
Contribution of Autophagy to Epithelial Mesenchymal Transition Induction during Cancer Progression

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
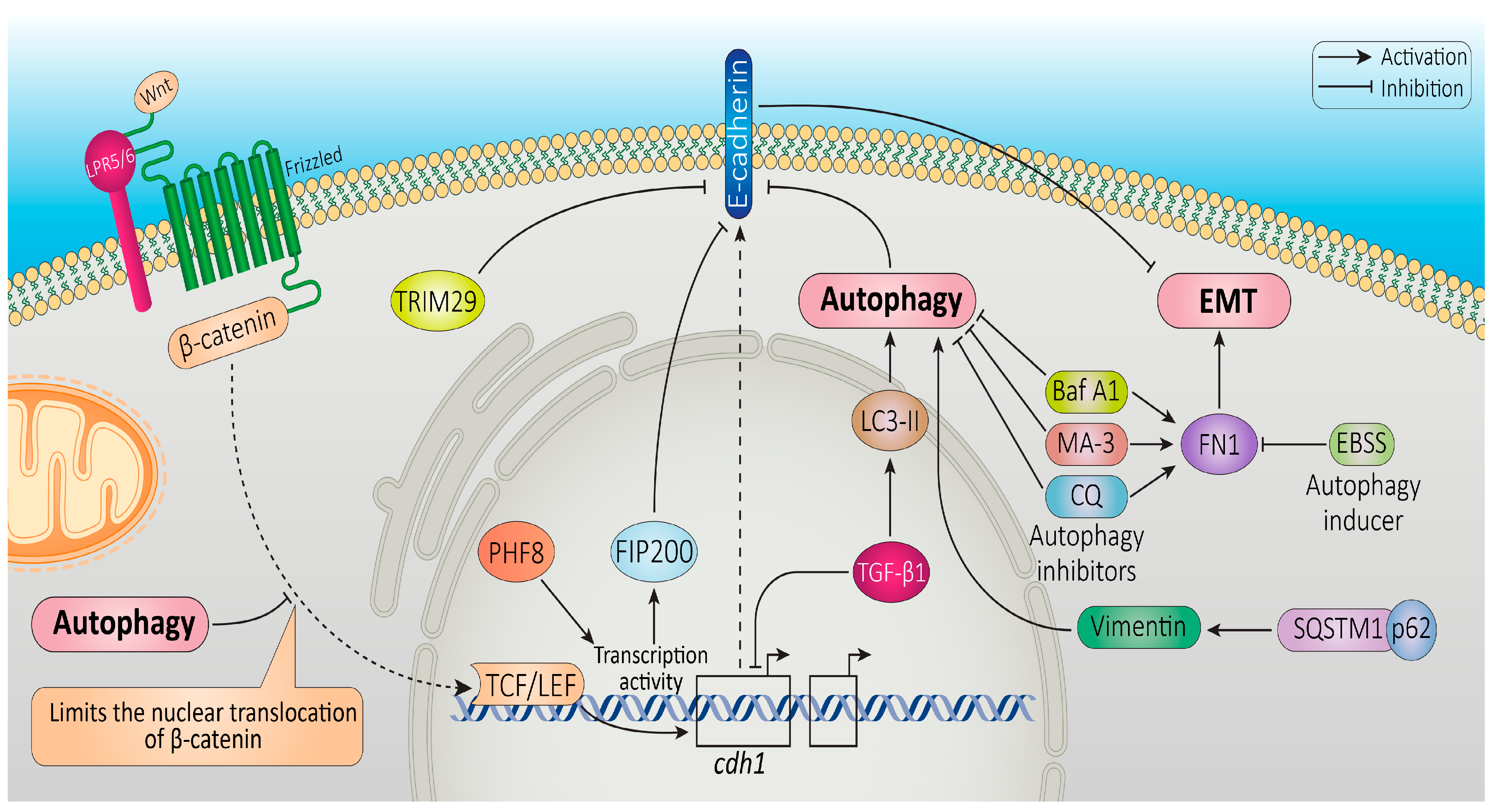{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction: The Fate of Normal and Transformed Cells
2. Autophagy and Cancer
2.1. Role of Autophagy in Cancer Development and Progression
2.2. Role of Autophagy in Cancer Stem Cell Maintenance
2.3. Autophagy as a Therapeutic Target in Cancer
3. Cell Transformation, EMT and Carcinogenesis
3.1. Morphological, Biochemical and Molecular Changes during EMT
3.2. Role of EMT in Tumor Invasion and Metastasis
3.3. EMT as a Target in Cancer Therapy and Drug Resistance
4. Autophagy-EMT Interplay
4.1. Common Signaling Pathways That Regulate Both Autophagy and EMT
4.2. Autophagy Degradation of EMT-Related Markers
4.3. Reciprocal Regulation between Autophagy and EMT-TFs
4.4. Clinical Relevance of Interplay between EMT and Autophagy
5. Conclusions
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Belmonte-Mateos, C.; Pujades, C. From Cell States to Cell Fates: How Cell Proliferation and Neuronal Differentiation Are Coordinated during Embryonic Development. Front. Neurosci. 2022, 15, 1752. [Google Scholar] [CrossRef] [PubMed]
- Haghverdi, L.; Ludwig, L.S. Single-Cell Multi-Omics and Lineage Tracing to Dissect Cell Fate Decision-Making. Stem Cell Rep. 2023, 18, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Blomen, V.A.; Boonstra, J. Cell Fate Determination during G1 Phase Progression. Cell. Mol. Life Sci. 2007, 64, 3084–3104. [Google Scholar] [CrossRef] [PubMed]
- Casey, M.J.; Stumpf, P.S.; MacArthur, B.D. Theory of Cell Fate. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1471. [Google Scholar] [CrossRef] [PubMed]
- Greulich, P.; Smith, R.; MacArthur, B.D. The Physics of Cell Fate. In Phenotypic Switching; Elsevier: Amsterdam, The Netherlands, 2020; pp. 189–206. [Google Scholar]
- Sun, W.; Yang, J. Functional Mechanisms for Human Tumor Suppressors. J. Cancer 2010, 1, 136. [Google Scholar] [CrossRef]
- Rossi, F.; Noren, H.; Jove, R.; Beljanski, V.; Grinnemo, K.-H. Differences and Similarities between Cancer and Somatic Stem Cells: Therapeutic Implications. Stem Cell Res. Ther. 2020, 11, 489. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the Hallmarks of Cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar]
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic Plasticity and Cell Fate Decisions in Cancer: Insights from Dynamical Systems Theory. Cancers 2017, 9, 70. [Google Scholar] [CrossRef]
- Wang, E.J.-Y.; Chen, I.; Kuo, B.Y.-T.; Yu, C.-C.; Lai, M.-T.; Lin, J.-T.; Lin, L.Y.-T.; Chen, C.-M.; Hwang, T.; Sheu, J.J.-C. Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development. Biomolecules 2022, 12, 1862. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Pukhalskaia, T.V.; Rizvanov, A.A.; Solovyeva, V. V Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells. Bioengineering 2022, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA Damage and the Balance between Survival and Death in Cancer Biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Participation of the ATR/CHK1 Pathway in Replicative Stress Targeted Therapy of High-Grade Ovarian Cancer. J. Hematol. Oncol. 2020, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Quilón, A.; Serrano-Benítez, A.; Ariel Lieberman, J.; Quintero, C.; Sánchez-Gutiérrez, D.; Escudero, L.M.; Cortés-Ledesma, F. ATM Specifically Mediates Repair of Double-Strand Breaks with Blocked DNA Ends. Nat. Commun. 2014, 5, 3347. [Google Scholar] [CrossRef]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA Damage Response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA Damage and Autophagy. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 158–166. [Google Scholar] [CrossRef]
- Liu, B.; Oltvai, Z.N.; Bayır, H.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Bahar, I. Quantitative Assessment of Cell Fate Decision between Autophagy and Apoptosis. Sci. Rep. 2017, 7, 17605. [Google Scholar] [CrossRef]
- Grasso, D.; Ropolo, A.; Vaccaro, M.I. Autophagy in Cell Fate and Diseases. Cell Death Autophagy Apoptosis Necrosis 2015, 1, 3–26. [Google Scholar]
- Cassidy, L.D.; Narita, M. Autophagy at the Intersection of Aging, Senescence, and Cancer. Mol. Oncol. 2022, 16, 3259–3275. [Google Scholar] [CrossRef]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef]
- Adelipour, M.; Saleth, L.R.; Ghavami, S.; Alagarsamy, K.N.; Dhingra, S.; Allameh, A. The Role of Autophagy in the Metabolism and Differentiation of Stem Cells. Biochim. Biophys. Acta BBA Molecular Basis Dis. 2022, 1868, 166412. [Google Scholar] [CrossRef]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The Role of Interaction between Autophagy and Apoptosis in Tumorigenesis. Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and Autophagy-Related Pathways in Cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The Spotlight for Cellular Stress Responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. FOXO Transcription Factors: Key Regulators of Cellular Quality Control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Karakaş, H.E.; GÖZÜAÇIK, D. Autophagy and Cancer. Turk. J. Biol. 2014, 38, 720–739. [Google Scholar] [CrossRef]
- Kenific, C.M.; Debnath, J. Cellular and Metabolic Functions for Autophagy in Cancer Cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER Stress: Mutual Crosstalk between Autophagy, Oxidative Stress and Inflammatory Response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, S.; Majello, B. Autophagy Roles in Genome Maintenance. Cancers 2020, 12, 1793. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhao, Y. Autophagy and DNA Damage Repair. Genome Instab. Dis. 2020, 1, 172–183. [Google Scholar] [CrossRef]
