
Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer


Abstract
Simple Summary
Abstract
1. Introduction
1.1. Androgen Receptor and Prostate Cancer
- AR overexpression.
- AR gene amplification.
- Point mutations in the ligand-binding domain of the AR, creating a larger androgen binding pocket and converting AR antagonists to agonists.
- AR splice variants (e.g., AR-V7) lacking the ligand-binding domain. These variants are constitutively active and activate downstream transcription independently of ligands. This explains the limited efficacy of current FDA-approved AR antagonists for castration-resistant prostate cancer patients with AR splice variances [1,14,15,16].
- The hypersensitive pathway [17].
- The promiscuous pathway [17].
- Coactivators and corepressors [17].
- The outlaw pathway [17].
1.2. Targeting Proteins for Degradation
1.3. Targeting AR for Degradation
- It degrades the AR rather than antagonizes its function, overcoming resistance caused by the reactivation of AR signaling through factors discussed in Section 1.1. This is evidenced by the superior performance of the enzalutamide-based PROTAC (ARCC-4) over enzalutamide in prostate tumor cells with point mutations (F876L and T877A) in the AR [21].
- It is “event-driven” rather than “occupancy-driven” in AR antagonists, potentially enhancing therapeutic potency while reducing systemic drug exposure.
- It can be synergistic with AR antagonists.
- Bifunctional AR PROTACs, which hijack the ubiquitin–proteasome system (Section 3).
- Hydrophobic tagged chimeric degraders, which simulate misfolded AR (Section 4).
- Monomeric AR degraders, which destabilize the AR via diverse mechanisms (Section 5).
- Molecular glues (Section 6).
- Autophagic degradation of AR (Section 7).
