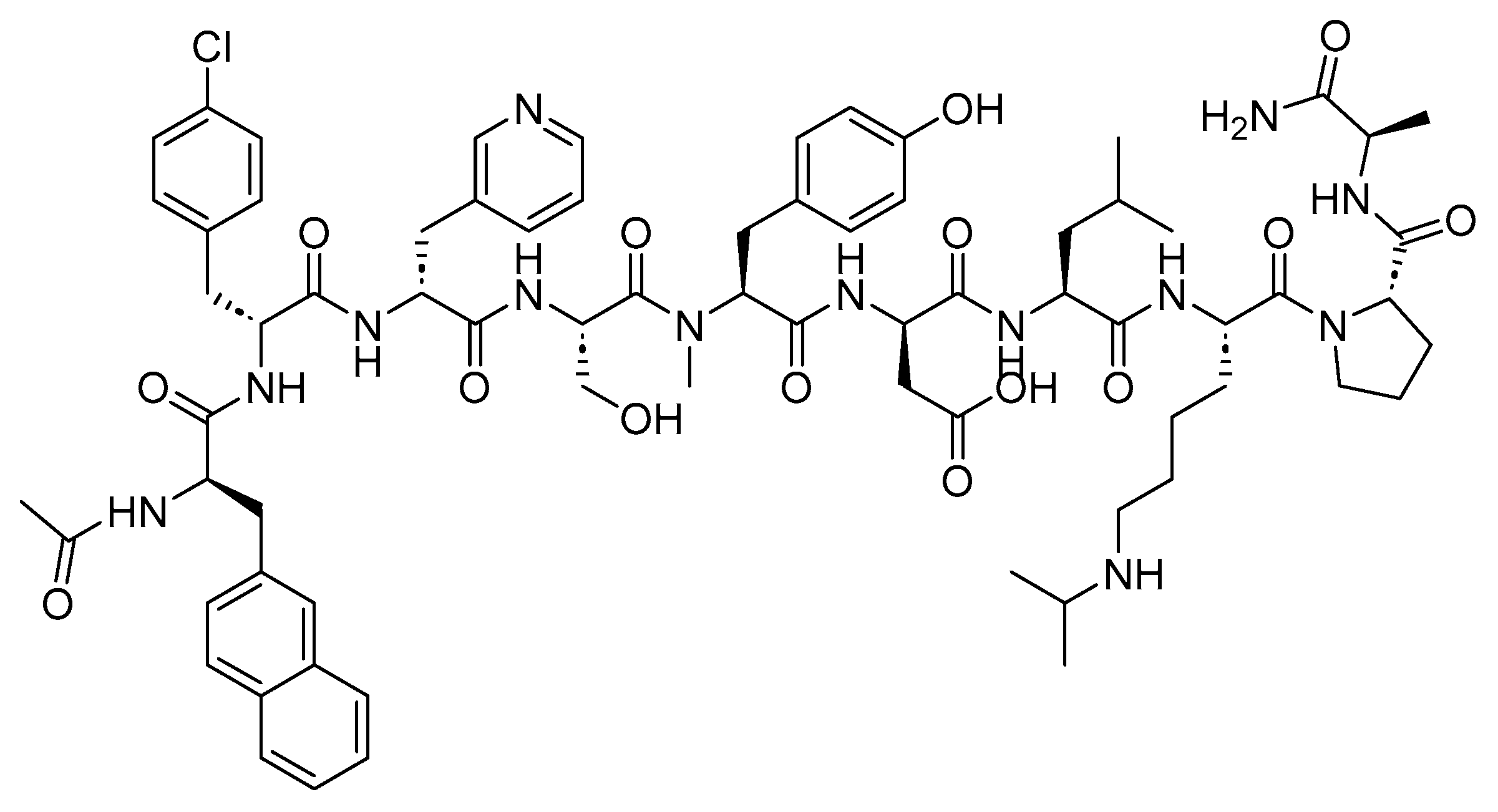
4.2. Lanreotide (Somatuline)
5. Antibody Drug Conjugate (ADCs)
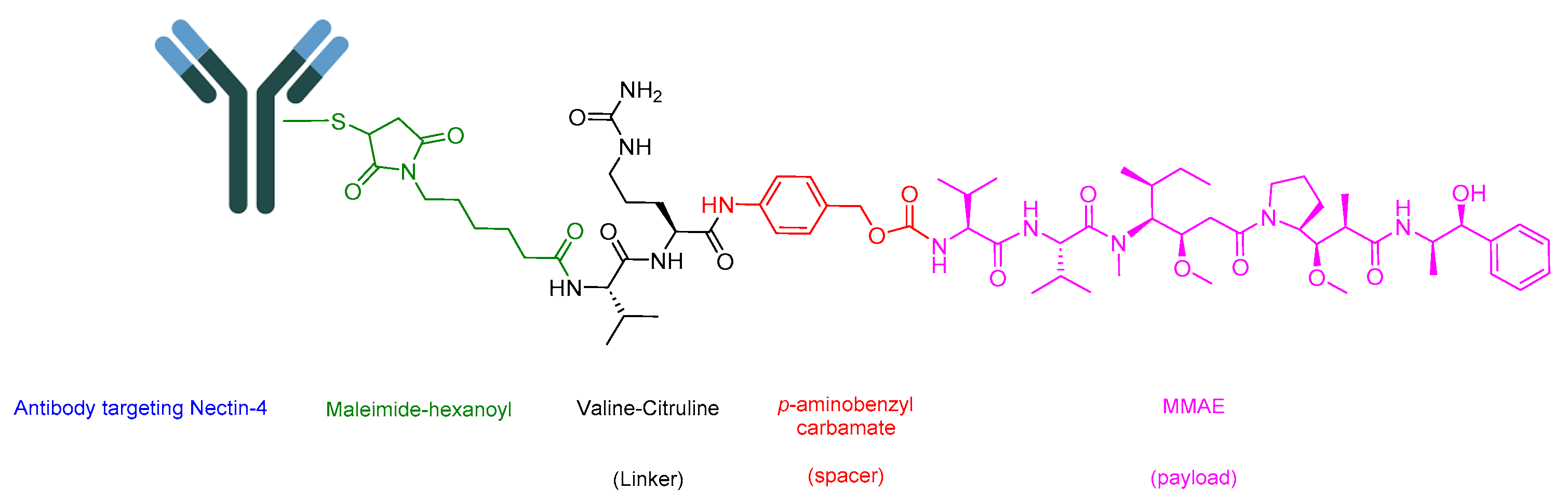
5.1. Enfortumab Vedotin-Ejfv (Padcev)
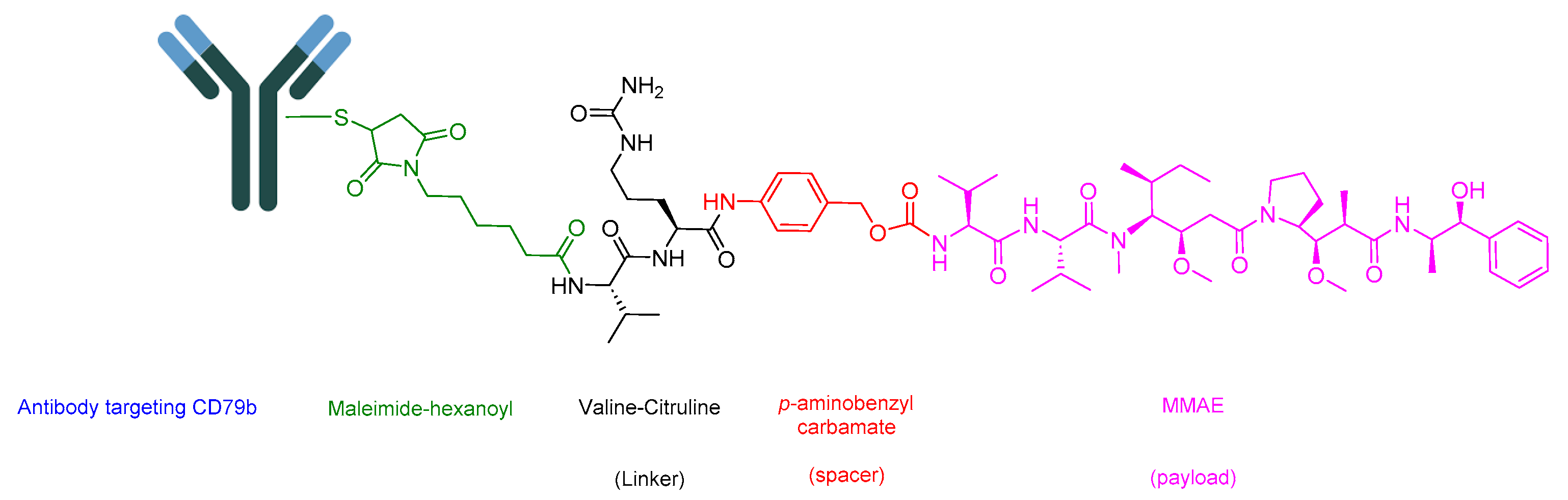
5.2. Polatuzumab Vedotin-Piiq (Polivy)
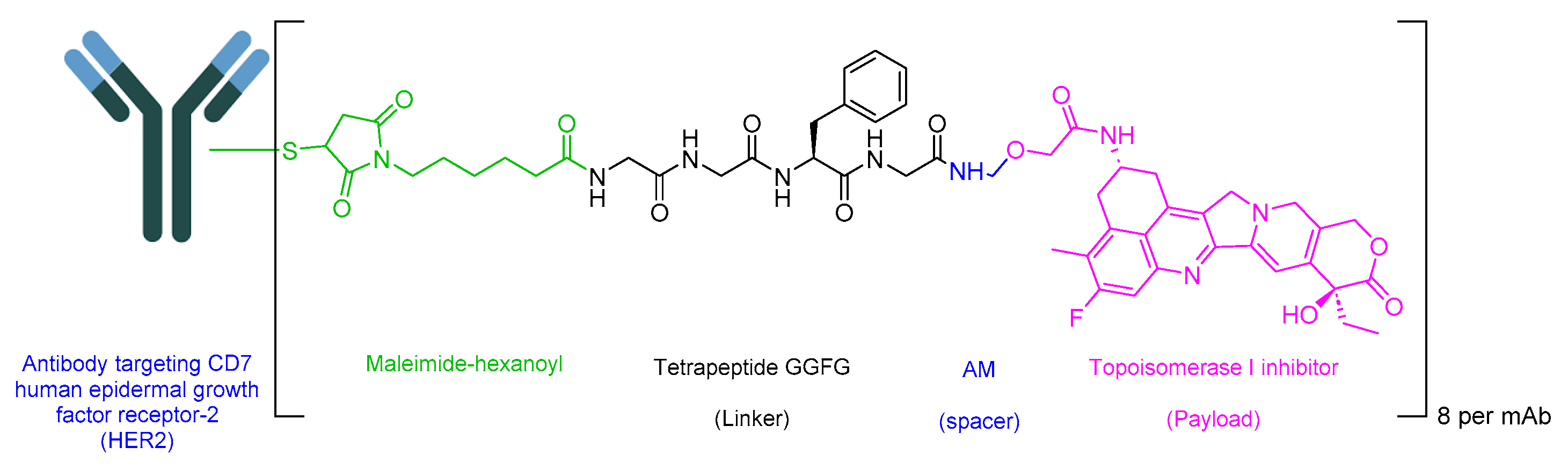
5.3. Fam-Trastuzumab Deruxtecan-Nxki (Enhertu)
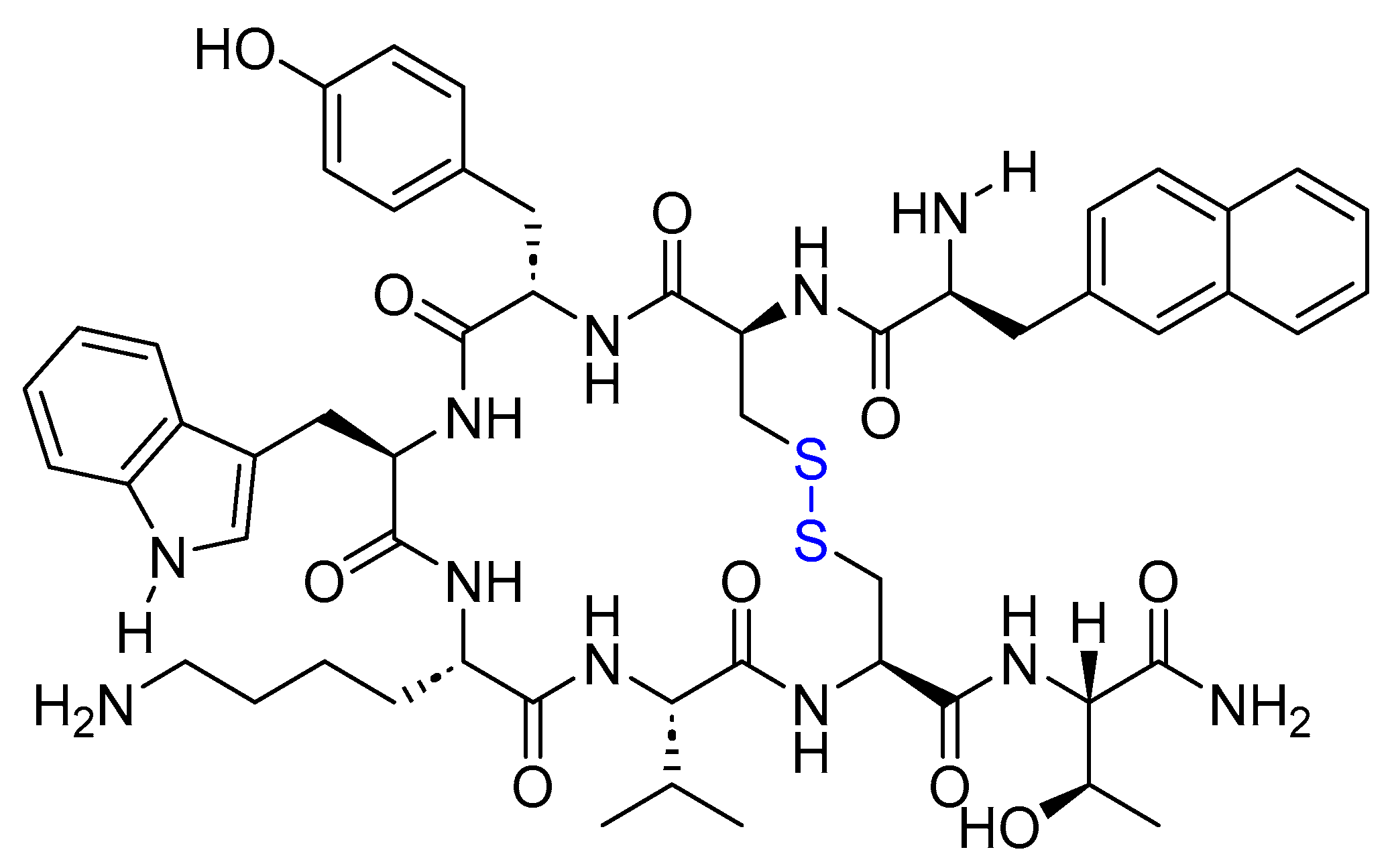
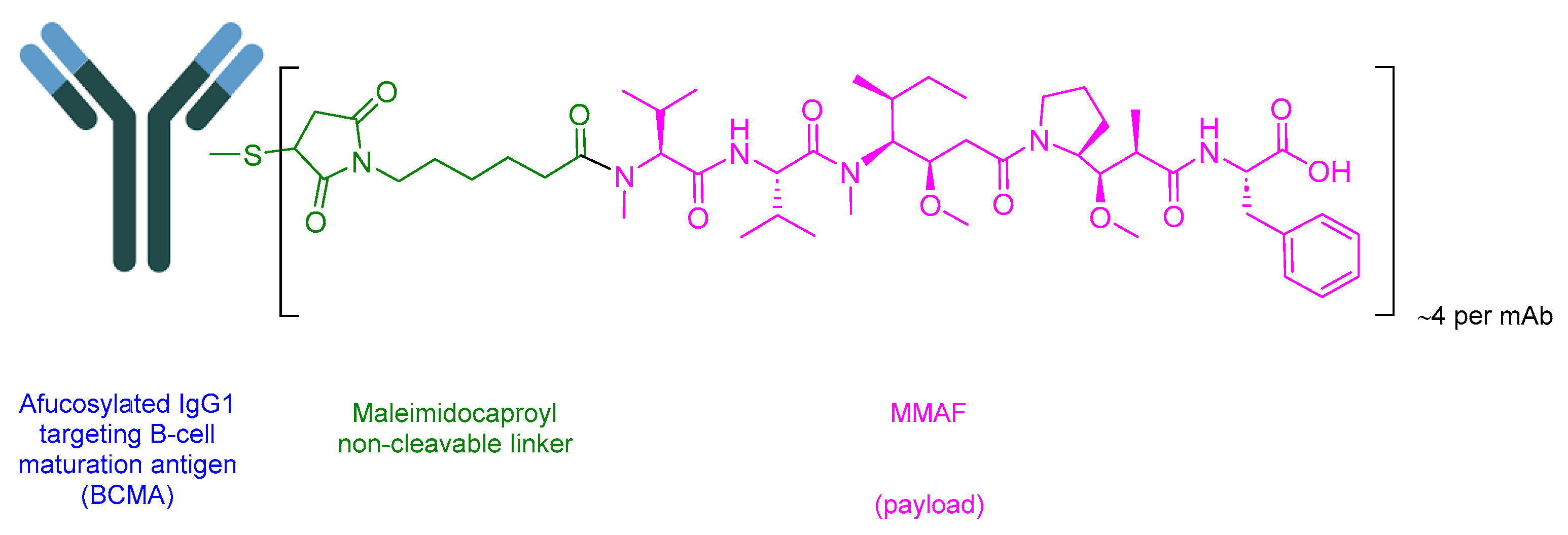
5.4. Belantamab Mafodotin-Blmf (Blenrep)
5.5. Tisotumab Vedotin-Tftv (Tivdak)
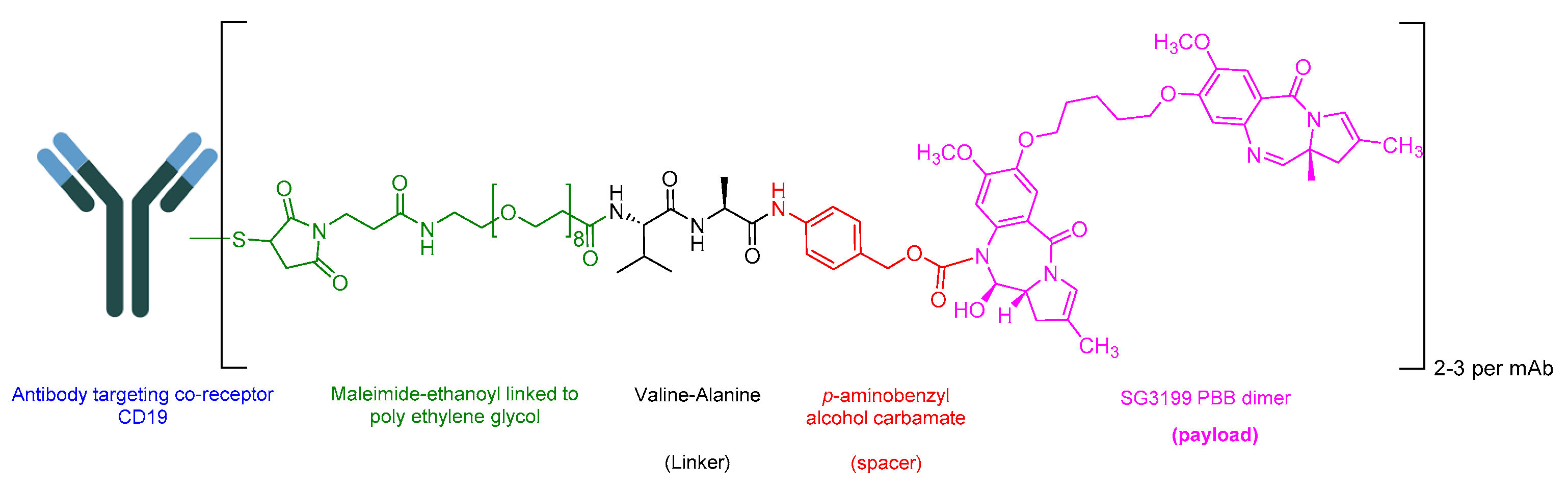
5.6. Loncastuximab Tesirine-Lpyl (Zynlonta)
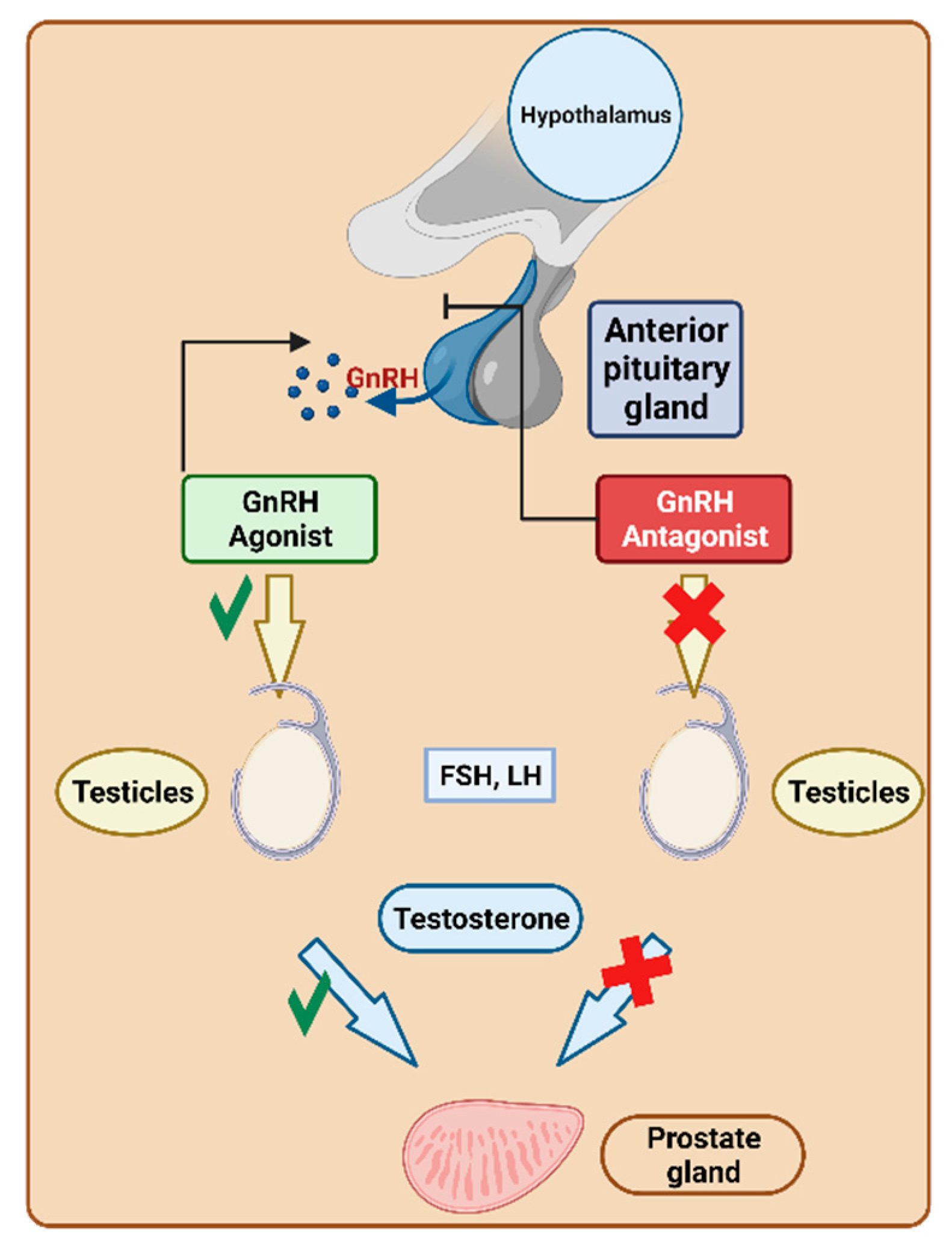
