
Poor Applicability of Currently Available Prognostic Scoring Systems for Prediction of Outcome in KIT D816V-Negative Advanced Systemic Mastocytosis

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
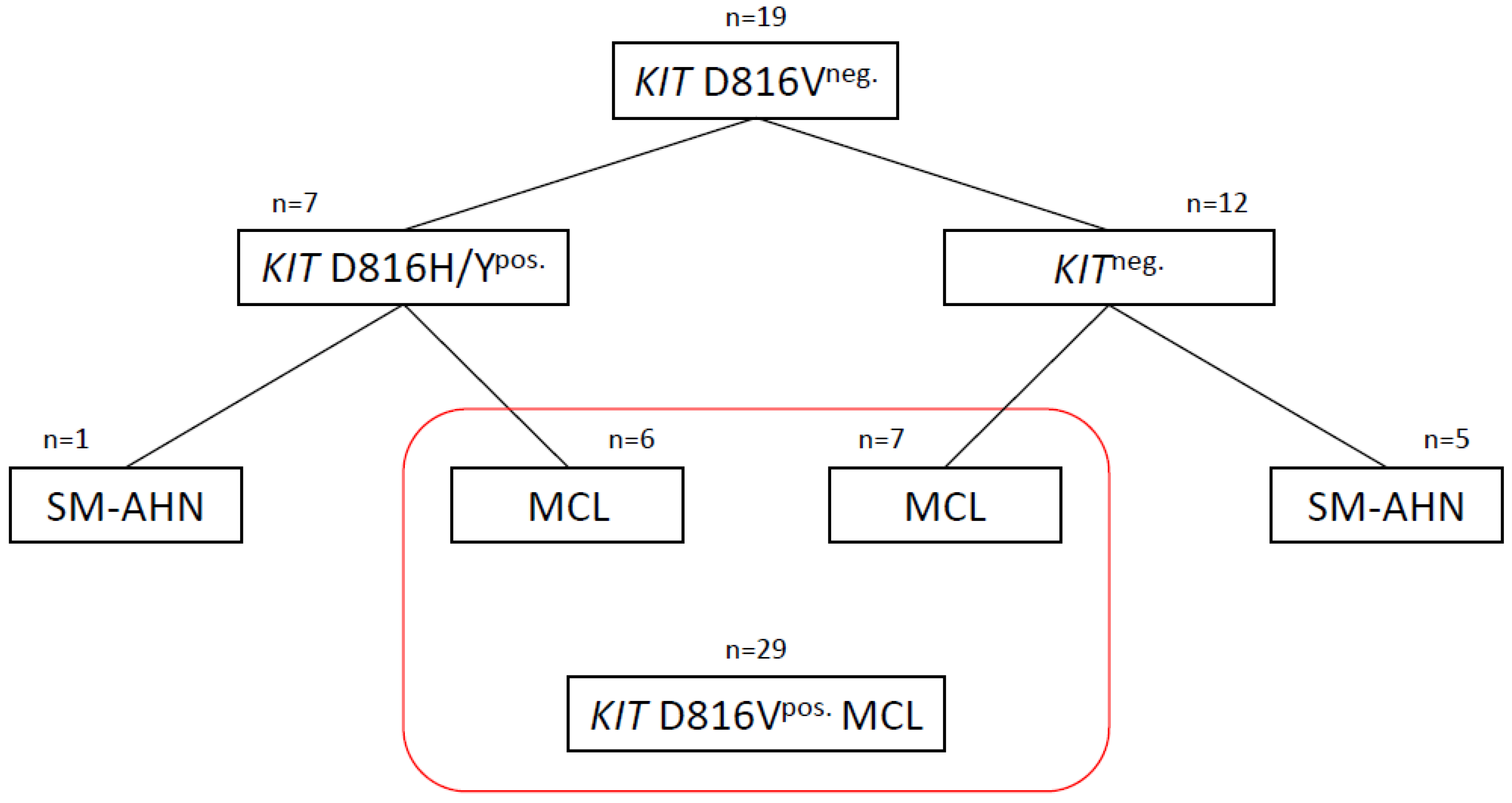
2. Patients and Methods