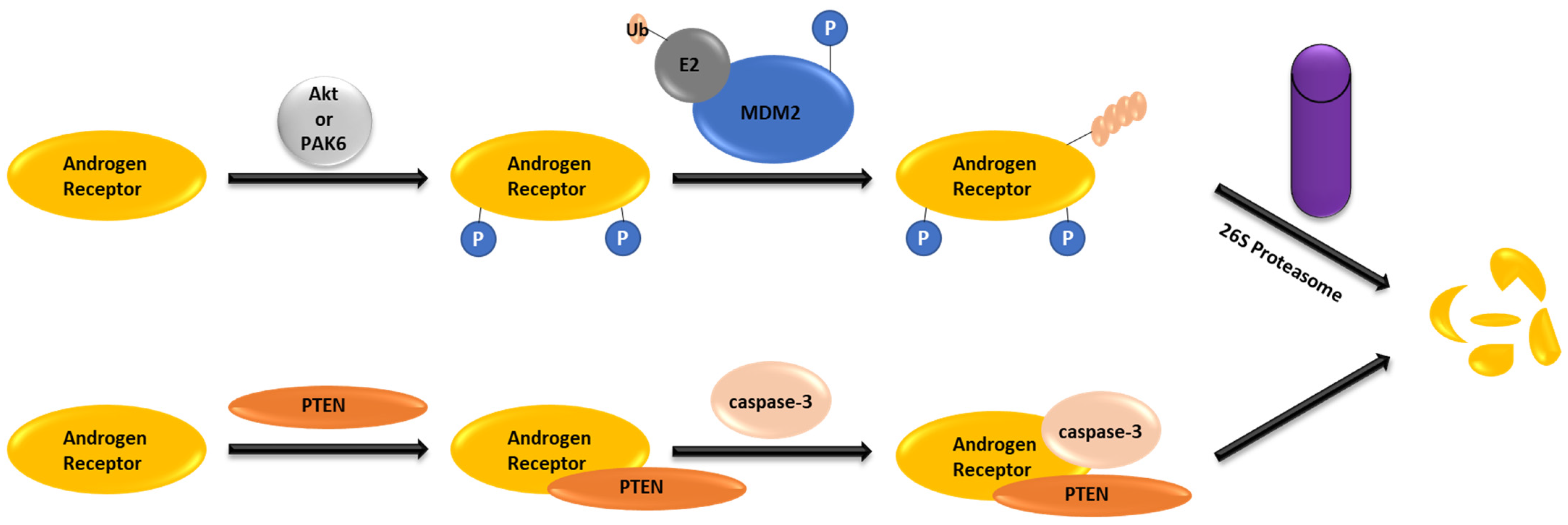
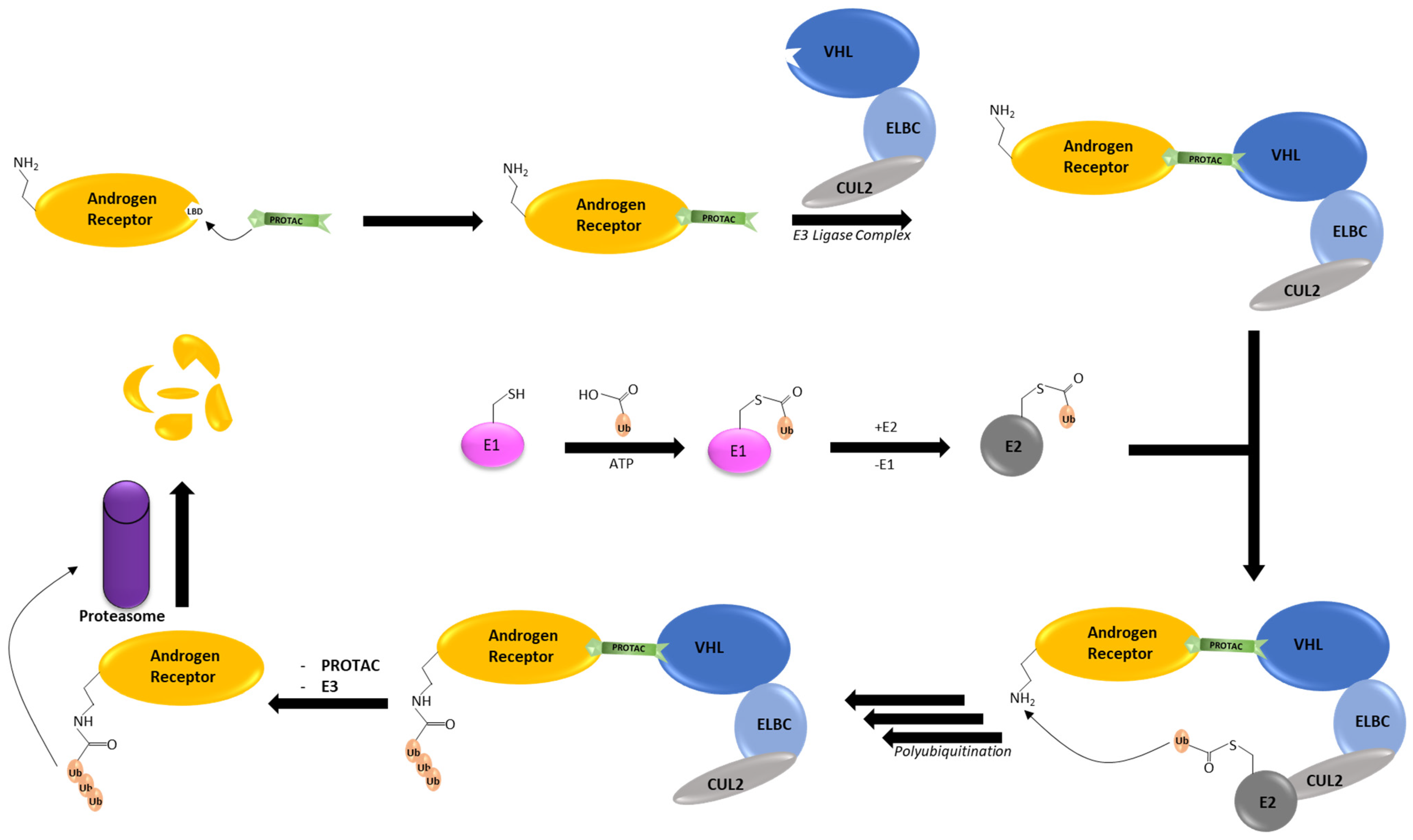
2. AR Degradation Pathways under the Physiological Conditions
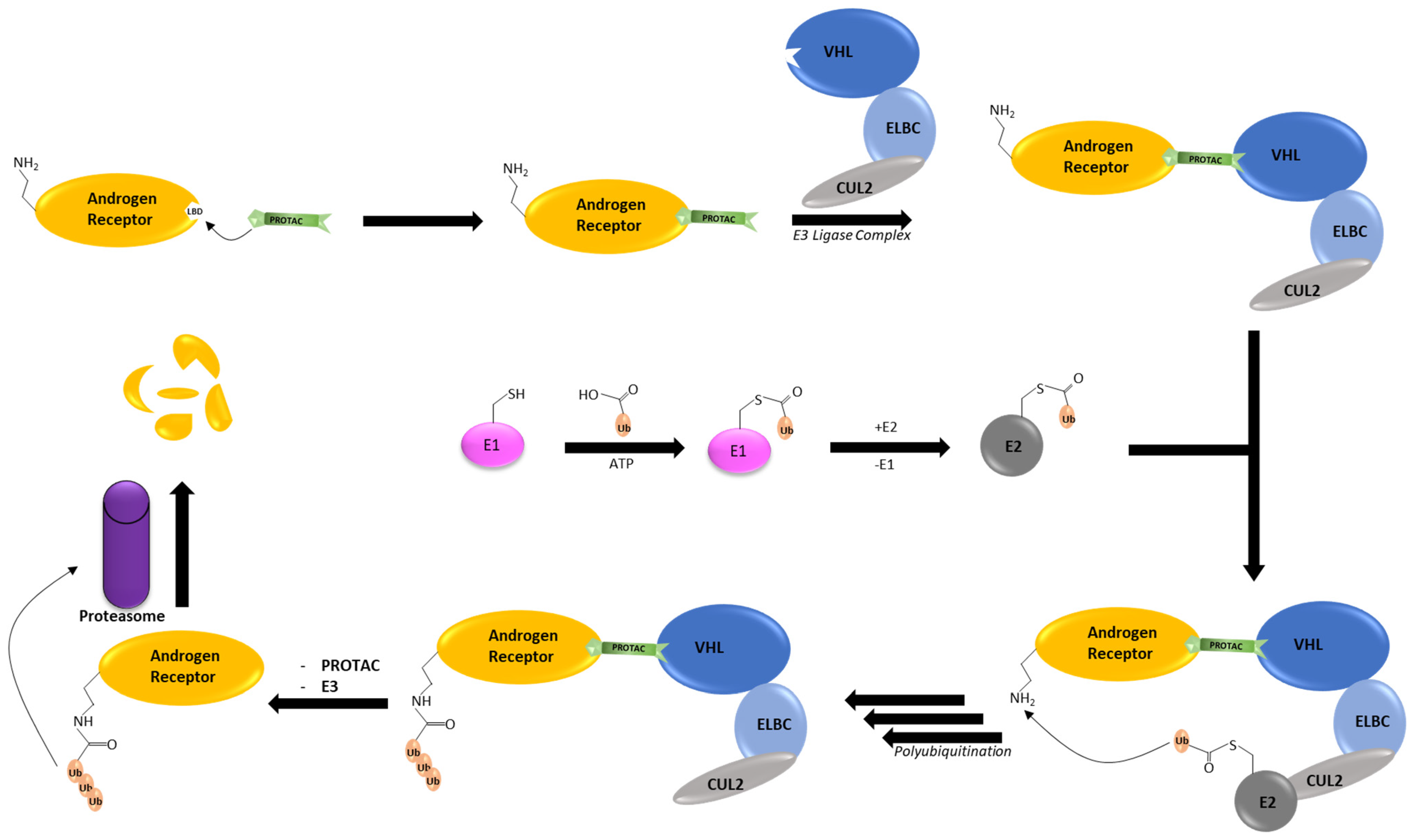
3. AR PROTACs
3.1. Advances in Clinical Studies
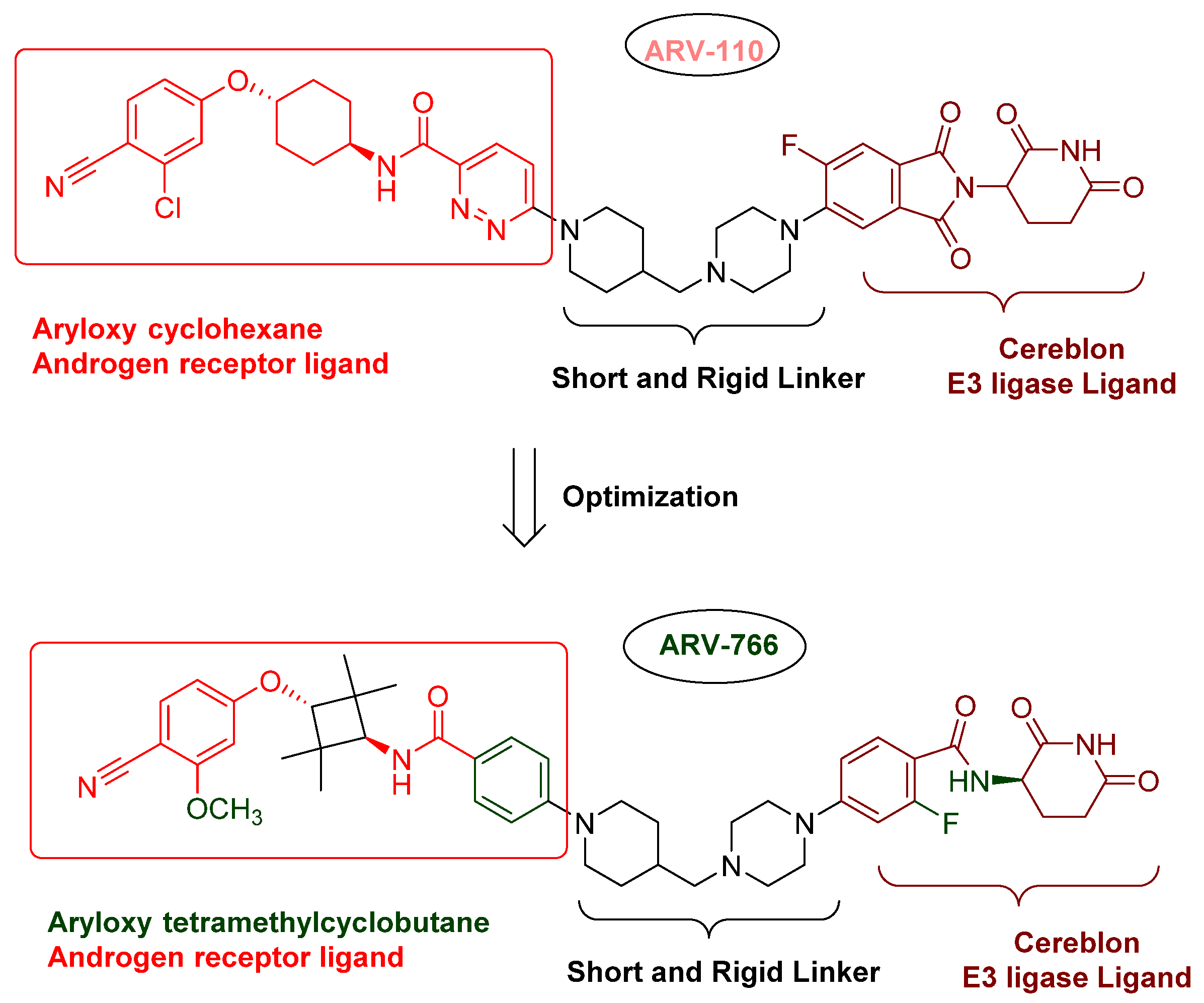
3.1.1. ARV-110 (Bavdegalutamide)
3.1.2. CC-94676 (AR-LDD, NCT04428788)
3.1.3. ARV-766 (NCT05067140)
3.1.4. HP-518 (NCT05252364)
3.1.5. AC176 (AC0176) (NCT05241613)
3.1.6. GT-20029 (NCT05428449)
3.2. Advances in AR Binders
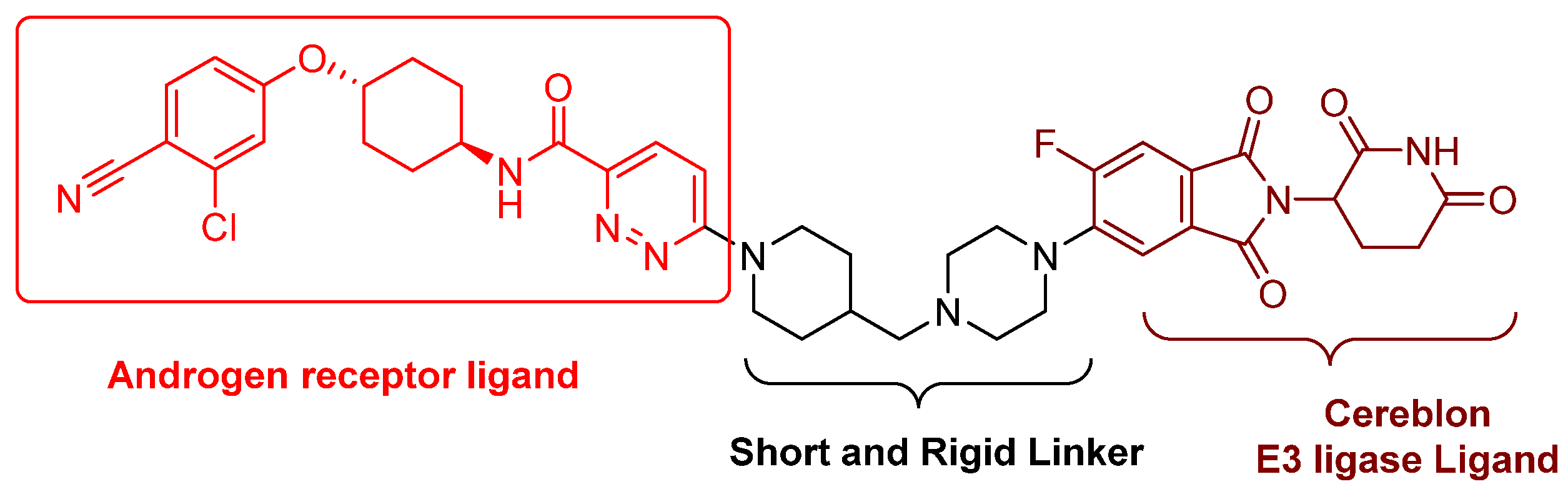
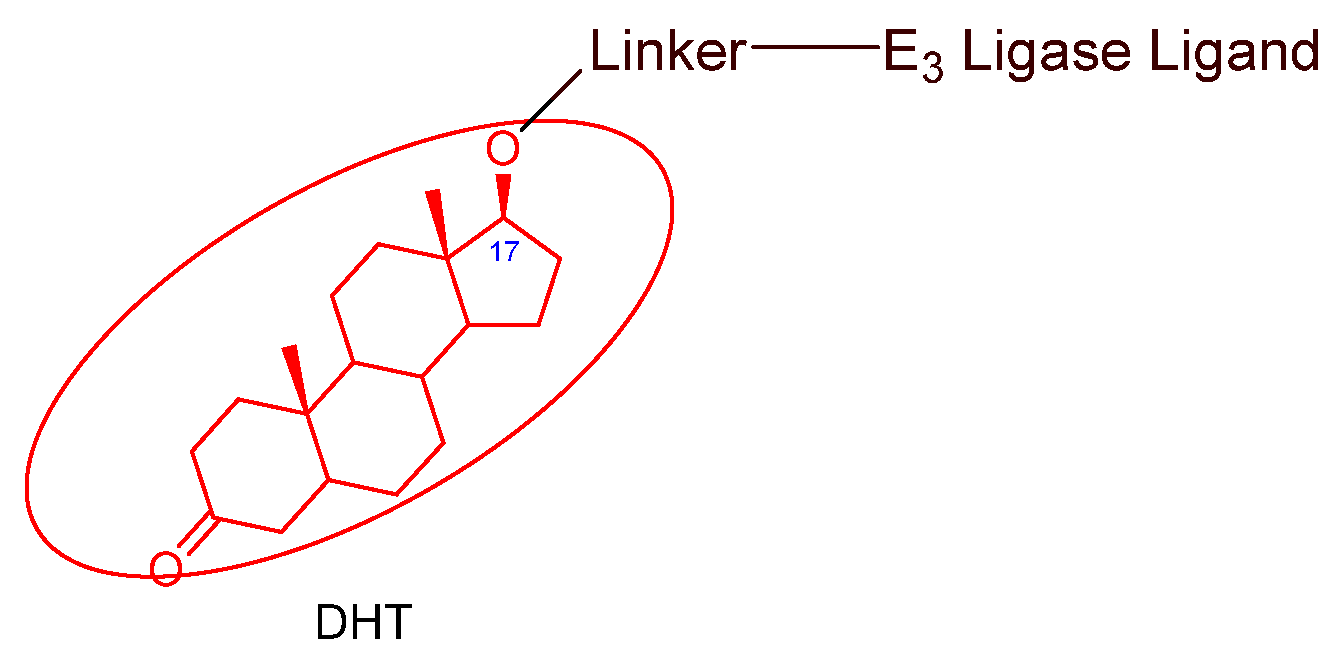
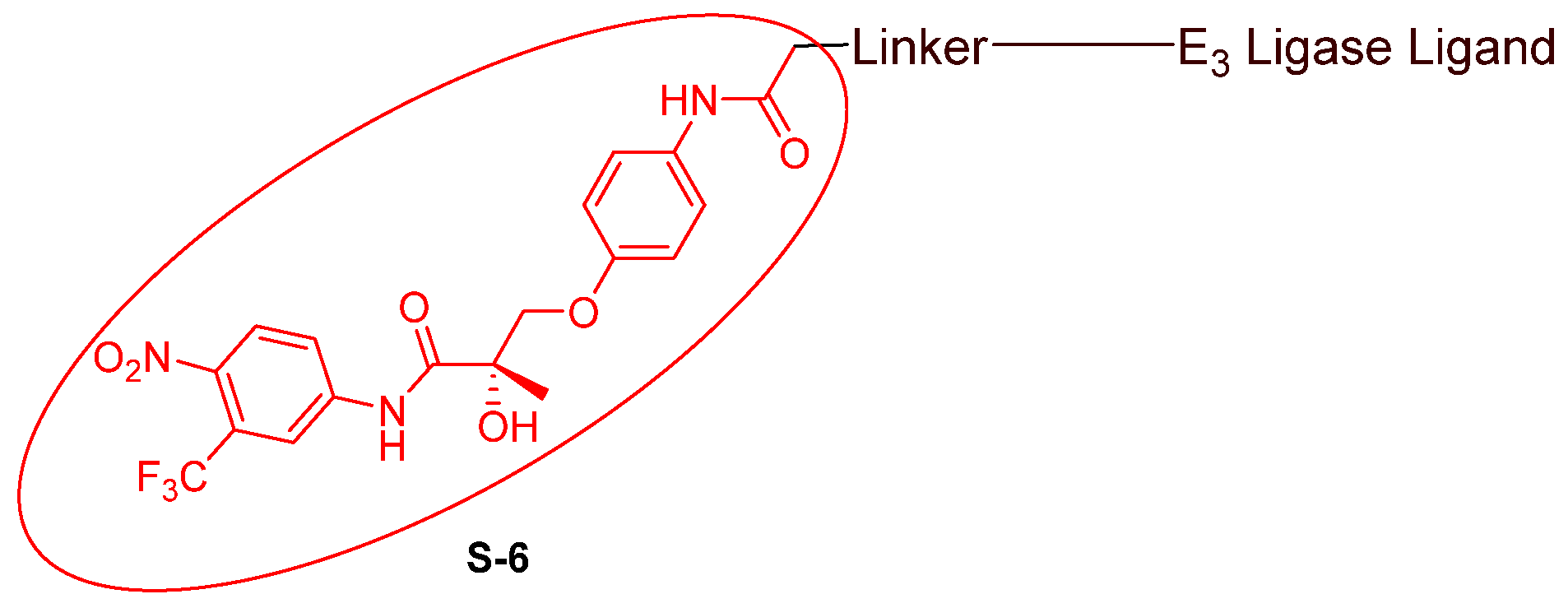
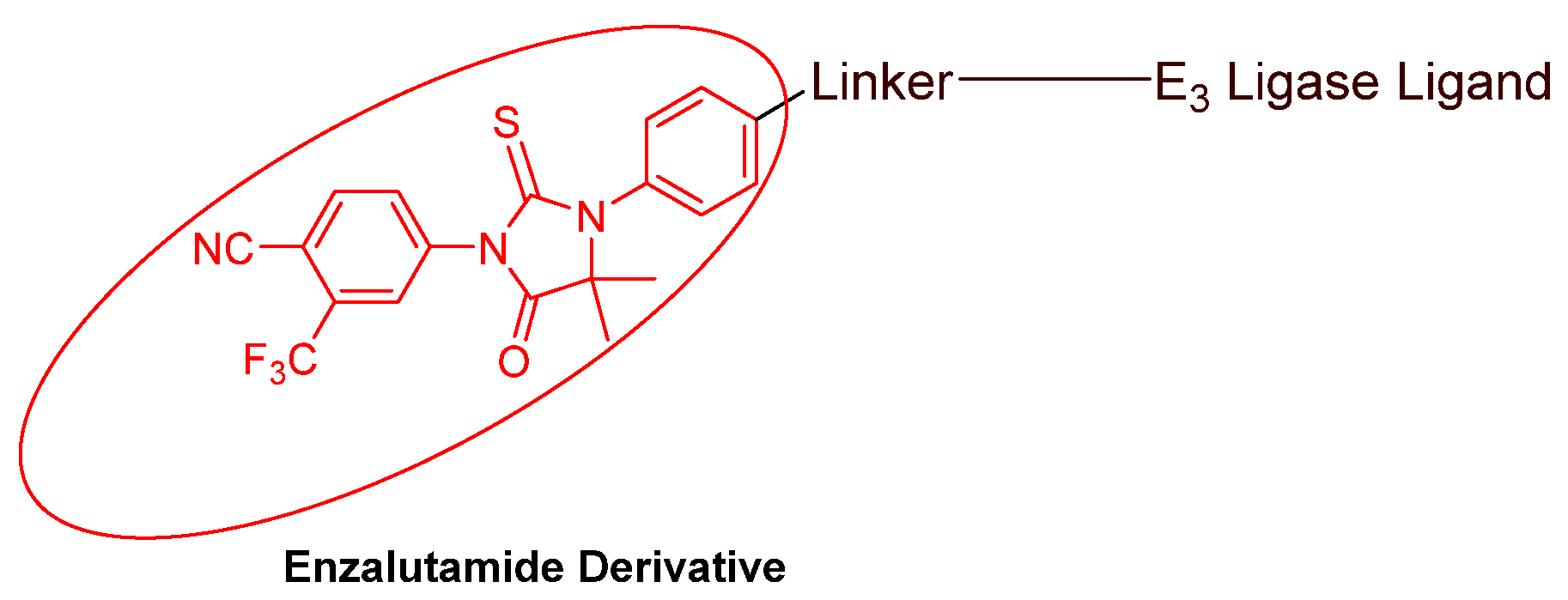
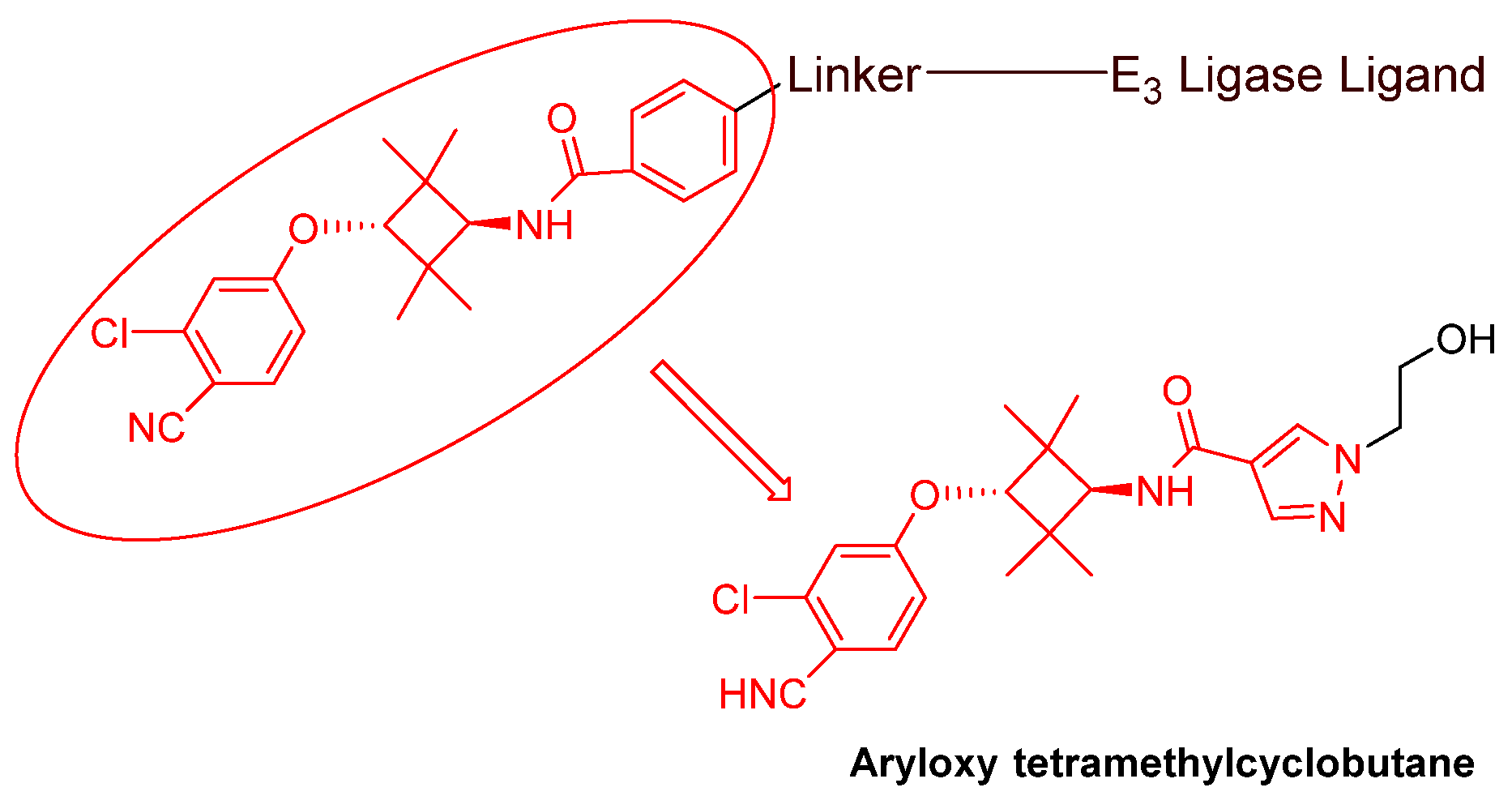
- Nonsteroidal AR antagonists binding to the ligand binding domain: enzalutamide (Figure 7) and aryloxy tetramethylcyclobutane (Figure 8). Enzalutamide is a currently marketed AR antagonist for castration-resistant prostate cancer. Enzalutamide-based PROTACs, such as ARCC-4, exemplify this class [21]. Aryloxy tetramethylcyclobutane was identified by Pfizer as a highly potent AR antagonist using cell-based high-throughput screening [44]. In addition to serving as the AR recruiter for ARC-766 developed by Arvinas, it has been successfully incorporated into a group of AR PROTACs, such as ARD-69, by Dr. Wang’s research group [45]. Aryloxy cyclohexane (Figure 3) and its amine derivative were used as AR binders in ARV-110 and ARD-2585 [46], which are two orally active PROTACs with exceptional potency. Technically, mutants and variants lacking the LBD render this group of AR degraders ineffective. However, treatment with ARV-110 led to about a 50% decrease in prostate-specific antigen in the treated patients with two mutations on the LBD.