6. Gonadotropin-Releasing Hormone (GnRH) Analogs
6.1. GnRH Agonists
6.1.1. Goserelin (Zoladex)
6.1.2. Leuprolide (Lupron)
6.1.3. Nafarelin (Synarel)
6.1.4. Trelstar (Triptorelin)
6.1.5. Histrelin (Supprelin LA)
6.2. GnRH Antagonists
6.2.1. Ganirelix (Antagon)
6.2.2. Cetrorelix (Cetrotide)
6.2.3. Abarelix (Plenaxis)
6.2.4. Degarelix (Firmagon)
7. Other Peptide-Based Anticancer Drugs
7.1. Bortezomib (Velcade)
7.2. Carfilzomib (Kyprolis)
7.3. Melphalan Flufenamide (Pepaxto)
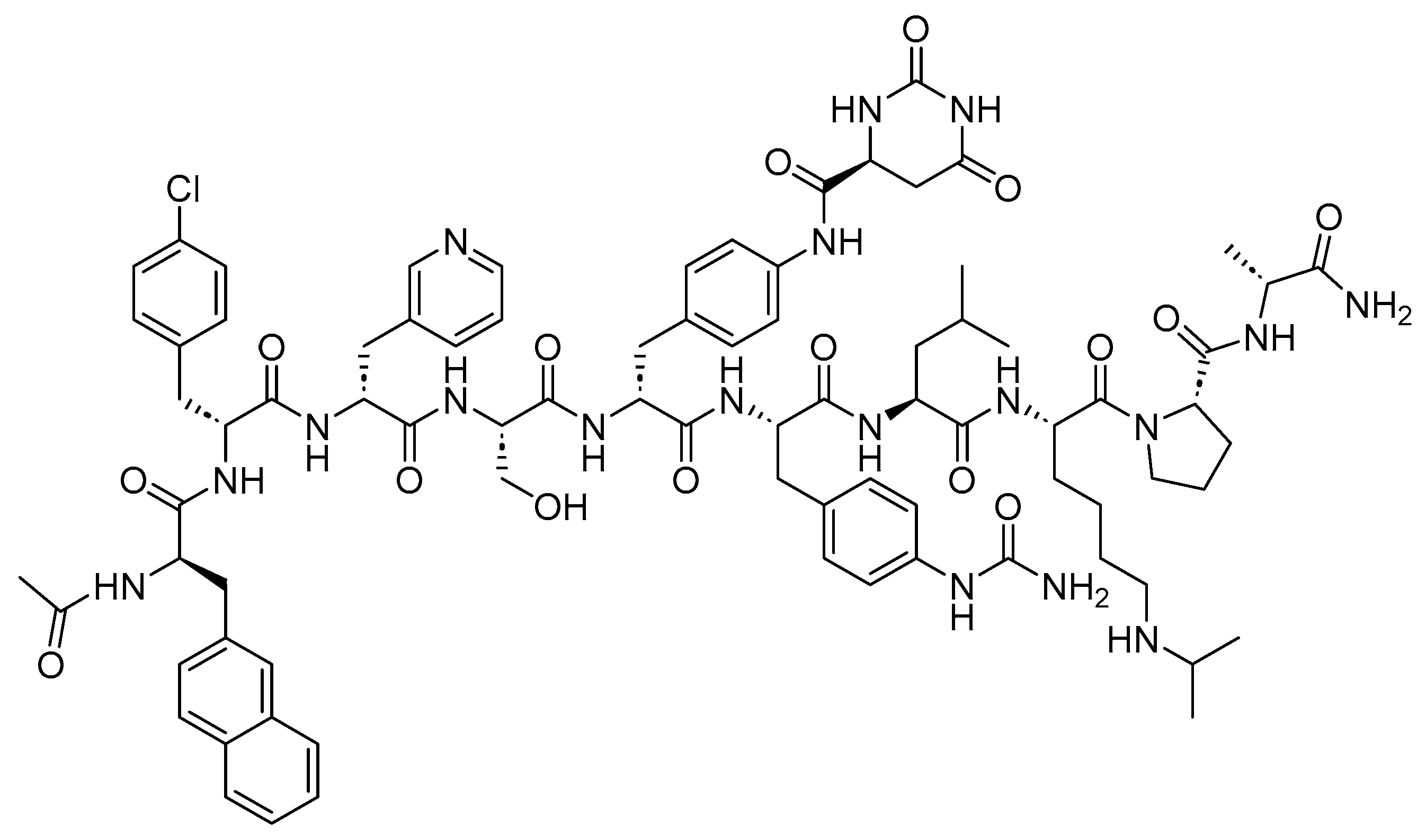
8. Peptide Drug Conjugates (PDCs)
9. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-targeted delivery systems based on peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef]