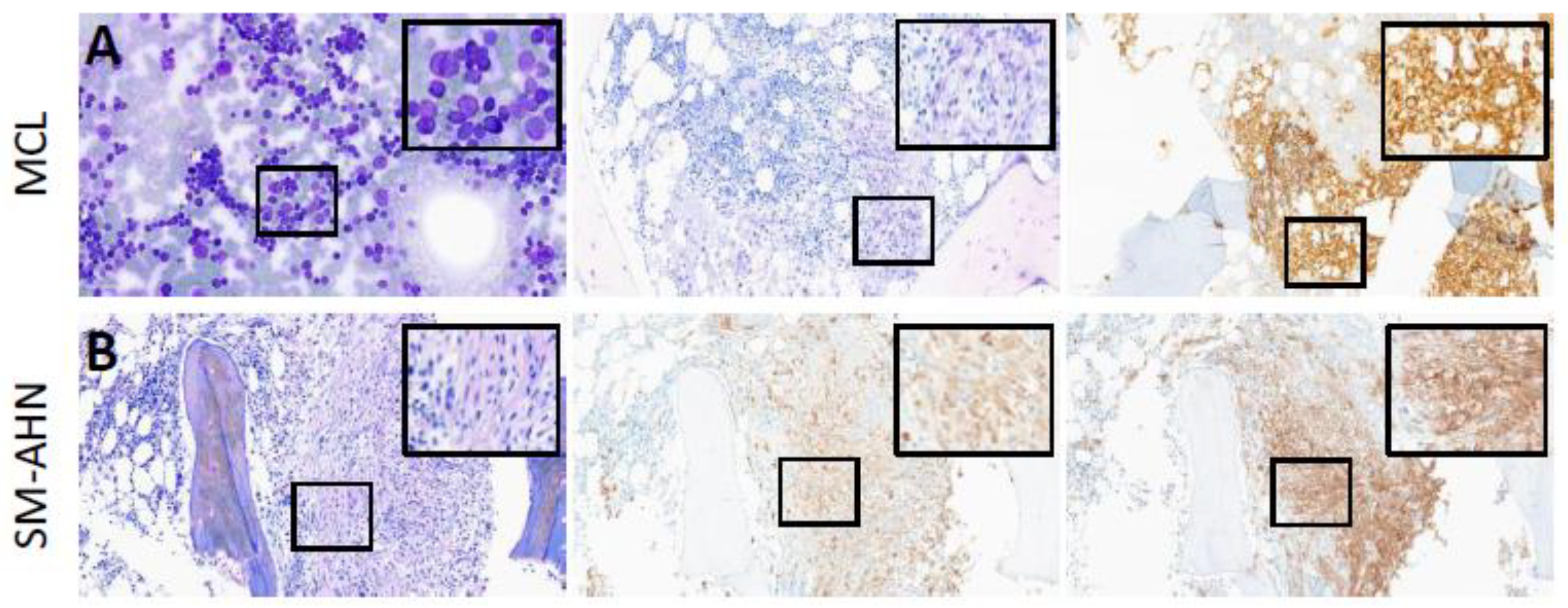
2.1. Cytomorphology and Histomorphology
2.2. Immunohistochemistry
2.3. Microdissection