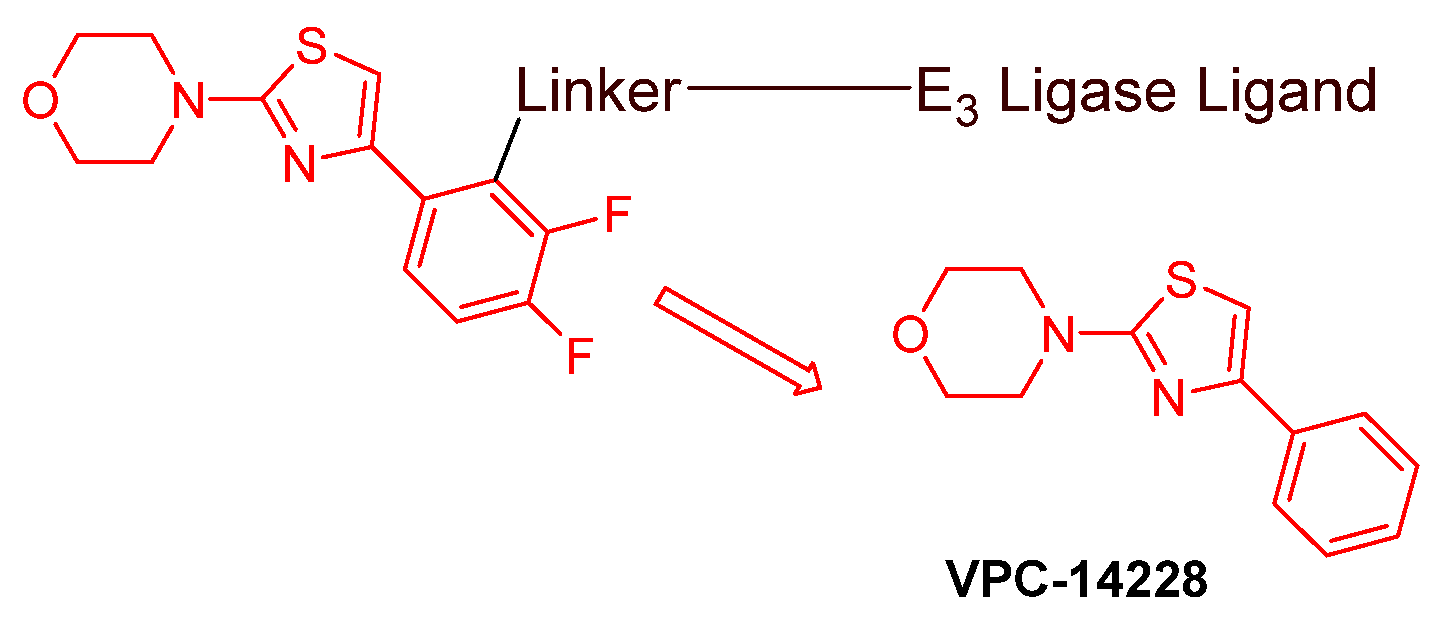
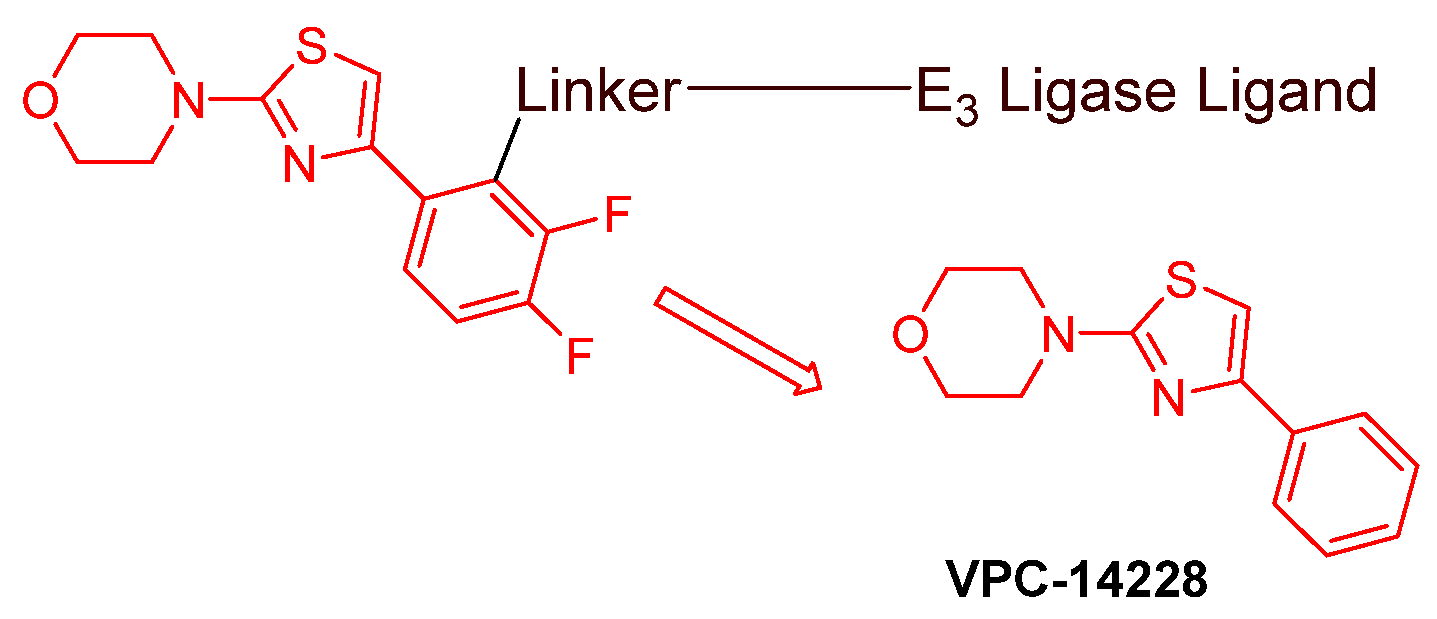
- AR DBD binders: VPC-14228. This group of AR ligands has less specificity for the AR due to the highly conserved DBD structure found in all nuclear receptors. MTX-23 is a reported AR PROTAC with VPC-14228 as an AR ligand that binds to DBD, enabling the degradation of ARV7 (Figure 9) [47]. The surface-exposed region on the AR DBD has been identified as an alternative druggable pocket for potential AR antagonists. VPC-14228 binds to this pocket through hydrogen bonding between its morpholine group and Tyr594, hydrophobic interactions between the thiazole ring and Val582/Phe583, and hydrophobic interactions between the phenyl ring and the aliphatic side chains of Arg609 and Lys610 [47].
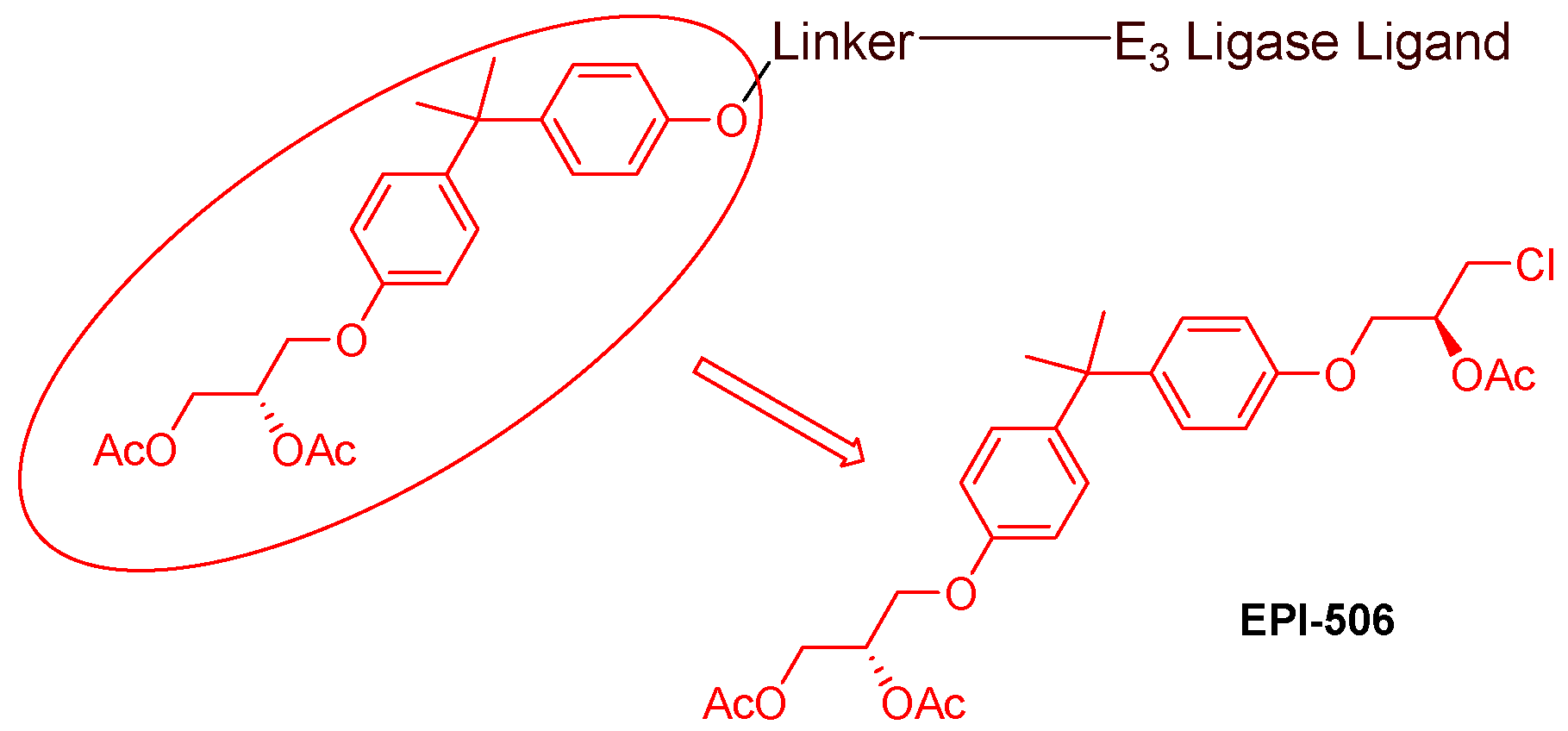
- AR antagonists binding to the N-terminal domain: The intrinsic disorder of the N-terminal domain poses a challenge to the rational design of N-terminal domain binders. The well-established AR N-terminal antagonist EPI-506 has been successfully used to create an AR PROTAC BWA-522 and derivatives (Figure 10) [48]. EPI-506 functions as a prodrug of EPI-002. Non-covalent interactions between EPI-002 and the transactivation unit 5 (Tau-5) domain of the N-terminal domain have been identified using NMR spectroscopy. This interaction involves a subset of three partially helical regions, namely, the R1 region (residues S341–G371), R2 region (residues L391–G414), and R3 (residues S426–G446) [49]. All-atom molecular dynamics computer simulations further elucidate that EPI-002 binds specifically to the R2 and R3 regions of Tau-5. Notably, amino acid residues W397 and W433 play the most significant role in mediating the interaction between EPI-002 and the R2 and R3 regions of Tau-5 [50].