- Monkkonen, T.; Debnath, J. Inflammatory Signaling Cascades and Autophagy in Cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef]
- Mommersteeg, M.C.; Simovic, I.; Yu, B.; van Nieuwenburg, S.A.V.; Bruno MJ, I.; Doukas, M.; Kuipers, E.J.; Spaander, M.C.W.; Peppelenbosch, M.P.; Castaño-Rodríguez, N. Autophagy Mediates ER Stress and Inflammation in Helicobacter Pylori-Related Gastric Cancer. Gut Microbes 2022, 14, 2015238. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhang, H.-L.; Li, D.-D.; Yang, K.-L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.-Y.; Ravichandran, S. Regulation of Glycolytic Metabolism by Autophagy in Liver Cancer Involves Selective Autophagic Degradation of HK2 (Hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R. Transcriptional Control of Autophagy–Lysosome Function Drives Pancreatic Cancer Metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, Identification, and Characterization of Cancer Stem Cells: A Review. J. Cell Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, H.; Liang, W.; Jiang, E.; Zhou, X.; Shao, Z.; Liu, K.; Shang, Z. Autophagy Regulates the Cancer Stem Cell Phenotype of Head and Neck Squamous Cell Carcinoma through the Noncanonical FOXO3/SOX2 Axis. Oncogene 2022, 41, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Guo, C.; Li, Y.; Ma, J. The Long Noncoding RNA TINCR Promotes Self-Renewal of Human Liver Cancer Stem Cells through Autophagy Activation. Cell Death Dis. 2022, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Brunel, A.; Hombourger, S.; Barthout, E.; Battu, S.; Kögel, D.; Antonietti, P.; Deluche, E.; Saada, S.; Durand, S.; Lalloué, F. Autophagy Inhibition Reinforces Stemness Together with Exit from Dormancy of Polydisperse Glioblastoma Stem Cells. Aging 2021, 13, 18106. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, Y.; Lee, S.; Oh, C.W.; Lee, E.J.; Ko, J.Y.; Park, J.H. Autophagy Inhibits Cancer Stemness in Triple-negative Breast Cancer via MiR-181a-mediated Regulation of ATG5 and/or ATG2B. Mol. Oncol. 2022, 16, 1857–1875. [Google Scholar] [CrossRef]
- Sharif, T.; Martell, E.; Dai, C.; Singh, S.K.; Gujar, S. Regulation of the Proline Regulatory Axis and Autophagy Modulates Stemness in TP73/P73 Deficient Cancer Stem-like Cells. Autophagy 2019, 15, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Macleod, K.F. Autophagy, Cancer Stem Cells and Drug Resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Yun, E.-J.; Kim, S.; Hsieh, J.-T.; Baek, S.T. Wnt/β-Catenin Signaling Pathway Induces Autophagy-Mediated Temozolomide-Resistance in Human Glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Yadav, V.K.; Chu, Y.C.; Ong, J.R.; Huang, T.-Y.; Lee, K.-F.; Lee, K.-H.; Yeh, C.-T.; Lee, W.-H. Hydroxychloroquine (HCQ) Modulates Autophagy and Oxidative DNA Damage Stress in Hepatocellular Carcinoma to Overcome Sorafenib Resistance via TLR9/SOD1/Hsa-MiR-30a-5p/Beclin-1 Axis. Cancers 2021, 13, 3227. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H. Protective Autophagy Decreases Osimertinib Cytotoxicity through Regulation of Stem Cell-like Properties in Lung Cancer. Cancer Lett. 2019, 452, 191–202. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Bi, F.; Jiang, Y.; Xu, Y.; An, Y.; Li, D.; Yang, Q. BRCA1 Affects the Resistance and Stemness of SKOV3-derived Ovarian Cancer Stem Cells by Regulating Autophagy. Cancer Med. 2019, 8, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A. Effect of Gemcitabine and Nab-Paclitaxel with or without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.P.; Tenner, L.; Sarantopoulos, J.; Morris, J.; Liu, Q.; Mendez, J.A.; Curiel, T.; Michalek, J.; Mahalingam, D. Modulation of Autophagy: A Phase II Study of Vorinostat plus Hydroxychloroquine versus Regorafenib in Chemotherapy-Refractory Metastatic Colorectal Cancer (MCRC). Br. J. Cancer 2022, 127, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The Clinical Value of Using Chloroquine or Hydroxychloroquine as Autophagy Inhibitors in the Treatment of Cancers: A Systematic Review and Meta-Analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET Plasticity in Cancer and Go-or-Grow Decisions in Quiescence: The Two Sides of the Same Coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef]
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.-H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef]
- Son, H.; Moon, A. Epithelial-Mesenchymal Transition and Cell Invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial Steps of Metastasis: Cell Invasion and Endothelial Transmigration. Mutat. Res. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A Multi-tool for Tumor Progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, W.; Wei, X. The Molecular Mechanisms and Therapeutic Strategies of EMT in Tumor Progression and Metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial–Mesenchymal Transition in Cancer Metastasis: Mechanisms, Markers and Strategies to Overcome Drug Resistance in the Clinic. Biochim. Biophys. Acta BBA Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Guilford, P.; Thiery, J.P. Early Events in Cell Adhesion and Polarity during Epithelial-Mesenchymal Transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Arnold, T.R.; Stephenson, R.E.; Dinshaw, K.M.; Miller, A.L. Maintenance of the Epithelial Barrier and Remodeling of Cell-Cell Junctions during Cytokinesis. Curr. Biol. 2016, 26, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed]