- Gronewold, A.; Horn, M.; Neundorf, I. Design and biological characterization of novel cell-penetrating peptides preferentially targeting cell nuclei and subnuclear regions. Beilstein J. Org. Chem. 2018, 14, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Curtius, T. Ueber einige neue der Hippursäure analog constituirte, synthetisch dargestellte Amidosäuren. J. Prakt. Chem. 1882, 26, 145–208. [Google Scholar] [CrossRef]
- Fischer, E.; Fourneau, E. Ueber einige Derivate des Glykocolls. Ber. Dtsch. Chem. Bunsenges 1901, 34, 2868–2877. [Google Scholar] [CrossRef]
- Scott, D.A.; Best, C.H. The Preparation of Insulin. Ind. Eng. Chem. 1925, 17, 238–240. [Google Scholar] [CrossRef]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front. Endocrinol. 2018, 9, 613. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. Exploring FDA-Approved Frontiers: Insights into Natural and Engineered Peptide Analogues in the GLP-1, GIP, GHRH, CCK, ACTH, and α-MSH Realms. Biomolecules 2024, 14, 264. [Google Scholar] [CrossRef]
- Peptide Therapeutics Market Size & Share Analysis—Growth Trends & Forecasts (2023–2028). Available online: https://www.mordorintelligence.com/industry-reports/peptide-therapeutics-market (accessed on 25 February 2024).