3.3. Advandes in E3 Ligase Binders
3.3.1. MDM2 and IAP Binders
3.3.2. VHL Binders
3.3.3. CRBN Binders
3.3.4. Emerging E2 Ligase Binders
3.4. Advances in Linkers
4. Hydrophobic Tagged Chimeric AR Degraders
5. Monomeric AR Degraders with Diverse Mechanisms
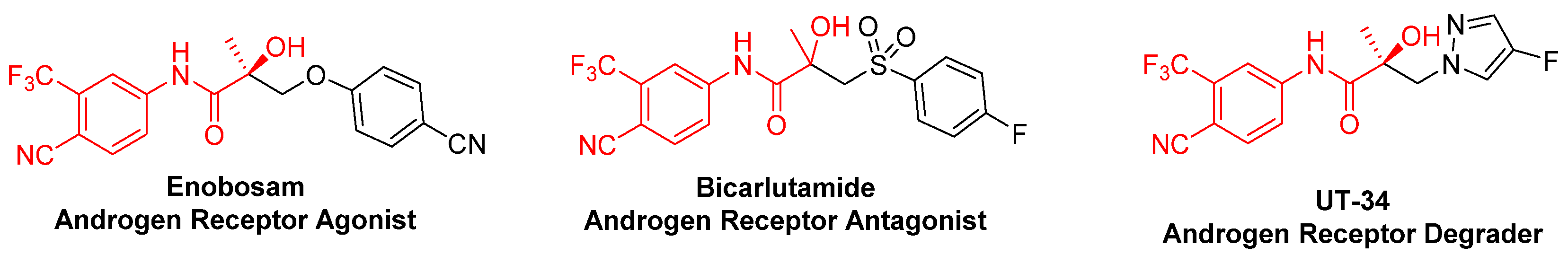
5.1. UT Series
5.2. AR Degraders via the Dissociation of Chaperone Protein
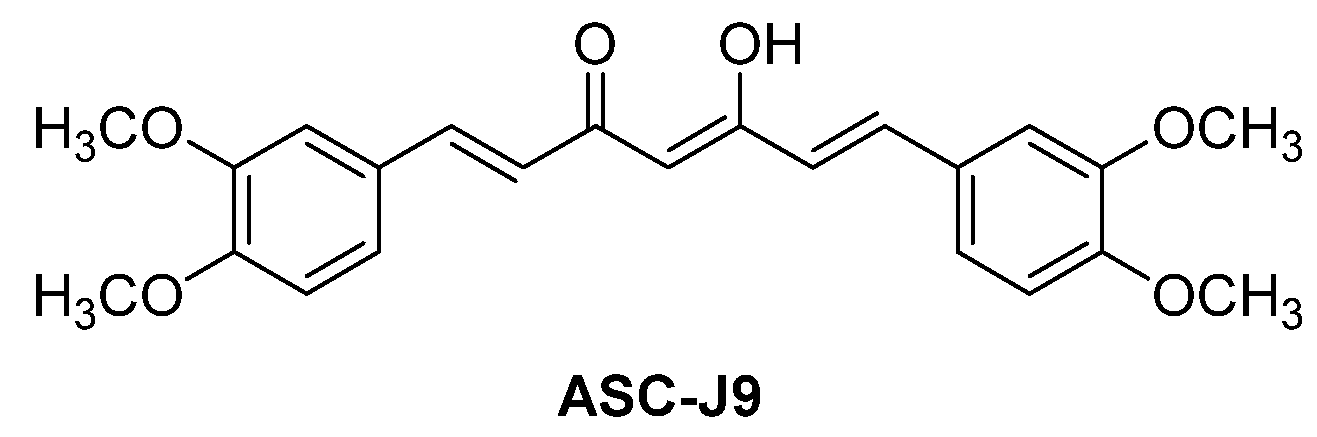
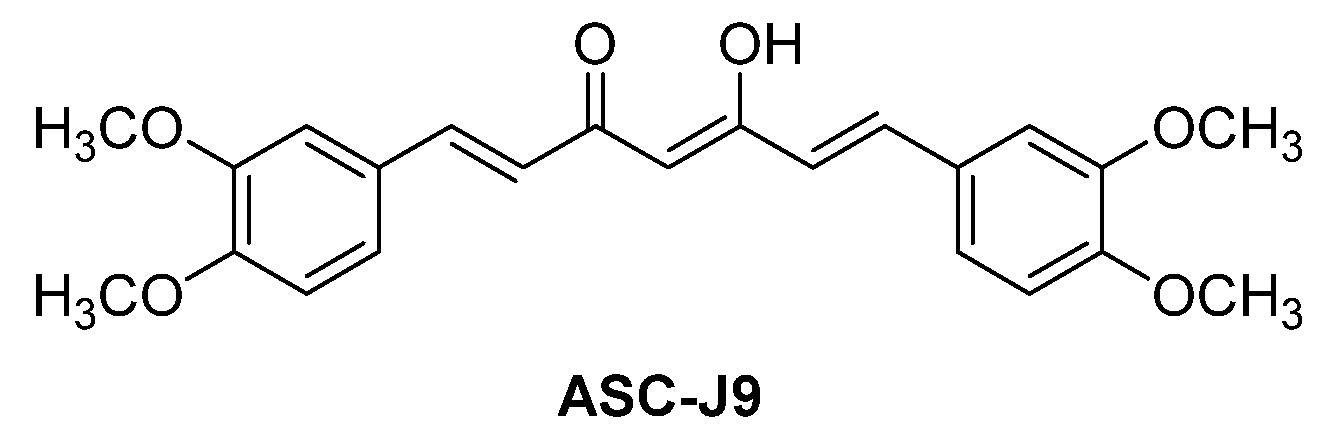
5.2.1. ASC-J9 (NCT01289574)
5.2.2. Niclosamide and ARVibs
5.2.3. Geldanamycin Analogs
5.3. Other Monomeric AR Degraders
6. Molecular Glues to Degrade AR
7. Autophagic Degradation of AR
8. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-I. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Maclean, H.E.; Warne, G.L.; Zajac, J.D. Localization of functional domains in the androgen receptor. J. Steroid Biochem. Mol. Biol. 1997, 62, 233–242. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2019, 380, 1235–1346. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, P.; Rivera, A.; Muthima, K.; Olveda, N.; Muchalski, H.; Chen, Q.-H. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules 2020, 25, 2448. [Google Scholar] [CrossRef]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Kwak, L.; Crowdis, J.P.; Sperger, J.M.; Zhao, S.G.; Xie, W.L.; Werner, L.; Lis, R.T.; Zhang, Z.W.; Wei, X.X.; et al. Phase II multicenter study of enzalutamide in metastatic castration-resistant prostate cancer to identify mechanisms driving resistance. Clin. Cancer Res. 2021, 27, 3610–3619. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. JNCI—J. Natl. Cancer Inst. 2017, 110, djx118. [Google Scholar] [CrossRef]
- Brooke, G.N.; Bevan, C.L. The role of androgen receptor mutations in prostate cancer progression. Curr. Genom. 2009, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Prekovic, S.; Van den Broeck, T.; Linder, S.; van Royen, M.E.; Houtsmuller, A.B.; Handle, F.; Joniau, S.; Zwart, W.; Claessens, F. Molecular underpinnings of enzalutamide resistance. Endocr.-Relat. Cancer 2018, 25, R545–R557. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Lu, C.X.; Luo, J.; Antonarakis, E.S. AR splicing variants and resistance to AR targeting agents. Cancers 2021, 13, 2563. [Google Scholar] [CrossRef] [PubMed]
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The androgen receptor in prostate cancer: Effect of structure, ligands and spliced variants on therapy. Biomedicines 2020, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Zhang, G.Y.; Wang, X.J.; Qi, Y.F.; Bai, S.S.; Li, D.Y.; Ma, T.F.; Sartor, O.; Flemington, E.K.; Zhang, H.T.; et al. Interplay between cytoplasmic and nuclear androgen receptor splice variants mediates castration resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; van Weerden, W.M.; Jenster, G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS ONE 2010, 5, e13500. [Google Scholar] [CrossRef] [PubMed]
- Nemes, A.; Tomuleasa, C.; Kacso, G. The androgen receptor remains a key player in metastatic hormone-refractory prostate cancer. Implications for new treatments. J. BUON 2014, 19, 357–364. [Google Scholar] [PubMed]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef]
- Ha, S.; Luo, G.; Xiang, H. A comprehensive overview of small-molecule androgen receptor degraders: Recent progress and future perspectives. J. Med. Chem. 2022, 65, 16128–16154. [Google Scholar] [CrossRef]
- Luan, H.; Xu, P.; Meng, Y.; Li, Z.; Bian, J. A critical update on the strategies towards modulators targeting androgen receptors. Bioorg. Med. Chem. 2020, 28, 115554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, C.; Xiao, X.; Zhou, J. Improving strategies in the development of protein-downregulation-based antiandrogens. ChemMedChem 2021, 16, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Chang, C. Expression and degradation of androgen receptor: Mechanism and clinical implication. J. Clin. Endocrinol. Metab. 2003, 88, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Collins, I.; Wang, H.; Caldwell, J.J.; Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem. J. 2017, 474, 1127–1147. [Google Scholar] [CrossRef] [PubMed]
- Sheflin, L.; Keegan, B.; Zhang, W.; Spaulding, S.W. Inhibiting Proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem. Biophys. Res. Commun. 2000, 276, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Wang, L.; Hu, Y.-C.; Altuwaijri, S.; Chang, C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002, 21, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Hu, Y.-C.; Lee, D.K.; Chang, C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol. Endocrinol. 2004, 18, 2409–2423. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins tot he Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- He, S.; Dong, G.; Cheng, J.; Wu, Y.; Sheng, C. Strategies for designing proteolysis targeting chimeras (PROTACs). Med. Res. Rev. 2022, 42, 1280–1342. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, T.; Simons, I.H.; Beier, A.K.; Ebersbach, C.; Aksoy, C.; Seed, R.I.; Stope, M.B.; Thomas, C.; Erb, H.H.H. A systematic comparison of antiandrogens identifies androgen receptor protein stability as an indicator for treatment response. Life 2021, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Coss, C.C.; Thiyagarajan, T.; Watts, K.; Hwang, D.-J.; He, Y.; Selth, L.A.; McEwan, I.J.; Duke, C.B.; Pagadala, J.; et al. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. 2017, 77, 6282–6298. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Han, X. Targeting androgen receptor degradation with PROTACs from bench to bedside. Biomed. Pharmacother. 2023, 158, 114112. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhi, Y.; Liu, B.; Yao, Q. Advancing strategies for proteolysis-targeting chimera design. J. Med. Chem. 2023, 66, 2308–2329. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Dougan, M.; Peck, R.A.; Burris, H.A. First-in-human Phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38 (Suppl. S15), 3500. [Google Scholar] [CrossRef]
- No authors listed. PROTAC shrinks mutated prostate tumors. Cancer Discov. 2022, 12, OF2. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef]