- Moch, M.; Schwarz, N.; Windoffer, R.; Leube, R.E. The Keratin–Desmosome Scaffold: Pivotal Role of Desmosomes for Keratin Network Morphogenesis. Cell Mol. Life Sci. 2020, 77, 543–558. [Google Scholar] [CrossRef]
- Tan, B.; Yatim, S.M.J.M.; Peng, S.; Gunaratne, J.; Hunziker, W.; Ludwig, A. The Mammalian Crumbs Complex Defines a Distinct Polarity Domain Apical of Epithelial Tight Junctions. Curr. Biol. 2020, 30, 2791–2804. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the Cytoskeleton, and Cancer Cell Invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Kuburich, N.A.; den Hollander, P.; Pietz, J.T.; Mani, S.A. Vimentin and Cytokeratin: Good Alone, Bad Together. Semin. Cancer Biol. 2022, 86, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Engler, A.J.; Slone, R.D.; Galante, L.L.; Schwarzbauer, J.E. Fibronectin Expression Modulates Mammary Epithelial Cell Proliferation during Acinar Differentiation. Cancer Res. 2008, 68, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Al Thani, H.; Rafii, A. Epithelial to Mesenchymal Transition in a Clinical Perspective. J. Oncol. 2015, 2015, 792182. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.; Yamada, S. N-Cadherin-Mediated Cell–Cell Adhesion Promotes Cell Migration in a Three-Dimensional Matrix. J. Cell Sci. 2012, 125, 3661–3670. [Google Scholar] [CrossRef]
- Talele, N.P.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of α-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef]
- Li, X.; Pei, D.; Zheng, H. Transitions between Epithelial and Mesenchymal States during Cell Fate Conversions. Protein Cell 2014, 5, 580–591. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G. New Signals from the Invasive Front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-Cadherin Switch Mediates Cancer Progression via TGF-β-Induced Epithelial-to-Mesenchymal Transition in Extrahepatic Cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885–1893. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Hussen, B.M.; Shoorei, H.; Mohaqiq, M.; Dinger, M.E.; Hidayat, H.J.; Taheri, M.; Ghafouri-Fard, S. The Impact of Non-Coding RNAs in the Epithelial to Mesenchymal Transition. Front. Mol. Biosci. 2021, 8, 665199. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial–Mesenchymal Transition and Its Transcription Factors. Biosci. Rep. 2022, 42, 499. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the Mesenchymal–Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Gerashchenko, T.S.; Novikov, N.M.; Krakhmal, N.V.; Zolotaryova, S.Y.; Zavyalova, M.V.; Cherdyntseva, N.V.; Denisov, E.V.; Perelmuter, V.M. Markers of Cancer Cell Invasion: Are They Good Enough? J. Clin. Med. 2019, 8, 1092. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Graziani, V.; Rodriguez-Hernandez, I.; Maiques, O.; Sanz-Moreno, V. The Amoeboid State as Part of the Epithelial-to-Mesenchymal Transition Programme. Trends Cell Biol. 2022, 32, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Chrisafis, G.; Wang, T.; Moissoglu, K.; Gasparski, A.N.; Ng, Y.; Weigert, R.; Lockett, S.J.; Mili, S. Collective Cancer Cell Invasion Requires RNA Accumulation at the Invasive Front. Proc. Natl. Acad. Sci. USA 2020, 117, 27423–27434. [Google Scholar] [CrossRef]
- Paňková, K.; Rösel, D.; Novotný, M.; Brábek, J. The Molecular Mechanisms of Transition between Mesenchymal and Amoeboid Invasiveness in Tumor Cells. Cell. Mol. Life Sci. 2010, 67, 63–71. [Google Scholar] [CrossRef]
- Lintz, M.; Muñoz, A.; Reinhart-King, C.A. The Mechanics of Single Cell and Collective Migration of Tumor Cells. J. Biomech. Eng. 2017, 139, 0210051–0210059. [Google Scholar] [CrossRef] [PubMed]
- Bergert, M.; Chandradoss, S.D.; Desai, R.A.; Paluch, E. Cell Mechanics Control Rapid Transitions between Blebs and Lamellipodia during Migration. Proc. Natl. Acad. Sci. USA 2012, 109, 14434–14439. [Google Scholar] [CrossRef] [PubMed]
- Lusby, R.; Dunne, P.; Tiwari, V.K. Tumour Invasion and Dissemination. Biochem. Soc. Trans. 2022, 50, 1245–1257. [Google Scholar] [CrossRef]
- Yeung, K.T.; Yang, J. Epithelial–Mesenchymal Transition in Tumor Metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Li, S.; Luo, W. Matrix Metalloproteinase 2 Contributes to Aggressive Phenotype, Epithelial-Mesenchymal Transition and Poor Outcome in Nasopharyngeal Carcinoma. Onco. Targets. Ther. 2019, 12, 5701. [Google Scholar] [CrossRef]
- Reunanen, N.; Kähäri, V. Matrix Metalloproteinases in Cancer Cell Invasion. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Nabeshima, K.; Inoue, T.; Shimao, Y.; Sameshima, T. Matrix Metalloproteinases in Tumor Invasion: Role for Cell Migration. Pathol. Int. 2002, 52, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-Induced Invadopodia Formation Promotes Tumor Metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Prekeris, R. The Regulation of MMP Targeting to Invadopodia during Cancer Metastasis. Front. Cell Dev. Biol. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix Metalloproteinase-Induced Epithelial-Mesenchymal Transition in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and Tumor Metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.-G.; Jaghi, N.Z.Z. EMT, Cancer Stem Cells and Autophagy; The Three Main Axes of Metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Wang, Y. The Importance of Epithelial-Mesenchymal Transition and Autophagy in Cancer Drug Resistance. Cancer Drug Resist. 2020, 3, 38. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer Drug Resistance Induced by EMT: Novel Therapeutic Strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J. EMT Transcription Factors Snail and Slug Directly Contribute to Cisplatin Resistance in Ovarian Cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D. The ZEB1 Pathway Links Glioblastoma Initiation, Invasion and Chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.-J.; Zhang, W.; Xu, X.-M.; Zhang, F.; Tao, W.-P.; Ye, J.-J.; Ge, W. Twist Mediates an Aggressive Phenotype in Human Colorectal Cancer Cells. Int. J. Oncol. 2016, 48, 1117–1124. [Google Scholar] [CrossRef]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription Factors That Mediate Epithelial–Mesenchymal Transition Lead to Multidrug Resistance by Upregulating ABC Transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial–Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-H.; Tsai, M.-F.; Su, K.-Y.; Wu, S.-G.; Huang, C.-P.; Yu, S.-L.; Yu, Y.-L.; Lan, C.-C.; Yang, C.-H.; Lin, S.-B. Slug Confers Resistance to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. Am. J. Respir. Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, H.; Yu, J.; Yu, H. Chemoresistance to Doxorubicin Induces Epithelial-Mesenchymal Transition via Upregulation of Transforming Growth Factor β Signaling in HCT116 Colon Cancer Cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef]
- Zhao, J. Cancer Stem Cells and Chemoresistance: The Smartest Survives the Raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug Resistance and Cancer Stem Cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Nan, K.-J. Activation of PI3 Kinase/Akt/HIF-1α Pathway Contributes to Hypoxia-Induced Epithelial-Mesenchymal Transition and Chemoresistance in Hepatocellular Carcinoma. Int. J. Oncol. 2012, 40, 461–468. [Google Scholar] [PubMed]
- Xu, Z.; Zhang, Y.; Dai, H.; Han, B. Epithelial–Mesenchymal Transition-Mediated Tumor Therapeutic Resistance. Molecules 2022, 27, 4750. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Gammon, L.; Liang, X.; Costea, D.E.; Mackenzie, I.C. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine 2016, 4, 138–145. [Google Scholar] [CrossRef]
- Yoshida, T.; Song, L.; Bai, Y.; Kinose, F.; Li, J.; Ohaegbulam, K.C.; Muñoz-Antonia, T.; Qu, X.; Eschrich, S.; Uramoto, H. ZEB1 Mediates Acquired Resistance to the Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0147344. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G. Dual Role of Autophagy in Hallmarks of Cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- López-Méndez, T.B.; Sánchez-Álvarez, M.; Trionfetti, F.; Pedraz, J.L.; Tripodi, M.; Cordani, M.; Strippoli, R.; González-Valdivieso, J. Nanomedicine for Autophagy Modulation in Cancer Therapy: A Clinical Perspective. Cell Biosci. 2023, 13, 44. [Google Scholar] [CrossRef]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef]
- Chen, H.-T.; Liu, H.; Mao, M.-J.; Tan, Y.; Mo, X.-Q.; Meng, X.-J.; Cao, M.-T.; Zhong, C.-Y.; Liu, Y.; Shan, H. Crosstalk between Autophagy and Epithelial-Mesenchymal Transition and Its Application in Cancer Therapy. Mol. Cancer 2019, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-W.; Law, B.Y.K.; Mok, S.W.F.; Leung, E.L.H.; Fan, X.X.; Coghi, P.S.; Zeng, W.; Leung, C.-H.; Ma, D.-L.; Liu, L. Autophagic Degradation of Epidermal Growth Factor Receptor in Gefitinib-Resistant Lung Cancer by Celastrol. Int. J. Oncol. 2016, 49, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Lai, T.H.; Ahmed, M.; Pham, T.M.; Elashkar, O.; Bahar, E.; Kim, D.R. Regulation of TGF-β 1-Induced EMT by Autophagy-Dependent Energy Metabolism in Cancer Cells. Cancers 2022, 14, 4845. [Google Scholar] [CrossRef] [PubMed]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Kim, D.R. Control of the Epithelial-to-Mesenchymal Transition and Cancer Metastasis by Autophagy-Dependent SNAI1 Degradation. Cells 2019, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- DePavia, A.; Jonasch, E.; Liu, X.-D. Autophagy Degrades Hypoxia Inducible Factors. Mol. Cell. Oncol. 2016, 3, e1104428. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wei, X.; Liu, Y.; Wang, M.-M.; Sui, Z.; Wang, X.; Zhu, W.; Wu, M.; Lu, C.; Fei, Y.-H. Autophagy Inhibition Mediated by MCOLN1/TRPML1 Suppresses Cancer Metastasis via Regulating a ROS-Driven TP53/P53 Pathway. Autophagy 2022, 18, 1932–1954. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and Epithelial–Mesenchymal Transition: An Intricate Interplay in Cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef]
- Singla, M.; Bhattacharyya, S. Autophagy as a Potential Therapeutic Target during Epithelial to Mesenchymal Transition in Renal Cell Carcinoma: An in Vitro Study. Biomed. Pharmacother. 2017, 94, 332–340. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef]
- Datta, S.; Choudhury, D.; Das, A.; Mukherjee, D.D.; Dasgupta, M.; Bandopadhyay, S.; Chakrabarti, G. Autophagy Inhibition with Chloroquine Reverts Paclitaxel Resistance and Attenuates Metastatic Potential in Human Nonsmall Lung Adenocarcinoma A549 Cells via ROS Mediated Modulation of β-Catenin Pathway. Apoptosis 2019, 24, 414–433. [Google Scholar] [CrossRef]
- Marsh, T.; Debnath, J. Autophagy Suppresses Breast Cancer Metastasis by Degrading NBR1. Autophagy 2020, 16, 1164–1165. [Google Scholar] [CrossRef]