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2023 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2024, 17, 243. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2022: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2023, 28, 1038. [Google Scholar] [CrossRef]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Sig. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Shi, R.; Song, Z.; Wu, J. Metabolic Control of Epilepsy: A Promising Therapeutic Target for Epilepsy. Front. Neurol. 2020, 11, 592514. [Google Scholar] [CrossRef] [PubMed]
- Davda, J.; Declerck, P.; Hu-Lieskovan, S.; Hickling, T.P.; Jacobs, I.A.; Chou, J.; Salek-Ardakani, S.; Kraynov, E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Gestwicki, J.E. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert. Rev. Mol. Med. 2012, 14, e16. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; De Bosscher, K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yu, J. Structure-based design for binding peptides in anti-cancer therapy. Biomaterials 2018, 156, 1–15. [Google Scholar] [CrossRef]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discov. 2022, 4, FDD75. [Google Scholar] [CrossRef]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef] [PubMed]
- Goserelin Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/19726-S018_ZOLADEX%203.6%20MG%20DEPOT_APPROV.PDF (accessed on 25 February 2024).
- Sun, X.; Li, Y.; Liu, T.; Li, Z.; Zhang, X.; Chen, X. Peptide-based imaging agents for cancer detection. Adv. Drug Deliv. Rev. 2017, 110–111, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, S.L. Phage display in molecular imaging and diagnosis of cancer. Chem. Rev. 2010, 110, 3196–3211. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, A.; de la Torre, B.G.; Albericio, F. Liquid-Phase Peptide Synthesis (LPPS): A Third Wave for the Preparation of Peptides. Chem. Rev. 2022, 122, 13516–13546. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Heston, W.D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell Biochem. 2004, 91, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Singh, P.; Topaloglu, O.; Isaacs, J.T.; Denmeade, S.R. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006, 66, 9171–9177. [Google Scholar] [CrossRef]
- Gallium 68 PSMA-11 Drug Label. 2020. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212642s000lbl.pdf (accessed on 25 February 2024).
- Gallium 68 PSMA-11 Approval Letter. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/212642Orig1s000ltr.pdf (accessed on 25 February 2024).
- Piflufolastat F 18 Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214793s000lbl.pdf (accessed on 25 February 2024).
- Piflufolastat F 18 Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214793Orig1s000Approv.pdf (accessed on 25 February 2024).
- Pluvicto Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215833s000lbl.pdf (accessed on 25 February 2024).
- Keam, S.J. Lutetium Lu 177 Vipivotide Tetraxetan: First Approval. Mol. Diagn. Ther. 2022, 26, 467–475. [Google Scholar] [CrossRef]
- Shah, H.; Ravi, P.; Sonpavde, G.; Jacene, H. Lutetium Lu 177 vipivotide tetraxetan for metastatic castration-resistant prostate cancer. Expert. Rev. Anticancer Therapy 2022, 22, 1163–1175. [Google Scholar] [CrossRef]
- Pluvicto Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215833Orig1s000ltr.pdf (accessed on 25 February 2024).
- Posluma Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216023s000lbl.pdf (accessed on 25 February 2024).
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2022 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2023, 16, 336. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Flotufolastat F 18: Diagnostic First Approval. Mol. Diagn. Ther. 2023, 27, 631–636. [Google Scholar] [CrossRef]
- Bergsma, H.; van Vliet, E.I.; Teunissen, J.J.M.; Kam, B.L.R.; de Herder, W.W.; Peeters, R.P.; Krenning, E.P.; Kwekkeboom, D.J. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best. Pract. Res. Clin. Gastroenterol. 2012, 26, 867–881. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Pillai, M.R.; Knapp, F.F. Lutetium-177 therapeutic radiopharmaceuticals: Linking chemistry, radiochemistry, and practical applications. Chem. Rev. 2015, 115, 2934–2974. [Google Scholar] [CrossRef] [PubMed]
- Depreotide Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2004/21012s007lbl.pdf (accessed on 25 February 2024).
- Wei-Jen, S.; Ramon, A.L.R.; Mullet, T.; Primo, P.M. 99mTc-Depreotide Chest SPECT Demonstrates Pulmonary Metastases from Renal Cell Carcinoma. J. Nucl. Med. Technol. 2004, 32, 19. [Google Scholar]
- Depreotide Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21012_Neotect_appltrs.pdf (accessed on 25 February 2024).
- Kane, S.M.; Padda, I.S.; Davis, D.D. Technetium-99m; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- 68Ga-DOTATATE Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208547s000lbl.pdf (accessed on 25 February 2024).
- Mojtahedi, A.; Thamake, S.; Tworowska, I.; Ranganathan, D.; Delpassand, E.S. The value of 68Ga-DOTATATE PET/CT in diagnosis and management of neuroendocrine tumors compared to current FDA approved imaging modalities: A review of literature. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 426–434. [Google Scholar]
- Menda, Y.; Ponto, L.L.B.; Schultz, M.K.; Zamba, G.K.D.; Watkins, G.L.; Bushnell, D.L.; Madsen, M.T.; Sunderland, J.J.; Graham, M.M.; O’Dorisio, T.M.; et al. Repeatability of Gallium-68 DOTATOC Positron Emission Tomographic Imaging in Neuroendocrine Tumors. Pancreas 2013, 42, 937–943. [Google Scholar] [CrossRef]
- 68Ga-DOTATATE Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/208547Orig1s004ltr.pdf (accessed on 25 February 2024).
- Lutathera Drug Label. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208700s000lbl.pdf (accessed on 25 February 2024).
- Kvols, L.K.; Reubi, J.C.; Horisberger, U.; Moertel, C.G.; Rubin, J.; Charboneau, J.W. The presence of somatostatin receptors in malignant neuroendocrine tumor tissue predicts responsiveness to octreotide. Yale J. Biol. Med. 1992, 65, 505–518, discussion 531-6. [Google Scholar]
- Rogoza, O.; Megnis, K.; Kudrjavceva, M.; Gerina-Berzina, A.; Rovite, V. Role of Somatostatin Signalling in Neuroendocrine Tumours. Int. J. Mol. Sci. 2022, 23, 1447. [Google Scholar] [CrossRef]
- Lutathera Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/208700Orig1s000ltr.pdf (accessed on 25 February 2024).
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef]
- Kam, B.L.; Teunissen, J.J.; Krenning, E.P.; de Herder, W.W.; Khan, S.; van Vliet, E.I.; Kwekkeboom, D.J. Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S103–S112. [Google Scholar] [CrossRef]
- 68Ga-DOTATOC Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210828s000lbl.pdf (accessed on 25 February 2024).
- 68Ga-DOTATOC Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210828Orig1s000ltr.pdf (accessed on 25 February 2024).
- Detectnet Drug Label. 2020. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213227s000lbl.pdf (accessed on 25 February 2024).
- Gutfilen, B.; Souza, S.A.; Valentini, G. Copper-64: A real theranostic agent. Drug Des. Dev. Ther. 2018, 12, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Johnbeck, C.B.; Knigge, U.; Loft, A.; Berthelsen, A.K.; Mortensen, J.; Oturai, P.; Langer, S.W.; Elema, D.R.; Kjaer, A. Head-to-Head Comparison of 64Cu-DOTATATE and 68Ga-DOTATOC PET/CT: A Prospective Study of 59 Patients with Neuroendocrine Tumors. J. Nucl. Med. 2017, 58, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Detectnet Aprroval Letter. 2020. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/213227Orig1s000ltr.pdf (accessed on 25 February 2024).