- Schneekloth, J.S., Jr.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.M.; Deshaies, R.J.; Sakamoto, K.M. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Linton, A.; Kephart, S.; Ornelas, M.; Pairish, M.; Gonzalez, J.; Greasley, S.; Nagata, A.; Burke, B.J.; Edwards, M.; et al. Discovery of aryloxy tetramethylcyclobutanes as novel androgen receptor antagonists. J. Med. Chem. 2011, 54, 7693–7704. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Zhao, L.; Han, X.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Kirchhoff, P.D.; Wang, L.; et al. Discovery of ARD-2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J. Med. Chem. 2021, 64, 13487–13509. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a novel PROTAC of androgen receptor splice variant-7 and androgen receptor, on CRPC resistant to second-line antiandrogen therapy. Mol. Cancer Ther. 2021, 20, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, C.; Yang, Z.; Zhang, S.; Hu, X.; Li, B.; Mao, M.; Wang, X.; Li, Z.; Ma, S.; et al. Discovery of BWA-522, a first-in-class and orally bioavailable PROTAC degrader of the androgen receptor targeting N-terminal domain for the treatment of prostate cancer. J. Med. Chem. 2023, 66, 11158–11186. [Google Scholar] [CrossRef]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.W.; Buzon, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; Garcia, J.; Bertoncini, C.W.; Estebanez-Perpina, E.; et al. EPI-001, a compound active against castration-resistant prostate cancer, targets transactivation unit 5 of the androgen receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef]
- Zhu, J.; Salvatella, X.; Robustelli, P. Small molecules targeting the disordered transactivation domain of the androgen receptor induce the formation of collapsed helical states. Nat. Commun. 2022, 13, 6390. [Google Scholar] [CrossRef]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. An emerging therapeutic modality in precision medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, Synthesis and Biological Evaluation of Nuclear Receptor degradation Inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef] [PubMed]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.B.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.H.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.R.; Han, L.Q.; Mao, S.J.; Xu, P.; Xu, X.X.; Zhao, R.B.; Wu, Z.H.; Zhong, K.; Yu, G.L.; Wang, X.L. Discovery of A031 as effective proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader for the treatment of prostate cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primay target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Takwale, A.D.; Jo, S.-H.; Jeon, Y.U.; Kim, H.S.; Shin, C.H.; Lee, H.K.; Ahn, S.; Lee, C.O.; Ha, J.D.; Kim, J.-H.; et al. Design and characterization of cereblon-mediated androgen receptor proteolysis-targeting chimeras. Eur. J. Med. Chem. 2020, 208, 112769. [Google Scholar] [CrossRef]
- Da, Y.; Liu, S.; Lin, P.; Wang, F.; Yan, R.; Shu, Y.; Lin, J. Design, synthesis, and biological evaluation of small molecule PROTACs for potential anticancer effects. Med. Chem. Res. 2020, 29, 334–340. [Google Scholar] [CrossRef]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Wang, L.; Matvekas, A.; et al. Strategies toward discovery of potent and orally bioavailable proteolysis targeting chimera degraders of androgen receptor for the treatment of prostate cancer. J. Med. Chem. 2021, 64, 12831–12854. [Google Scholar] [CrossRef]
- Han, X.; Zhao, L.; Xiang, W.; Miao, B.; Qin, C.; Wang, M.; Xu, T.; McEachern, D.; Lu, J.; Wang, Y.; et al. Discovery of ARD-2051 as a potent and orally efficacious proteolysis targeting chimera (PROTAC) degrader of androgen receptor for the treatment of advanced prostate cancer. J. Med. Chem. 2023, 66, 8822–8843. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Rooney, T.P.C.; Bayle, E.D.; Mirza, T.; Willems, H.M.G.; Clarke, J.H.; Andrews, S.P.; Skidmore, J. Systematic investigation of the permeability of androgen receptor PROTACs. ACS Med. Chem. Lett. 2020, 11, 1539–1547. [Google Scholar] [CrossRef]
- Kim, G.Y.; Song, C.W.; Yang, Y.-S.; Lee, N.-R.; Yoo, H.-S.; Son, S.H.; Lee, S.J.; Park, J.S.; Lee, J.K.; Inn, K.-S.; et al. Chemical degradation of androgen receptor (AR) using bicalutamide analog-thalidomide PROTACs. Molecules 2021, 26, 2525. [Google Scholar] [CrossRef] [PubMed]
- Ishoey, M.; Chorn, S.; Singh, N.; Jaeger, M.G.; Brand, M.; Paulk, J.; Bauer, S.; Erb, M.A.; Parapatics, K.; Muller, A.C.; et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 2018, 13, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Forte, N.; Dovala, D.; Hesse, M.J.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Nomura, D.K. Targeted protein degradation through E2 recruitment. ACS Chem. Biol. 2023, 18, 897–904. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current strategies for the design of PROTAC linkers: A critical review. Explor. Target. Antitumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef] [PubMed]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the role of linker design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Xie, H.; Liang, J.J.; Wang, Y.L.; Hu, T.X.; Wang, J.Y.; Yang, R.H.; Yan, J.K.; Zhang, Q.R.; Xu, X.; Liu, H.M.; et al. The design, synthesis and anti-tumor mechanism study of new androgen receptor degrader. Eur. J. Med. Chem. 2020, 204, 112512. [Google Scholar] [CrossRef]
- He, Q.; Zhao, X.; Wu, D.; Jia, S.; Liu, C.; Cheng, Z.; Huang, F.; Chen, Y.; Lu, T.; Lu, S. Hydrophobic tag-based protein degradation: Development, opportunity and challenge. Eur. J. Med. Chem. 2023, 260, 115741. [Google Scholar] [CrossRef]
- Ponnusamy, S.; He, Y.L.; Hwang, D.J.; Thiyagarajan, T.; Houtman, R.; Bocharova, V.; Sumpter, B.G.; Fernandez, E.; Johnson, D.; Du, Z.Y.; et al. Orally bioavailable androgen receptor degrader, potential next-generation therapeutic for enzalutamide-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.-J.; He, Y.; Ponnusamy, S.; Mohler, M.L.; Thiyagarajan, T.; McEwan, I.J.; Narayanan, R.; Miller, D.D. New generation of selective androgen receptor degraders: Our initial design, synthesis, and biological evaluation of new compounds with enzalutamide-resistant prostate cancer activity. J. Med. Chem. 2019, 62, 491–511. [Google Scholar] [CrossRef] [PubMed]
- He, Y.L.; Hwang, D.J.; Ponnusamy, S.; Thiyagarajan, T.; Mohler, M.L.; Narayanan, R.; Miller, D.D. Pyrazol-1-yl-propanamide as SARD and Pan-Antagonists for the treatment of enzalutamide-resistant prostate cancer. J. Med. Chem. 2020, 63, 12642–12665. [Google Scholar] [CrossRef] [PubMed]
- He, Y.L.; Hwang, D.J.; Ponnusamy, S.; Thiyagarajan, T.; Mohler, M.L.; Narayanan, R.; Miller, D.D. Exploration and biological evaluation of basic heteromonocyclic propanamide derivatives as SARDs for the treatment of enzalutamide-resistant prostate cancer. J. Med. Chem. 2021, 64, 11045–11062. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, T.; Ponnusamy, S.; Hwang, D.-J.; He, Y.; Asemota, S.; Young, K.L.; Johnson, D.L.; Bocharova, V.; Zhou, W.; Jain, A.K.; et al. Inhibiting androgen receptor splice variants with cysteine-selective irreversible covalent inhibitors to treat prostate cancer. Proc. Natl. Acad. Sci. USA 2023, 120, e2211832120. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Xiao, Z.; Ishida, J.; Nagai, M.; Wang, H.-K.; Itokawa, H.; Su, C.-Y.; Shih, C.; Chiang, T.; Chang, E.; et al. Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J. Med. Chem. 2002, 45, 5037–5042. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.M.; Chang, Y.J.; Yu, I.C.; Yeh, S.Y.; Wu, C.C.; Miyamoto, H.; Merry, D.E.; Sobue, G.; Chen, L.M.; Chang, S.S.; et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat. Med. 2007, 13, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.P.; Huang, C.K.; Chang, Y.J.; Chung, C.Y.; Yamashita, S.; Li, L.; Lee, S.O.; Yeh, S.Y.; Chang, C.S. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am. J. Pathol. 2013, 182, 460–473. [Google Scholar] [CrossRef]