- Marsh, T.; Tolani, B.; Debnath, J. The Pleiotropic Functions of Autophagy in Metastasis. J. Cell Sci. 2021, 134, jcs247056. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Lorzadeh, S.; Ghavami, S. Autophagy and Cancer Metastasis: A Trojan Horse. J. Investig. Med. 2021, 69, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Subramani, R.; Gonzalez, E.; Arumugam, A.; Nandy, S.; Gonzalez, V.; Medel, J.; Camacho, F.; Ortega, A.; Bonkoungou, S.; Narayan, M. Nimbolide Inhibits Pancreatic Cancer Growth and Metastasis through ROS-Mediated Apoptosis and Inhibition of Epithelial-to-Mesenchymal Transition. Sci. Rep. 2016, 6, 19819. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.-J.; Zhou, Z.-W.; Zhu, D.-J.; Ju, Y.-L.; Wu, J.-H.; Ouyang, M.-Z.; Chen, X.-W.; Zhou, S.-F. Alisertib Induces Cell Cycle Arrest, Apoptosis, Autophagy and Suppresses EMT in HT29 and Caco-2 Cells. Int. J. Mol. Sci. 2015, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Zi, D.; Zhou, Z.-W.; Yang, Y.-J.; Huang, L.; Zhou, Z.-L.; He, S.-M.; He, Z.-X.; Zhou, S.-F. Danusertib Induces Apoptosis, Cell Cycle Arrest, and Autophagy but Inhibits Epithelial to Mesenchymal Transition Involving PI3K/Akt/MTOR Signaling Pathway in Human Ovarian Cancer Cells. Int. J. Mol. Sci. 2015, 16, 27228–27251. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F. Autophagy Induction Impairs Migration and Invasion by Reversing EMT in Glioblastoma Cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, D.; Yu, J.; Dong, H.; Zhang, J.; Yang, S. Targeting Autophagy in Cancer Stem Cells as an Anticancer Therapy. Cancer Lett. 2017, 393, 33–39. [Google Scholar] [CrossRef]
- Sun, N.-Y.; Yang, M.-H. Metabolic Reprogramming and Epithelial-Mesenchymal Plasticity: Opportunities and Challenges for Cancer Therapy. Front. Oncol. 2020, 10, 792. [Google Scholar] [CrossRef]
- Saxena, M.; Balaji, S.A.; Deshpande, N.; Ranganathan, S.; Pillai, D.M.; Hindupur, S.K.; Rangarajan, A. AMP-Activated Protein Kinase Promotes Epithelial-Mesenchymal Transition in Cancer Cells through Twist1 Upregulation. J. Cell Sci. 2018, 131, jcs208314. [Google Scholar] [CrossRef]
- Bonavida, B. Linking Autophagy and the Dysregulated NFκB/SNAIL/YY1/RKIP/PTEN Loop in Cancer: Therapeutic Implications. Crit. Rev. Oncog. 2018, 23, 307–320. [Google Scholar] [CrossRef]
- Coelho, B.P.; Fernandes, C.F. de L.; Boccacino, J.M.; Souza, M.C. da S.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.L.R. Multifaceted WNT Signaling at the Crossroads between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef] [PubMed]
- Said, N.A.B.M.; Williams, E.D. Growth Factors in Induction of Epithelial-Mesenchymal Transition and Metastasis. Cells Tissues Organs 2010, 193, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Hou, X.; Xia, J.; Qian, X.; Miele, L.; Sarkar, F.H.; Wang, Z. Emerging Roles of PDGF-D in EMT Progression during Tumorigenesis. Cancer Treat. Rev. 2013, 39, 640–646. [Google Scholar] [CrossRef]
- Bao, Y.; Ding, Z.; Zhao, P.; Li, J.; Chen, P.; Zheng, J.; Qian, Z. Autophagy Inhibition Potentiates the Anti-EMT Effects of Alteronol through TGF-β/Smad3 Signaling in Melanoma Cells. Cell Death Dis. 2020, 11, 223. [Google Scholar] [CrossRef] [PubMed]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Elashkar, O.; Kim, D.R. Cross Talk between Autophagy and Oncogenic Signaling Pathways and Implications for Cancer Therapy. Biochim. Biophys. Acta BBA Rev. Cancer 2021, 1876, 188565. [Google Scholar] [CrossRef] [PubMed]
- De Iuliis, V.; Marino, A.; Caruso, M.; Capodifoglio, S.; Flati, V.; Marynuk, A.; Marricareda, V.; Ursi, S.; Lanuti, P.; Talora, C. Autophagy Processes Are Dependent on EGF Receptor Signaling. Oncotarget 2018, 9, 30289. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-H.; Park, H.-S.; Lee, D.-H.; Jo, J.-H.; Heo, K.-S.; Myung, C.-S. Regulation of Autophagy by Controlling Erk1/2 and MTOR for Platelet-Derived Growth Factor-BB-Mediated Vascular Smooth Muscle Cell Phenotype Shift. Life Sci. 2021, 267, 118978. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, W.; Wei, H.; Wang, T.; Zheng, K.; Yang, C.; Feng, R.; Xu, R.; Fu, Y.; Li, C. Ferritinophagy-Mediated Ferroptosis and Activation of Keap1/Nrf2/HO-1 Pathway Were Conducive to EMT Inhibition of Gastric Cancer Cells in Action of 2, 2-Di-Pyridineketone Hydrazone Dithiocarbamate Butyric Acid Ester. Oxid. Med. Cell Longev. 2022, 2022, 3920664. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Wu, R.; Bi, L.; Zhang, C.; Chen, H.; Yang, Y. Antioral Squamous Cell Carcinoma Effects of Carvacrol via Inhibiting Inflammation, Proliferation, and Migration Related to Nrf2/Keap1 Pathway. Biomed Res. Int. 2021, 2021, 6616547. [Google Scholar] [CrossRef]
- Kozak, J.; Forma, A.; Czeczelewski, M.; Kozyra, P.; Sitarz, E.; Radzikowska-Büchner, E.; Sitarz, M.; Baj, J. Inhibition or Reversal of the Epithelial-Mesenchymal Transition in Gastric Cancer: Pharmacological Approaches. Int. J. Mol. Sci. 2020, 22, 277. [Google Scholar] [CrossRef]
- Li, C.-W.; Xia, W.; Huo, L.; Lim, S.-O.; Wu, Y.; Hsu, J.L.; Chao, C.-H.; Yamaguchi, H.; Yang, N.-K.; Ding, Q. Epithelial–Mesenchymal Transition Induced by TNF-α Requires NF-ΚB–Mediated Transcriptional Upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef]
- Tyciakova, S.; Valova, V.; Svitkova, B.; Matuskova, M. Overexpression of TNFα Induces Senescence, Autophagy and Mitochondrial Dysfunctions in Melanoma Cells. BMC Cancer 2021, 21, 507. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; De Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-KappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Souquère, S.; Pierron, G.; Codogno, P. NF-ΚB Activation Represses Tumor Necrosis Factor-α-Induced Autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Codogno, P. Regulation of Autophagy by NF-KappaB Transcription Factor and Reactives Oxygen Species. Autophagy 2007, 3, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, Z.; Hao, Y.; Zhao, Y.; Qian, F.; Shi, Y.; Li, P.; Liu, C.; Yu, P. HIF-1α Induces the Epithelial-Mesenchymal Transition in Gastric Cancer Stem Cells through the Snail Pathway. Oncotarget 2017, 8, 9535. [Google Scholar] [CrossRef]