2.4. Mutation Analysis
2.5. Comparison to Control Group
2.6. Statistical Analyses
3. Results
3.1. KIT D816H/Ypos. Patients
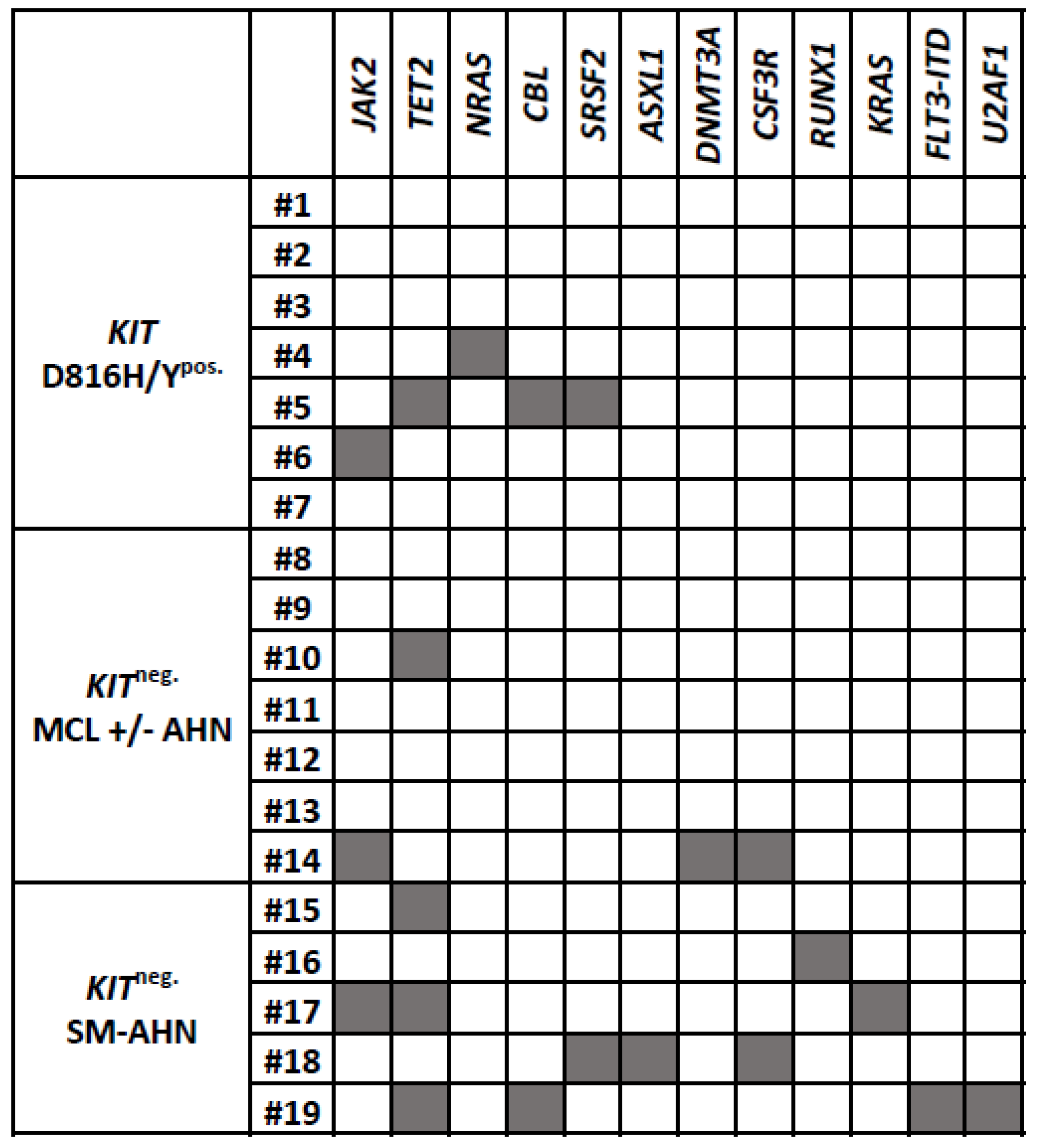
3.1.1. Molecular Findings
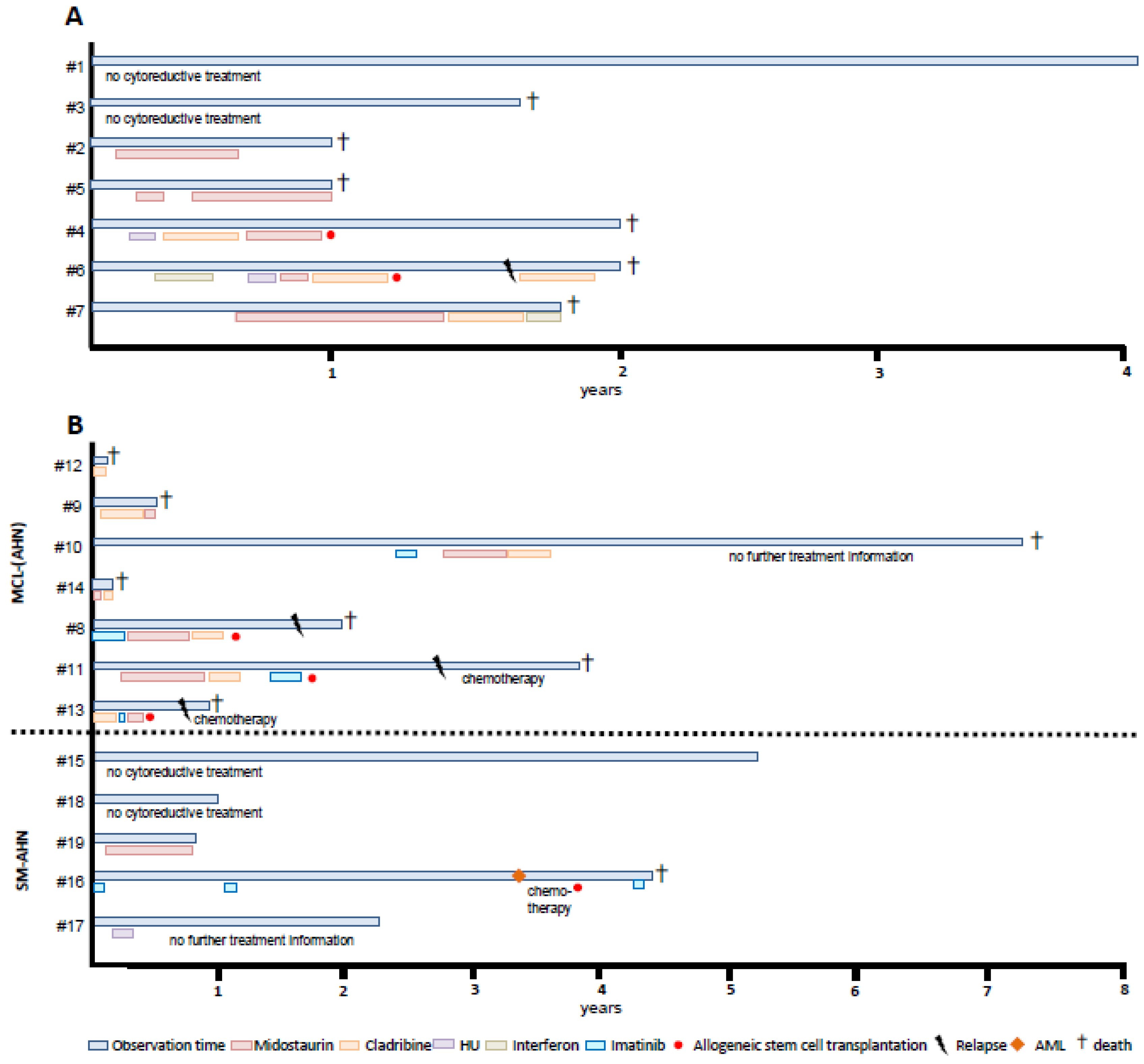
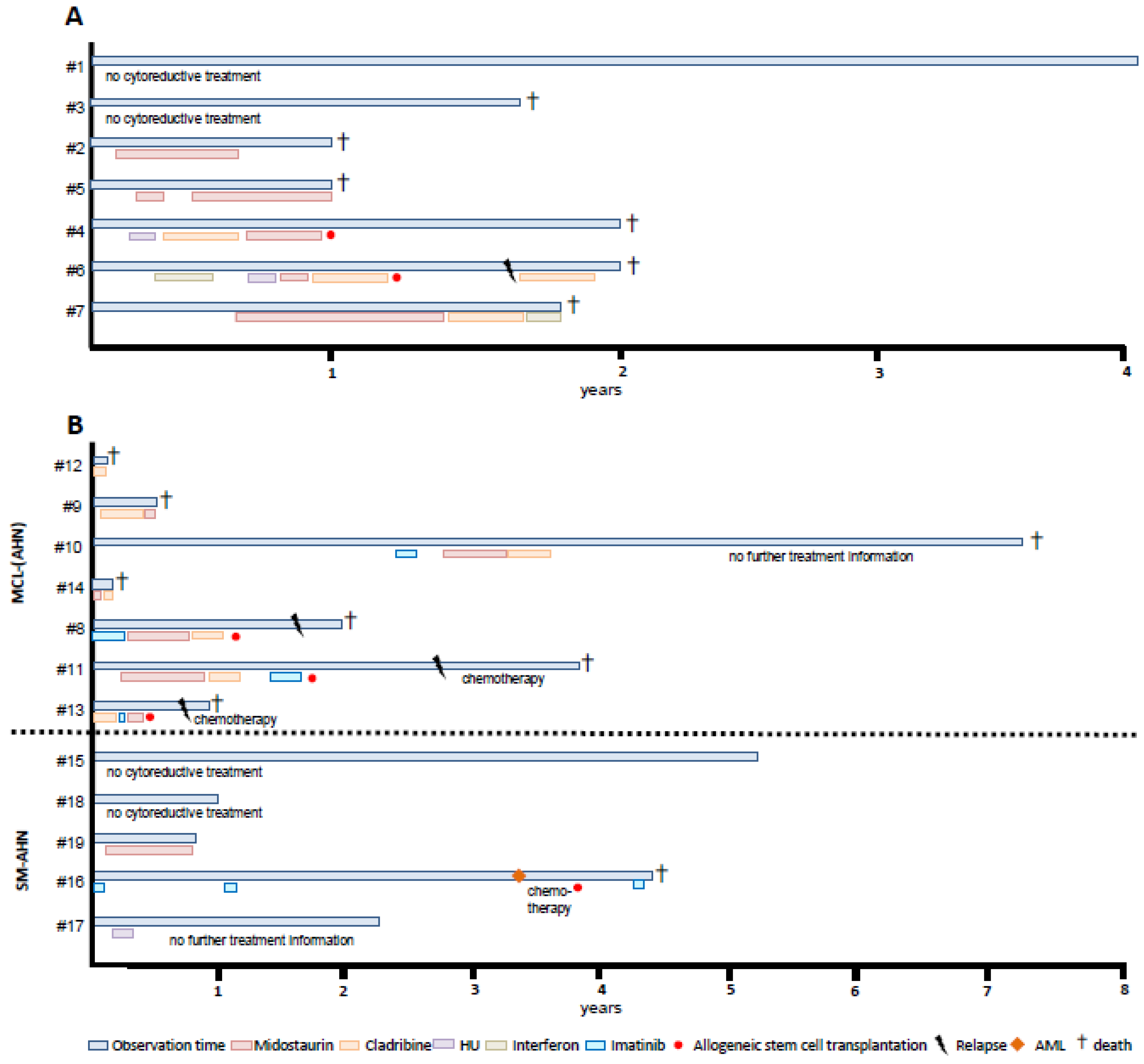