- Chou, F.J.; Chen, Y.Y.; Chen, D.; Niu, Y.J.; Li, G.H.; Keng, P.; Yeh, S.Y.; Chang, C.S. Preclinical study using androgen receptor (AR) degradation enhancer to increase radiotherapy efficacy via targeting radiation-increased AR to better suppress prostate cancer progression. EBioMedicine 2019, 40, 504–516. [Google Scholar] [CrossRef]
- Liu, C.F.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide Inhibits Androgen Receptor Variants Expression and Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.M.; Ning, S.; Yang, J.C.; Lou, W.; Lombard, A.P.; Zhao, J.; Wu, C.-Y.; Yu, A.; Evans, C.P.; et al. ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling. Oncogene 2021, 40, 5379–5392. [Google Scholar] [CrossRef]
- Solit, D.B.; Zheng, F.Z.F.; Drobnjak, M.; Münster, P.N.; Higgins, B.; Verbel, D.; Heller, G.; Tong, W.; Cordon-Cardo, C.; Agus, D.B.; et al. 17-allylamino-17-demythoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 2002, 8, 986–993. [Google Scholar]
- Heath, E.I.; Hillman, D.W.; Vaishampayan, U.; Sheng, S.; Sarkar, F.; Harper, F.; Gaskins, M.; Pitot, H.C.; Tan, W.; Ivy, S.P.; et al. A phase II trial of 17-allyamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008, 14, 7940–7946. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Lin, Z.; Zhao, B.; Wang, Y.; Peng, R.; Wang, M.; Lu, C.; Shi, G.; Shen, Y. 17-DMCHAG, a new geldanamycin derivative, inhibits prostate cancer cells through Hsp90 inhibition and surviving downregulation. Cancer Lett. 2015, 362, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, P.; Hu, M.; Yang, L.; Yan, G.; Xu, R.; Deng, Y.; Li, X.; Chen, Y. Discovery and biological evaluation of darolutamide derivatives as inhibitors and down-regulators of wild-type AR and the mutants. Eur. J. Med. Chem. 2019, 182, 111608. [Google Scholar] [CrossRef] [PubMed]
- Bastos, D.A.; Antonarakis, E.S. Galeterone for the treatment of advanced prostate cancer: The evidence to date. Drug Des. Dev. Ther. 2016, 10, 2289–2297. [Google Scholar]
- Gerry, C.J.; Schreiber, S.L. Unifying principles of bifunctional, proximity-inducing small molecules. Nat. Chem. Biol. 2020, 16, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Q.; Ding, Y.; He, S.P.; Sheng, C.Q. Molecular glues for targeted protein degradation: From serendipity to rational discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Thankan, R.S.; Purushottamachar, R.; Weber, D.J.; Njar, V.C.O. Targeted degradation of androgen receptor by VNPP433-3β in castration-resistant prostate cancer cells implicates interaction with E3 ligase MDM2 resulting in ubiquitin-proteasomal degradation. Cancers 2023, 15, 1198. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasone and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Kommander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Costales, M.G.; Aikawa, H.; Li, Y.; Childs-Disney, J.L.; Abegg, D.; Hoch, D.G.; Pradeep Belagapudi, S.; Nakai, Y.; Khan, T.; Wang, K.W.; et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Shourideh, M.; Goodrich, D.W.; Koochekpour, S. Riluzole Induces AR Degradation via Endoplasmic Reticulum Stress Pathway in Androgen-dependent and Castration-resistant Prostate Cancer Cells. Prostate 2019, 79, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hu, Q.; Zhang, X.; Sun, R.; Liu, Z.; Wu, S.; Tian, S.; Ma, X.; Dai, Z.; Yang, X.; et al. DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs. Nat. Commun. 2022, 13, 7133. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Y.; Wang, Z.; Li, H.; Dong, C.; Zhang, X. Recent advances in Pro-PROTAC development to address on-target off-tumor toxicity. J. Med. Chem. 2023, 66, 8428–8440. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, Y.; Li, Y.; Ni, Q.; Li, J. Radiotherapy-triggered proteolysis targeting chimera prodrug activation in tumors. J. Am. Chem. Soc. 2023, 145, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.; Tang, W.; Hu, Q. Proteolysis-targeting chimera (PROTAC) delivery system: Advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022, 51, 5330–5350. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Wang, T.; Luo, M.; Chen, Y.; Chen, C.; Ronai, Z.; Zhou, Y.; Ruppin, E.; Han, L. Expanding PROTACtable genome universe of E3 ligases. Nat. Commun. 2023, 14, 6509. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degrader | Other Name | Treatment | Phase | Sponsor | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| ARV-110 | bavdegalutamide | Prostate cancer | I and II | Arvinas Inc. | NCT03888612 |
| ARV-766 | luxdegalutamide | Prostate cancer | I and II | Arvinas Inc. | NCT05067140 |
| CC-94676 | AR-LDD | Prostate cancer | I | Bristol Myers Squibb | NCT04428788 |
| HP-518 | – | Prostate cancer | I | Hinova Pharmaceuticals Inc. | NCT05252364 |
| AC176 | AC0176 | Prostate cancer | I | Accutar Biotechnology Inc. | NCT05241613 |
| GT-20029 | – | Acne vulgaris and androgenetic alopecia | I | Suzhou Kintor Pharmaceutical, Inc. | NCT05428449 |
| UT-34 | ONCT-534 GTx-534 | Prostate cancer | I & II | Oncternal Therapeutics, Inc. | NCT05917470 |
| ASC-J9 | – | Acne vulgaris | II | AndroScience Corp. | NCT00525499 |
| 17-AAG | – | Prostate cancer | II | National Cancer Institute | NCT00118092 |
| Galeterone | – | Prostate Cancer with AR-V7 | III | LTN Pharmaceuticals, Inc. | NCT02438007 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.-H.; Munoz, E.; Ashong, D. Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer. Cancers 2024, 16, 663. https://doi.org/10.3390/cancers16030663
Chen Q-H, Munoz E, Ashong D. Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer. Cancers. 2024; 16(3):663. https://doi.org/10.3390/cancers16030663
Chicago/Turabian StyleChen, Qiao-Hong, Erick Munoz, and Dennis Ashong. 2024. "Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer" Cancers 16, no. 3: 663. https://doi.org/10.3390/cancers16030663
APA StyleChen, Q.-H., Munoz, E., & Ashong, D. (2024). Insight into Recent Advances in Degrading Androgen Receptor for Castration-Resistant Prostate Cancer. Cancers, 16(3), 663. https://doi.org/10.3390/cancers16030663