- Wang, P.; Long, M.; Zhang, S.; Cheng, Z.; Zhao, X.; He, F.; Liu, H.; Ming, L. Hypoxia Inducible Factor-1α Regulates Autophagy via the P27-E2F1 Signaling Pathway. Mol. Med. Rep. 2017, 16, 2107–2112. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Withdrawal: Mitochondrial Autophagy Is an HIF-1-Dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2023, 299, 105125. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Y.; Lin, M.; Huang, B.; Lin, J. Qingjie Fuzheng Granule Inhibits EMT and Induces Autophagy in Colorectal Cancer via MTOR Signaling Pathways. Evidence-Based Complement. Altern. Med. 2021, 2021, 9950499. [Google Scholar] [CrossRef]
- Shen, H.; Yin, L.; Deng, G.; Guo, C.; Han, Y.; Li, Y.; Cai, C.; Fu, Y.; Liu, S.; Zeng, S. Knockdown of Beclin-1 Impairs Epithelial-mesenchymal Transition of Colon Cancer Cells. J. Cell Biochem. 2018, 119, 7022–7031. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, H.-Y.; Du, Z.-X.; Li, C.; An, M.-X.; Zong, Z.-H.; Liu, B.-Q.; Wang, H.-Q. Induction of Epithelial-Mesenchymal Transition (EMT) by Beclin 1 Knockdown via Posttranscriptional Upregulation of ZEB1 in Thyroid Cancer Cells. Oncotarget 2016, 7, 70364. [Google Scholar] [CrossRef][Green Version]
- Niklaus, N.J.; Tokarchuk, I.; Zbinden, M.; Schläfli, A.M.; Maycotte, P.; Tschan, M.P. The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells 2021, 10, 1447. [Google Scholar] [CrossRef]
- Hashemi, M.; Nadafzadeh, N.; Imani, M.H.; Rajabi, R.; Ziaolhagh, S.; Bayanzadeh, S.D.; Norouzi, R.; Rafiei, R.; Koohpar, Z.K.; Raei, B. Targeting and Regulation of Autophagy in Hepatocellular Carcinoma: Revisiting the Molecular Interactions and Mechanisms for New Therapy Approaches. Cell Commun. Signal. 2023, 21, 32. [Google Scholar] [CrossRef]
- Ye, R.; Dai, N.; He, Q.; Guo, P.; Xiang, Y.; Zhang, Q.; Hong, Z.; Zhang, Q. Comprehensive Anti-Tumor Effect of Brusatol through Inhibition of Cell Viability and Promotion of Apoptosis Caused by Autophagy via the PI3K/Akt/MTOR Pathway in Hepatocellular Carcinoma. Biomed. Pharmacother. 2018, 105, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Yang, Q.; Liu, Z.; Chang, X.; Yang, H.; Liu, J.; Li, Z.; Zuo, D. FAT4 Activation Inhibits Epithelial-Mesenchymal Transition (EMT) by Promoting Autophagy in H2228/Cer Cells. Med. Oncol. 2022, 40, 64. [Google Scholar] [CrossRef]
- Hashemi, M.; Hasani, S.; Hajimazdarany, S.; Mirmazloomi, S.R.; Makvandy, S.; Zabihi, A.; Goldoost, Y.; Gholinia, N.; Kakavand, A.; Tavakolpournegari, A. Non-Coding RNAs Targeting Notch Signaling Pathway in Cancer: From Proliferation to Cancer Therapy Resistance. Int. J. Biol. Macromol. 2022, 222, 1151–1167. [Google Scholar] [CrossRef]
- Rojas-Sanchez, G.; Cotzomi-Ortega, I.; Pazos-Salazar, N.G.; Reyes-Leyva, J.; Maycotte, P. Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive. Biology 2019, 8, 71. [Google Scholar] [CrossRef]
- Chockley, P.J.; Keshamouni, V.G. Immunological Consequences of Epithelial–Mesenchymal Transition in Tumor Progression. J. Immunol. 2016, 197, 691–698. [Google Scholar] [CrossRef]
- Vuletić, A.; Mirjačić Martinović, K.; Tišma Miletić, N.; Zoidakis, J.; Castellvi-Bel, S.; Čavić, M. Cross-Talk between Tumor Cells Undergoing Epithelial to Mesenchymal Transition and Natural Killer Cells in Tumor Microenvironment in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 750022. [Google Scholar] [CrossRef]
- Cui, B.; Lin, H.; Yu, J.; Yu, J.; Hu, Z. Autophagy and the Immune Response. In Autophagy: Biology and Diseases; Basic Science; Springer: Berlin/Heidelberg, Germany, 2019; pp. 595–634. [Google Scholar]
- Hubbi, M.E.; Hu, H.; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-Mediated Autophagy Targets Hypoxia-Inducible Factor-1α (HIF-1α) for Lysosomal Degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.-W. DEDD Interacts with PI3KC3 to Activate Autophagy and Attenuate Epithelial–Mesenchymal Transition in Human Breast Cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef]
- Liu, X.; Meng, L.; Li, X.; Li, D.; Liu, Q.; Chen, Y.; Li, X.; Bu, W.; Sun, H. Regulation of FN1 Degradation by the P62/SQSTM1-Dependent Autophagy–Lysosome Pathway in HNSCC. Int. J. Oral Sci. 2020, 12, 34. [Google Scholar] [CrossRef]
- Damiano, V.; Spessotto, P.; Vanin, G.; Perin, T.; Maestro, R.; Santarosa, M. The Autophagy Machinery Contributes to E-Cadherin Turnover in Breast Cancer. Front. Cell Dev. Biol. 2020, 8, 545. [Google Scholar] [CrossRef]
- Zhou, W.; Gong, L.; Wu, Q.; Xing, C.; Wei, B.; Chen, T.; Zhou, Y.; Yin, S.; Jiang, B.; Xie, H. PHF8 Upregulation Contributes to Autophagic Degradation of E-Cadherin, Epithelial-Mesenchymal Transition and Metastasis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 215. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Wang, H.; Rao, P.; Zhao, Y.; Xie, J.; Cao, Q.; Wang, Y.; Wang, Y.M.; Lee, V.W.; Alexander, S.I. Autophagy Links β-Catenin and Smad Signaling to Promote Epithelial-Mesenchymal Transition via Upregulation of Integrin Linked Kinase. Int. J. Biochem. Cell Biol. 2016, 76, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Jiao, L.; Wang, Y.; Yu, Y.; Ming, L. SIRT1 Induces Epithelial-Mesenchymal Transition by Promoting Autophagic Degradation of E-Cadherin in Melanoma Cells. Cell Death Dis. 2018, 9, 136. [Google Scholar] [CrossRef]