- Zhao, W.; Han, S.; Qiu, N.; Feng, W.; Lu, M.; Zhang, W.; Wang, M.; Zhou, Q.; Chen, S.; Xu, W.; et al. Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 2022, 32, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C. Molecular pharmacology of somatostatin receptor subtypes. J. Endocrinol. Investig. 1997, 20, 348–367. [Google Scholar] [CrossRef] [PubMed]
- Octreotide Approval Letter and Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/021008a_appltr_prntlbl.pdf (accessed on 25 February 2024).
- Prelević, G.M.; Wurzburger, M.I.; Balint-Perić, L.; Nesić, J.S. Inhibitory effect of sandostatin on secretion of luteinising hormone and ovarian steroids in polycystic ovary syndrome. Lancet 1990, 336, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Battershill, P.E.; Clissold, S.P. Octreotide. Drugs 1989, 38, 658–702. [Google Scholar] [CrossRef] [PubMed]
- Mycapssa Drug Label. Available online: https://label.mycapssa.com/wp-content/uploads/sites/4/2020/06/prescribinginformation.pdf (accessed on 25 February 2024).
- Mycapssa Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/208232Orig1s000ltr.pdf (accessed on 25 February 2024).
- Lanreotide Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215395s000lbl.pdf (accessed on 25 February 2024).
- Lanreotide Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022074s000_Approv.pdf (accessed on 25 February 2024).
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Dal Corso, A.; Cazzamalli, S.; Gebleux, R.; Mattarella, M.; Neri, D. Protease-Cleavable Linkers Modulate the Anticancer Activity of Noninternalizing Antibody-Drug Conjugates. Bioconjug. Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Jackson, D.; Atkinson, J.; Guevara, C.I.; Zhang, C.; Kery, V.; Moon, S.J.; Virata, C.; Yang, P.; Lowe, C.; Pinkstaff, J.; et al. In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS ONE 2014, 9, e83865. [Google Scholar] [CrossRef]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Raines, R.T. Catalysis of imido group hydrolysis in a maleimide conjugate. Bioorg. Med. Chem. Lett. 2007, 17, 6286–6289. [Google Scholar] [CrossRef]
- Xu, Z.; Guo, D.; Jiang, Z.; Tong, R.; Jiang, P.; Bai, L.; Chen, L.; Zhu, Y.; Guo, C.; Shi, J.; et al. Novel HER2-Targeting Antibody-Drug Conjugates of Trastuzumab Beyond T-DM1 in Breast Cancer: Trastuzumab Deruxtecan(DS-8201a) and (Vic-)Trastuzumab Duocarmazine (SYD985). Eur. J. Med. Chem. 2019, 183, 111682. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [PubMed]
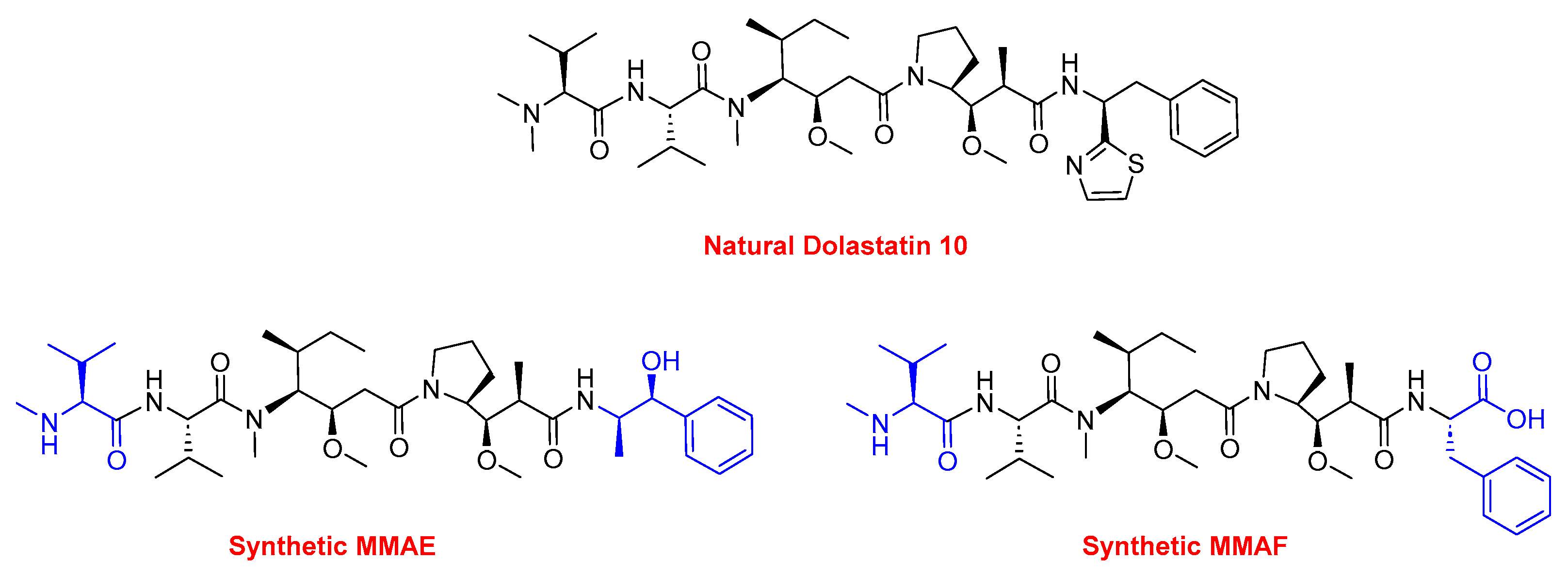
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloyd-Williams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. Antineoplastic agents. Part 189. The absolute configuration and synthesis of natural (-)-dolastatin 10. J. Am. Chem. Soc. 1989, 111, 5463–5465. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Y.; Hua, H.; Li, D.; Tang, C. Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry. Mar. Drugs 2021, 19, 363. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.; Jonnalagadda, V.; Bhakta, S.; Jammalamadaka, V.; Junutula, J.R. Exploration of the antibody–drug conjugate clinical landscape. mAbs 2023, 15, 2229101. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents 337. Synthesis of dolastatin 10 structural modifications. Anticancer. Drug Des. 1995, 10, 529–544. [Google Scholar]
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin. Investig. Drugs. 2019, 28, 821–826. [Google Scholar] [CrossRef]
- PADCEV Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 25 February 2024).
- Hanna, K.S. Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs 2019, 80, 1–7. [Google Scholar] [CrossRef]
- PADCEV Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761137Orig1s000ltr.pdf (accessed on 25 February 2024).
- Sehn, L.H.; Matasar, M.J.; Flowers, C.R.; Kamdar, M.; McMillan, A.K.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin Plus Bendamustine with Rituximab in Relapsed/Refractory Diffuse Large B-Cell Lymphoma: Updated Results of a Phase Ib/II Randomized Study. Blood 2019, 134 (Suppl. S1), 4081. [Google Scholar] [CrossRef]
- Polivy Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 25 February 2024).
- Onrust, S.V.; Lamb, H.M.; Barman Balfour, J.A. Rituximab. Drugs 1999, 58, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Muss, H.B.; Bundy, B.; DiSaia, P.J.; Homesley, H.D.; Fowler, W.C.; Creasman, W., Jr.; Yordan, E. Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 1985, 55, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. The POLARIX Study: Polatuzumab Vedotin with Rituximab, Cyclophosphamide, Doxorubicin, and Prednisone (pola-R-CHP) Versus Rituximab, Cyclophosphamide, Doxorubicin, Vincristine and Prednisone (R-CHOP) Therapy in Patients with Previously Untreated Diffuse Large B-Cell Lymphoma. Blood 2021, 138, LBA-1. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Masri, N.; Dalle, I.A.; El-Cheikh, J.; Ghanem, H.; Bazarbachi, A. Polatuzumab Vedotin: Current Role and Future Applications in the Treatment of Patients with Diffuse Large B-Cell Lymphoma. Clin. Hematol. Int. 2021, 3, 21–26. [Google Scholar] [CrossRef]
- Polivy Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761121Orig1s000ltr.pdf (accessed on 25 February 2024).