3.1.2. Treatment
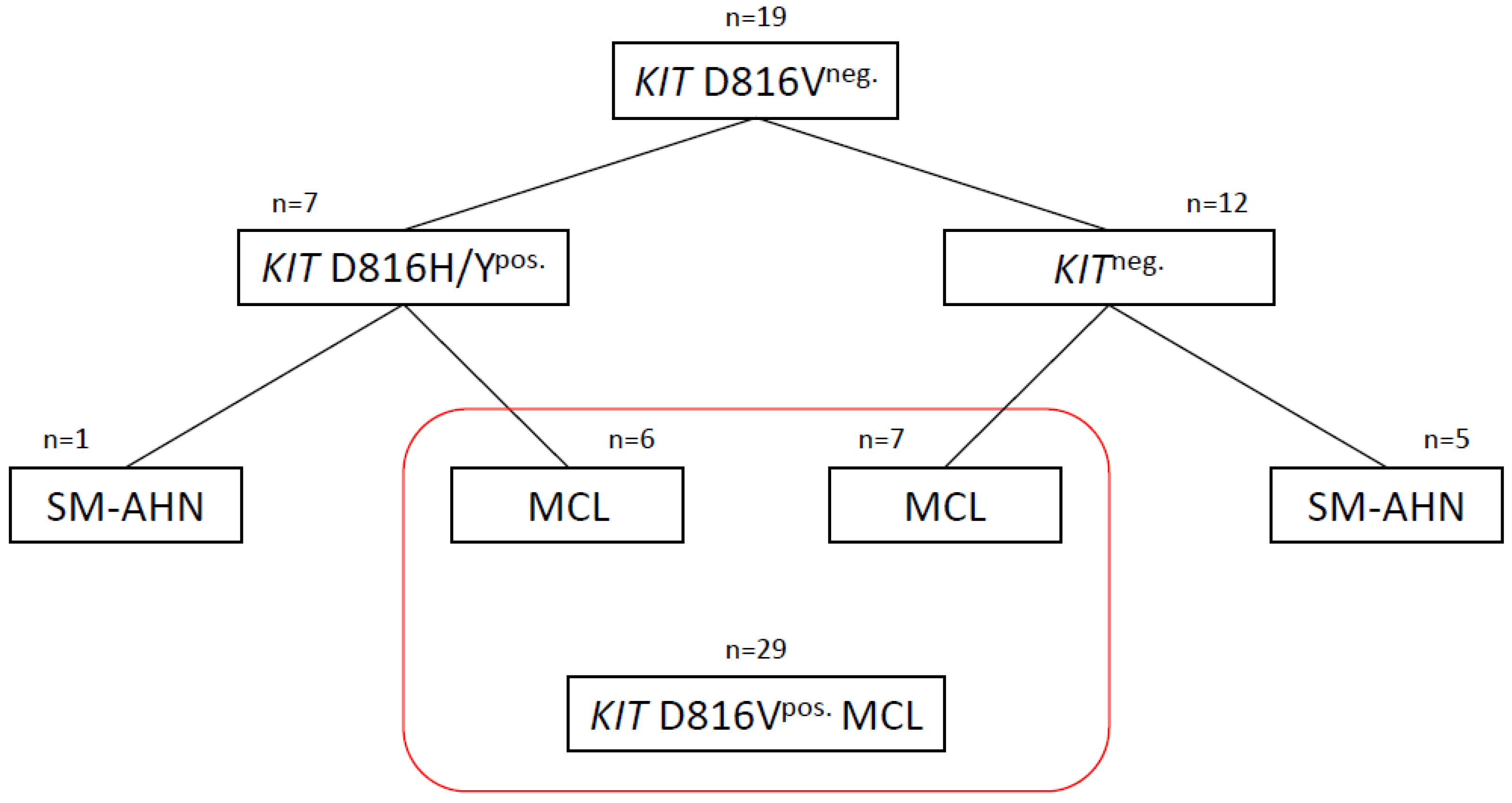
3.2. KITneg. Patients
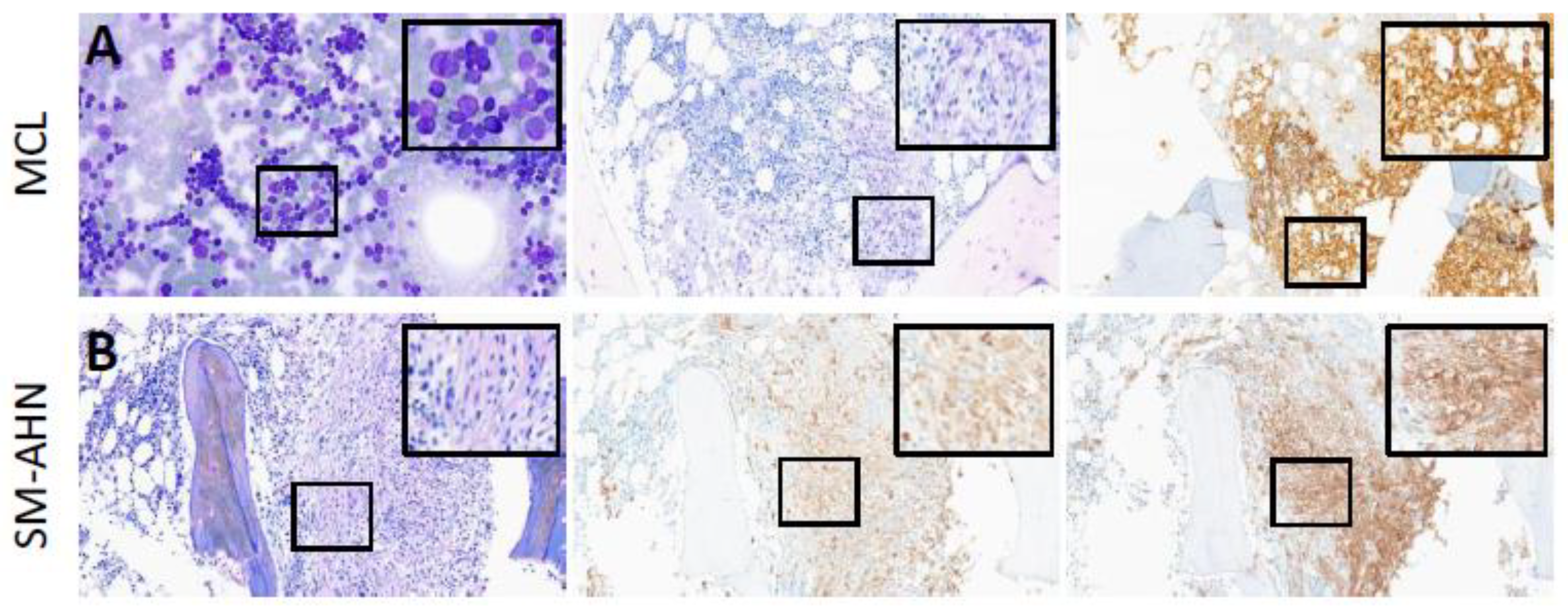