- Rossi, L.; Battistelli, C.; de Turris, V.; Noce, V.; Zwergel, C.; Valente, S.; Moioli, A.; Manzione, A.; Palladino, M.; Bordoni, V. HDAC1 Inhibition by MS-275 in Mesothelial Cells Limits Cellular Invasion and Promotes MMT Reversal. Sci. Rep. 2018, 8, 8492. [Google Scholar] [CrossRef]
- Zhan, Y.; Gong, K.; Chen, C.; Wang, H.; Li, W. P38 MAP Kinase Functions as a Switch in MS-275-Induced Reactive Oxygen Species-Dependent Autophagy and Apoptosis in Human Colon Cancer Cells. Free Radic. Biol. Med. 2012, 53, 532–543. [Google Scholar] [CrossRef]
- Xu, W.; Chen, B.; Ke, D.; Chen, X. TRIM29 Mediates Lung Squamous Cell Carcinoma Cell Metastasis by Regulating Autophagic Degradation of E-Cadherin. Aging 2020, 12, 13488. [Google Scholar] [CrossRef]
- Arnold, A.; Tronser, M.; Sers, C.; Ahadova, A.; Endris, V.; Mamlouk, S.; Horst, D.; Möbs, M.; Bischoff, P.; Kloor, M. The Majority of β-Catenin Mutations in Colorectal Cancer Is Homozygous. BMC Cancer 2020, 20, 1038. [Google Scholar]
- Allameh, A.; Dadkhah, A.; Rahbarizadeh, F.; Ashrafi-Helan, J.; Fatemi, F. Effect of Dietary Caraway Essential Oils on Expression of β-Catenin during 1, 2-Dimethylhydrazine-Induced Colonic Carcinogenesis. J. Nat. Med. 2013, 67, 690–697. [Google Scholar] [CrossRef]
- Fröhlich, J.; Rose, K.; Hecht, A. Transcriptional Activity Mediated by β-CATENIN and TCF/LEF Family Members Is Completely Dispensable for Survival and Propagation of Multiple Human Colorectal Cancer Cell Lines. Sci. Rep. 2023, 13, 287. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signalling: Function, Biological Mechanisms, and Therapeutic Opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.-B.; Jung, S.H.; Choi, H.J. β-Catenin Activation down-Regulates Cell-Cell Junction-Related Genes and Induces Epithelial-to-Mesenchymal Transition in Colorectal Cancers. Sci. Rep. 2019, 9, 18440. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.M.; Batson, J.; Malik, K.; Paraskeva, C. Autolysosomal Β-catenin Degradation Regulates Wnt-autophagy-p62 Crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wang, J.; Wang, W.; Tian, Y.; XiangWei, W.; Chen, P.; Ma, K.; Zhou, C. Autophagy Eliminates Cytoplasmic β-Catenin and NICD to Promote the Cardiac Differentiation of P19CL6 Cells. Cell. Signal. 2014, 26, 2299–2305. [Google Scholar] [CrossRef] [PubMed]
- Colella, B.; Faienza, F.; Carinci, M.; D’Alessandro, G.; Catalano, M.; Santoro, A.; Cecconi, F.; Limatola, C.; Di Bartolomeo, S. Autophagy Induction Impairs Wnt/β-Catenin Signalling through β-Catenin Relocalisation in Glioblastoma Cells. Cell. Signal. 2019, 53, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Spada, S.; Tocci, A.; Di Modugno, F.; Nisticò, P. Fibronectin as a Multiregulatory Molecule Crucial in Tumor Matrisome: From Structural and Functional Features to Clinical Practice in Oncology. J. Exp. Clin. Cancer Res. 2021, 40, 102. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 Promotes Melanoma Proliferation and Metastasis by Inhibiting Apoptosis and Regulating EMT. Onco. Targets. Ther. 2019, 12, 3207. [Google Scholar] [CrossRef]
- Tang, X.; Tang, Q.; Yang, X.; Xiao, Z.-A.; Zhu, G.; Yang, T.; Yang, Q.; Zhang, Y.; Li, S. FN1 Promotes Prognosis and Radioresistance in Head and Neck Squamous Cell Carcinoma: From Radioresistant HNSCC Cell Line to Integrated Bioinformatics Methods. Front. Genet. 2022, 13, 1017762. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin Contributes to Epithelial-Mesenchymal Transition Cancer Cell Mechanics by Mediating Cytoskeletal Organization and Focal Adhesion Maturation. Oncotarget 2015, 6, 15966. [Google Scholar] [CrossRef] [PubMed]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Qi, J.-L.; He, J.-R.; Liu, C.-B.; Jin, S.-M.; Yang, X.; Bai, H.-M.; Ma, Y.-B. SQSTM1/P62 Regulate Breast Cancer Progression and Metastasis by Inducing Cell Cycle Arrest and Regulating Immune Cell Infiltration. Genes Dis. 2022, 9, 1332–1344. [Google Scholar] [CrossRef]
- Li, S.-S.; Xu, L.-Z.; Zhou, W.; Yao, S.; Wang, C.-L.; Xia, J.-L.; Wang, H.-F.; Kamran, M.; Xue, X.-Y.; Dong, L. P62/SQSTM1 Interacts with Vimentin to Enhance Breast Cancer Metastasis. Carcinogenesis 2017, 38, 1092–1103. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist Is a Transcriptional Repressor of E-Cadherin Gene Expression in Breast Cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef]
- Zou, J.; Liu, Y.; Li, B.; Zheng, Z.; Ke, X.; Hao, Y.; Li, X.; Li, X.; Liu, F.; Zhang, Z. Autophagy Attenuates Endothelial-to-Mesenchymal Transition by Promoting Snail Degradation in Human Cardiac Microvascular Endothelial Cells. Biosci. Rep. 2017, 37, BSR20171049. [Google Scholar] [CrossRef]
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy Regulates Hepatocyte Identity and Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transitions Promoting Snail Degradation. Cell Death Dis. 2015, 6, e1880. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, H.; Liu, D.; Hill, C.; Ertay, A.; Li, J.; Zou, Y.; Miller, P.; White, E.; Downward, J. Autophagy Inhibition Specifically Promotes Epithelial-Mesenchymal Transition and Invasion in RAS-Mutated Cancer Cells. Autophagy 2019, 15, 886–899. [Google Scholar] [CrossRef]
- Li, Y.; Ma, J.; Qian, X.; Wu, Q.; Xia, J.; Miele, L.; H. Sarkar, F.; Wang, Z. Regulation of EMT by Notch Signaling Pathway in Tumor Progression. Curr. Cancer Drug Targets 2013, 13, 957–962. [Google Scholar] [CrossRef]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch Signalling Pathway of Cancer Stem Cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef]