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorg. Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef]
- Enhertu Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761139s000lbl.pdf (accessed on 25 February 2024).
- Enhertu Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761139Orig1s000ltr.pdf (accessed on 25 February 2024).
- Blenrep Drug Label. 2020. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 25 February 2024).
- McMillan, A.; Warcel, D.; Popat, R. Antibody-drug conjugates for multiple myeloma. Expert Opin. Biol. Ther. 2021, 21, 889–901. [Google Scholar] [CrossRef]
- Blenrep Approval Letter. 16 January 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/761158Orig1s000ltr.pdf (accessed on 25 February 2024).
- TIVDAK Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208Orig1s000lbledt.pdf (accessed on 25 February 2024).
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef]
- TIVDAK Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761208Orig1s000_Corrected_ltr.pdf (accessed on 25 February 2024).
- Wang, Y.; Fan, S.; Zhong, W.; Zhou, X.; Li, S. Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload. Int. J. Mol. Sci. 2017, 18, 1860. [Google Scholar] [CrossRef]
- Zynlonta Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf (accessed on 25 February 2024).
- Zynlonta Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761196Orig1s000ltr.pdf (accessed on 25 February 2024).
- Hurine, J.A.; Lambalk, C.B. Gonadotropin-releasing-hormone-receptor antagonists. Lancet 2001, 358, 1793–1803. [Google Scholar] [CrossRef]
- Carolsfeld, J.; Powell, J.F.; Park, M.; Fischer, W.H.; Craig, A.G.; Chang, J.P.; Rivier, J.E.; Sherwood, N.M. Primary structure and function of three gonadotropin-releasing hormones, including a novel form, from an ancient teleost, herring. Endocrinology 2000, 141, 505–512. [Google Scholar] [CrossRef]
- Olberg, D.E.; Hausner, S.H.; Bauer, N.; Klaveness, J.; Indrevoll, B.; Andressen, K.W.; Dahl, M.; Levy, F.O.; Sutcliffe, J.L.; Haraldsen, I. Radiosynthesis of high affinity fluorine-18 labeled GnRH peptide analogues: In vitro studies and in vivo assessment of brain uptake in rats. MedChemComm. 2015, 6, 708–714. [Google Scholar] [CrossRef]
- Melloni, C.; Nelson, A. Effect of Androgen Deprivation Therapy on Metabolic Complications and Cardiovascular Risk. J. Cardiovasc. Trans. Res. 2020, 13, 451–462. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Y.; Xu, B.; Cai, L.; Feng, S.; Liu, Y.; Zhu, Z.; Yu, Q.; Guo, H. Safety, Pharmacokinetics, and Pharmacodynamics of SHR7280, a Non-peptide GnRH Antagonist in Premenopausal Women with Endometriosis: A Randomized, Double-Blind, Placebo-Controlled Phase 1 Study. Clin. Pharmacokinet. 2023, 62, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Crowley, W.F., Jr. Gonadotropin-Releasing Hormone and its Analogs. Annu. Rev. Med. 1994, 45, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Al-Inany, H.G.; Youssef, M.A.; Ayeleke, R.O.; Brown, J.; Lam, W.S.; Broekmans, F.J. Gonadotrophin-releasing hormone antagonists for assisted reproductive technology. Cochrane Database Syst. Rev. 2016, 4, Cd001750. [Google Scholar] [CrossRef] [PubMed]
- Jurincic, C.D.; Horlbeck, R.; Klippel, K.F. Combined treatment (goserelin plus flutamide) versus monotherapy (goserelin alone) in advanced prostate cancer: A randomized study. Semin. Oncol. 1991, 18 (Suppl. S6), 21–25. [Google Scholar] [PubMed]
- Goserelin Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/019726s050s051s052lbl.pdf (accessed on 25 February 2024).
- Leuprolide Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/019010s033,019732s031s035s036,020517s024s028s029lbl.pdf (accessed on 25 February 2024).
- Hoda, M.R.; Kramer, M.W.; Merseburger, A.S.; Cronauer, M.V. Androgen deprivation therapy with Leuprolide acetate for treatment of advanced prostate cancer. Expert Opin. Pharmacother. 2017, 18, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Chaudhry, Z.T.; Al-Hendy, A. Successes and failures of uterine leiomyoma drug discovery. Expert Opin. Drug Discov. 2018, 13, 169–177. [Google Scholar] [CrossRef]
- Wilson, A.C.; Meethal, S.V.; Bowen, R.L.; Atwood, C.S. Leuprolide acetate: A drug of diverse clinical applications. Expert Opin. Investig. Drugs 2007, 16, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Leuprolide Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/019943_Lupron%20Depot_APPROV.pdf (accessed on 25 February 2024).
- Nafarelin Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/019886s030lbl.pdf (accessed on 25 February 2024).
- Nafarelin Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/019886s013_synarel_appltr.pdf (accessed on 25 February 2024).
- Triptorelin Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022437Orig1s000lbl.pdf (accessed on 25 February 2024).
- Leone Roberti Maggiore, U.; Scala, C.; Remorgida, V.; Venturini, P.L.; Del Deo, F.; Torella, M.; Colacurci, N.; Salvatore, S.; Ferrari, S.; Papaleo, E.; et al. Triptorelin for the treatment of endometriosis. Expert Opin. Pharmacother. 2014, 15, 1153–1179. [Google Scholar] [CrossRef]
- Triptorelin Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022437Orig1s000ltr.pdf (accessed on 25 February 2024).
- Histrelin Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022058s000_Lbl.pdf (accessed on 25 February 2024).
- Djavan, B.; Schlegel, P.; Salomon, G.; Eckersberger, E.; Sadri, H.; Graefen, M. Analysis of testosterone suppression in men receiving histrelin, a novel GnRH agonist for the treatment of prostate cancer. Can. J. Urol. 2010, 17, 5265–5271. [Google Scholar]
- Histrelin Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022058s000_Approv.pdf (accessed on 25 February 2024).
- Choi, D. Evolutionary Viewpoint on GnRH (gonadotropin-releasing hormone) in Chordata—Amino Acid and Nucleic Acid Sequences. Dev. Reprod. 2018, 22, 119–132. [Google Scholar] [CrossRef]
- Ganirelix Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21057_Antagon_prntlbl.pdf (accessed on 25 February 2024).
- Gillies, P.S.; Faudas, D.; Barman Balfour, J.A.; Perry, C.M. Ganirelix. Drugs 2000, 59, 107–111. [Google Scholar] [CrossRef]
- Ganirelix Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21057_Antagon_Approv.pdf (accessed on 25 February 2024).
- Cetrorelix Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21-197_Cetrotide_prntlbl.pdf (accessed on 25 February 2024).
- Findeklee, S.; Diedrich, P.K. Cetrorelix in reproductive medicine. F&S Rep. 2023, 4 (Suppl. S2), 62–64. [Google Scholar] [CrossRef]
- Cetrorelix Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21-197_Cetrotide_Approv.pdf (accessed on 25 February 2024).
- Abarelix Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-320_Plenaxis_Prntlbl.pdf (accessed on 25 February 2024).
- Debruyne, F.M. Gonadotropin-releasing hormone antagonist in the management of prostate cancer. Rev. Urol. 2004, 6 (Suppl. 7), S25–S32. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1472892/ (accessed on 25 February 2024).