- You, W.-K.; Schuetz, T.J.; Lee, S.H. Targeting the DLL/Notch Signaling Pathway in Cancer: Challenges and Advances in Clinical Development. Mol. Cancer Ther. 2023, 22, 3–11. [Google Scholar] [CrossRef]
- Zada, S.; Hwang, J.S.; Lai, T.H.; Pham, T.M.; Ahmed, M.; Elashkar, O.; Kim, W.; Kim, D.R. Autophagy-Mediated Degradation of NOTCH1 Intracellular Domain Controls the Epithelial to Mesenchymal Transition and Cancer Metastasis. Cell Biosci. 2022, 12, 17. [Google Scholar] [CrossRef]
- Yang, M.; Zhao, H.; Guo, L.; Zhang, Q.; Zhao, L.; Bai, S.; Zhang, M.; Xu, S.; Wang, F.; Wang, X. Autophagy-Based Survival Prognosis in Human Colorectal Carcinoma. Oncotarget 2015, 6, 7084. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, S.; Xue, H.; Zhao, E.; Jiang, H.; Hao, C. The Roles of Beclin 1 Expression in Gastric Cancer: A Marker for Carcinogenesis, Aggressive Behaviors and Favorable Prognosis, and a Target of Gene Therapy. Front. Oncol. 2020, 10, 613679. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, Y.K.; Kim, H.; Lee, J.; Oh, M.J.; Kim, S.B.; Kim, M.; Kim, K.H.; Yoon, H.J.; Lee, M. Snail Acetylation by Autophagy-derived Acetyl-coenzyme A Promotes Invasion and Metastasis of KRAS-LKB1 Co-mutated Lung Cancer Cells. Cancer Commun. 2022, 42, 716–749. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bonavida, B. A New Linkage between the Tumor Suppressor RKIP and Autophagy: Targeted Therapeutics. Crit. Rev. Oncog. 2018, 23, 613679. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.-Y.; Valapala, M. Regulation of Autophagy by the Glycogen Synthase Kinase-3 (GSK-3) Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 1709. [Google Scholar] [CrossRef]
- Guo, X.; Li, Z.; Zhu, X.; Zhan, M.; Wu, C.; Ding, X.; Peng, K.; Li, W.; Ma, X.; Lv, Z. A Coherent FOXO3-SNAI2 Feed-Forward Loop in Autophagy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118285119. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y. An FBXW7-ZEB2 Axis Links EMT and Tumour Microenvironment to Promote Colorectal Cancer Stem Cells and Chemoresistance. Oncogenesis 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Qian, X.; Ding, Y.; Chen, Q.; Ye, F.; Ye, Y.; Hou, Y.; Yu, J.; Zhao, L. C-Fos Regulated by TMPO/ERK Axis Promotes 5-FU Resistance via Inducing NANOG Transcription in Colon Cancer. Cell Death Dis. 2024, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Metge, B.J.; Mitra, A.; Chen, D.; Shevde, L.A.; Samant, R.S. N-Myc and STAT Interactor Regulates Autophagy and Chemosensitivity in Breast Cancer Cells. Sci. Rep. 2015, 5, 11995. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Li, Z.; Dong, Y.; Huang, H.; Yang, B.; Zhao, E.; Chen, Y.; Yang, L.; Lu, J. An EMT-Related Genes Signature as a Prognostic Biomarker for Patients with Endometrial Cancer. BMC Cancer 2023, 23, 879. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Z.; Yin, J.; Li, Y.; Yin, Y.; Lei, X.; Zhu, W. Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer. Front. Oncol. 2021, 11, 566539. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xia, S.; Zhao, Z.; Deng, L.; Wang, H.; Yang, D.; Hu, Y.; Ji, J.; Huang, D.; Xin, T. EMP3 as a Prognostic Biomarker Correlates with EMT in GBM. BMC Cancer 2024, 24, 89. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Chiachio, M.; Molina-Crespo, Á.; Ramos-Nebot, C.; Martinez-Val, J.; Martinez, L.; Gassner, K.; Llobet, F.J.; Soriano, M.; Hernandez, A.; Cordani, M. Gasdermin B Over-Expression Modulates HER2-Targeted Therapy Resistance by Inducing Protective Autophagy through Rab7 Activation. J. Exp. Clin. Cancer Res. 2022, 41, 285. [Google Scholar] [CrossRef]
- Shen, X.; Deng, Y.; Chen, L.; Liu, C.; Li, L.; Huang, Y. Modulation of Autophagy Direction to Enhance Antitumor Effect of Endoplasmic-Reticulum-Targeted Therapy: Left or Right? Adv. Sci. 2023, 10, 2301434. [Google Scholar] [CrossRef]
- Pangilinan, C.; Xu, X.; Herlyn, M.; Liang, C. Autophagy Paradox: Strategizing Treatment Modality in Melanoma. Curr. Treat. Options Oncol. 2023, 24, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jing, F.-J.; Xu, W.; Li, X.; Li, X.; Sun, J.-L.; Xing, X.-M.; Zhou, C.-K.; Jing, F.-B. Ubenimex Induces Autophagy Inhibition and EMT Suppression to Overcome Cisplatin Resistance in GC Cells by Perturbing the CD13/EMP3/PI3K/AKT/NF-ΚB Axis. Aging 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strippoli, R.; Niayesh-Mehr, R.; Adelipour, M.; Khosravi, A.; Cordani, M.; Zarrabi, A.; Allameh, A. Contribution of Autophagy to Epithelial Mesenchymal Transition Induction during Cancer Progression. Cancers 2024, 16, 807. https://doi.org/10.3390/cancers16040807
Strippoli R, Niayesh-Mehr R, Adelipour M, Khosravi A, Cordani M, Zarrabi A, Allameh A. Contribution of Autophagy to Epithelial Mesenchymal Transition Induction during Cancer Progression. Cancers. 2024; 16(4):807. https://doi.org/10.3390/cancers16040807
Chicago/Turabian StyleStrippoli, Raffaele, Reyhaneh Niayesh-Mehr, Maryam Adelipour, Arezoo Khosravi, Marco Cordani, Ali Zarrabi, and Abdolamir Allameh. 2024. "Contribution of Autophagy to Epithelial Mesenchymal Transition Induction during Cancer Progression" Cancers 16, no. 4: 807. https://doi.org/10.3390/cancers16040807
APA StyleStrippoli, R., Niayesh-Mehr, R., Adelipour, M., Khosravi, A., Cordani, M., Zarrabi, A., & Allameh, A. (2024). Contribution of Autophagy to Epithelial Mesenchymal Transition Induction during Cancer Progression. Cancers, 16(4), 807. https://doi.org/10.3390/cancers16040807