- Abarelix Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-320_Plenaxis_Approv.pdf (accessed on 25 February 2024).
- Degarelix Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/022201s000_Lbl.pdf (accessed on 25 February 2024).
- Degarelix Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/022201s000_Approv.pdf (accessed on 25 February 2024).
- Bortezomib Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_prntlbl.pdf (accessed on 25 February 2024).
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Schwartz, R.; Davidson, T. Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology 2004, 18 (Suppl. S11), 14–21. [Google Scholar]
- Bortezomib Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_Approv.pdf (accessed on 25 February 2024).
- Carfilzomib Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202714Orig1s000LBL.pdf (accessed on 25 February 2024).
- Carfilzomib Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202714Orig1s000Approv.pdf (accessed on 25 February 2024).
- Pepaxto Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214383s000lbl.pdf (accessed on 25 February 2024).
- Dhillon, S. Melphalan Flufenamide (Melflufen): First Approval. Drugs 2021, 81, 963–969. [Google Scholar] [CrossRef]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res./Rev. Genet. Toxicol. 1994, 318, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Phillips, D.H. DNA adducts from chemotherapeutic agents. Mutat. Res. 1996, 355, 13–40. [Google Scholar] [CrossRef]
- Pepaxto Approval Letter. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/214383Orig1s000ltr.pdf (accessed on 25 February 2024).
- Olivier, T.; Prasad, V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 18, 101374. [Google Scholar] [CrossRef] [PubMed]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide-drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2023, 13, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Lombardi, L.; Williams, D.R.; Albericio, F. Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals 2022, 15, 1283. [Google Scholar] [CrossRef] [PubMed]
- Balogh, B.; Ivánczi, M.; Nizami, B.; Beke-Somfai, T.; Mándity, I.M. ConjuPepDB: A database of peptide–drug conjugates. Nucleic Acids Res. 2021, 49, D1102–D1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, J.; Xia, M.; Yin, L.; Zhang, L.; Liu, X.; Cheng, Y. Peptide-drug conjugates: A new paradigm for targeted cancer therapy. Eur. J. Med. Chem. 2024, 265, 116119. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef]
- Fetse, J.; Kandel, S.; Mamani, U.-F.; Cheng, K. Recent advances in the development of therapeutic peptides. Trends Pharmacol. Sci. 2023, 44, 425–441. [Google Scholar] [CrossRef]
- Al Musaimi, O.; Williams, D.R. Methods of Chemical Synthesis of Peptides; I.P. Office, Ed.; Imperial College Innovations Limited: London, UK, 2023. [Google Scholar]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2023: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2024, 29, 585. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide (Trade Name) | Indication | Therapeutic Target | Route | FDA Approval Year |
|---|---|---|---|---|
| 68Ga-PSMA-11 (68Ga gozetotide) | A diagnostic employed for detecting PET PSMA-positive lesions in males diagnosed with prostate cancer | PSMA | IV | 2020 |
| Piflufolastat F 18 (Pylarify) | A diagnostic employed for detecting PET PSMA-positive lesions in males diagnosed with prostate cancer | PSMA | IV | 2021 |
| Lutetium 177Lu Vipivotide Tetraxetan (Pluvicto) | Treatment of PSMA-positive metastatic castration-resistant prostate cancer (mCRPC) in adult patients | PSMA and the neighboring cells | IV | 2022 |
| Flotufolastat F 18 (Posluma) | A diagnostic employed for detecting PET PSMA-positive lesions in males diagnosed with prostate cancer | PSMA | IV | 2023 |
| Peptide (Trade Name) | Indication | Therapeutic Target | Route | FDA Approval Year |
|---|---|---|---|---|
| Depreotide (Neotect) | Scintigraphic imaging | Somatostatin receptor | IV | 1999 |
| 68Ga-DOTATATE (Netspot) | Scintigraphic imaging | Somatostatin receptor | IV | 2016 |
| 177Lu-DOTATATE (Lutathera) | To treat somatostatin receptor-positive GEP-NETs, including foregut, midgut, and hindgut NETs. | Somatostatin receptor | IV | 2018 |
| 68Ga-DOTATOC | Scintigraphic imaging | Somatostatin receptor | IV | 2019 |
| 64Cu-DOTATATE (Detectnet) | Scintigraphic imaging | Somatostatin receptor | IV | 2020 |
| Peptide (Trade Name) | Indication | Therapeutic Target | Route | FDA Approval Year |
|---|---|---|---|---|
| Enfortumab Vedotin-Ejfv (Padcev) | Urothelial cancer (a cancer of the bladder and urinary tract). | Nectin-4 receptor | IV | 2019 |
| Polatuzumab vedotin-piiq (Polivy) | 1. DLBCL patients whose cancer has returned or has stopped responding to other treatments and who cannot have a bone-marrow transplantation. 2. Relapsed or refractory DLBCL, NOS, after undergoing at least two prior therapies interventions. | CD79b | IV | 2019 |
| Fam-trastuzumab deruxtecan-nxki (Enhertu) | Unresectable or metastatic HER2-positive breast cancer and concomitantly has unresectable or metastatic NSCLC. | Human epidermal growth factor receptor-2 (HER2) | IV | 2019 |
| Belantamab Mafodotin-Blmf (Blenrep) | Relapsed or refractory multiple myeloma. | B-cell maturation antigen (BCMA) | IV | 2020 |
| Tisotumab Vedotin-Tftv (TIVDAK) | Recurrent or metastatic cervical cancer with disease progression following chemotherapy. | Tissue factor TF011 | IV | 2021 |
| Loncastuximab Tesirine-Lpyl (Zynlonta) | Relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including DLBCL. | CD19 | IV | 2021 |
| Peptide (Trade Name) | Indication | Therapeutic Target | Route | FDA Approval Year |
|---|---|---|---|---|
| Agonists | ||||
| Goserelin (Zoladex) | Managing carcinoma of the prostate, addressing endometriosis, and providing palliative treatment for advanced breast cancer | GnRH | SC | 1989 |
| Leuprolide (Lupron) | Palliative treatment for prostate cancer, uterine leiomyomata, endometriosis, and central precocious puberty | GnRH | IM | 1995 |
| Nafarelin (Synarel) | To address endometriosis, including the alleviation of pain and the reduction of endometriotic lesions | GnRH | Nasal solution | 1998 |
| Trelstar (triptorelin) | Palliative treatment of advanced prostate cancer | GnRH | IM | 2000 |
| Histrelin (Supprelin LA) | Treatment of central precocious puberty (CPP) in children | GnRH | SC | 2007 |
| GnRH | ||||
| Antagonists | ||||
| Ganirelix (Antagon) | A fertility medication designed to prevent premature LH surges or ovulation in women undergoing fertility treatment with controlled ovarian hyperstimulation | GnRH | SC | 1999 |
| Cetrorelix (Cetrotide) | To inhibit premature ovulation | GnRH | SC | 2000 |
| Abarelix (Plenaxis) | Palliative treatment of advanced prostate cancer | GnRH | IM | 2003 |
| Degarelix (Firmagon) | Advanced prostate cancer | GnRH | SC | 2008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Musaimi, O. Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989. Cancers 2024, 16, 1032. https://doi.org/10.3390/cancers16051032
Al Musaimi O. Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989. Cancers. 2024; 16(5):1032. https://doi.org/10.3390/cancers16051032
Chicago/Turabian StyleAl Musaimi, Othman. 2024. "Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989" Cancers 16, no. 5: 1032. https://doi.org/10.3390/cancers16051032
APA StyleAl Musaimi, O. (2024). Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989. Cancers, 16(5), 1032. https://doi.org/10.3390/cancers16051032