3.2.1. Cytomorphology and Histomorphology
3.2.2. Molecular Findings
3.2.3. Treatment
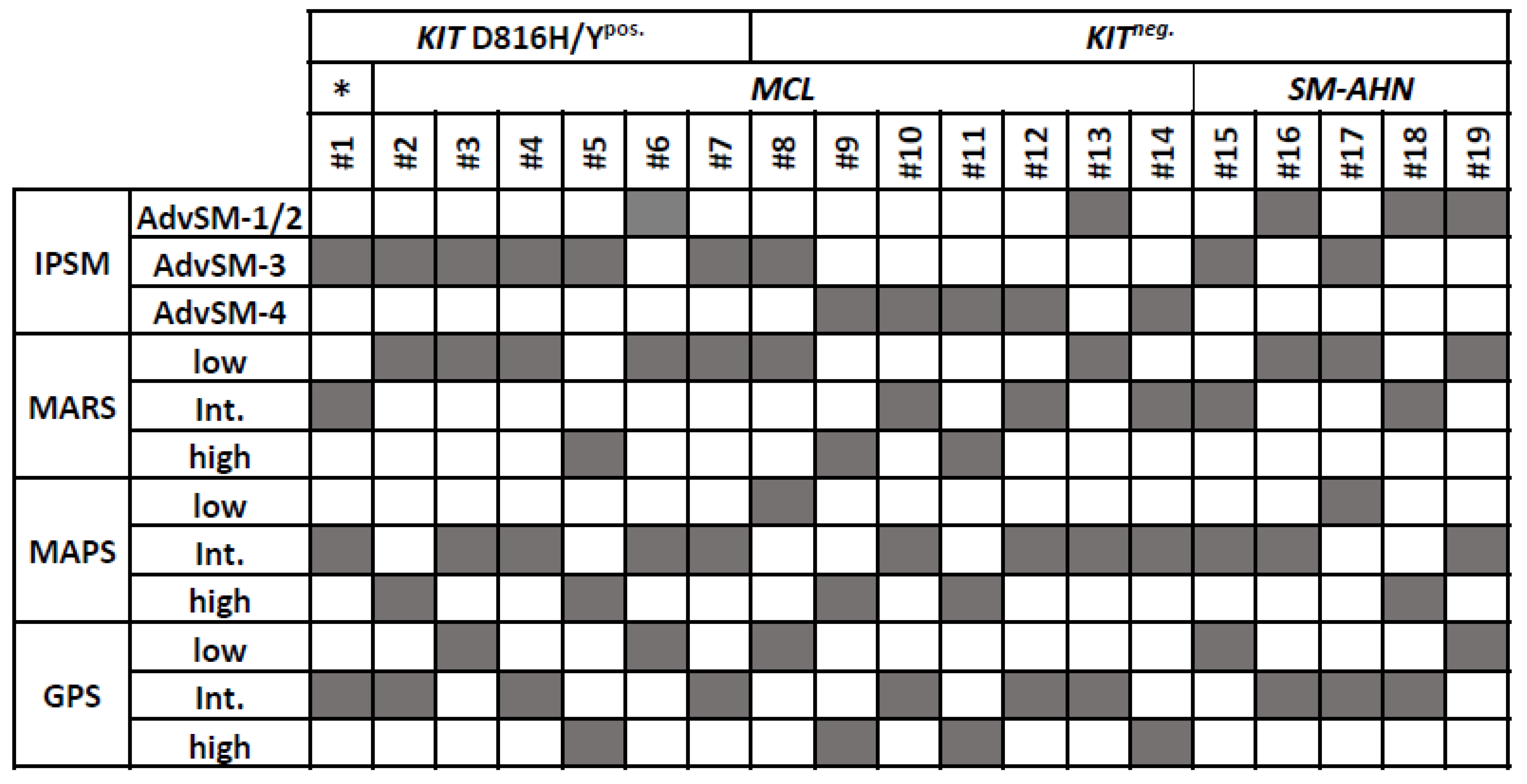
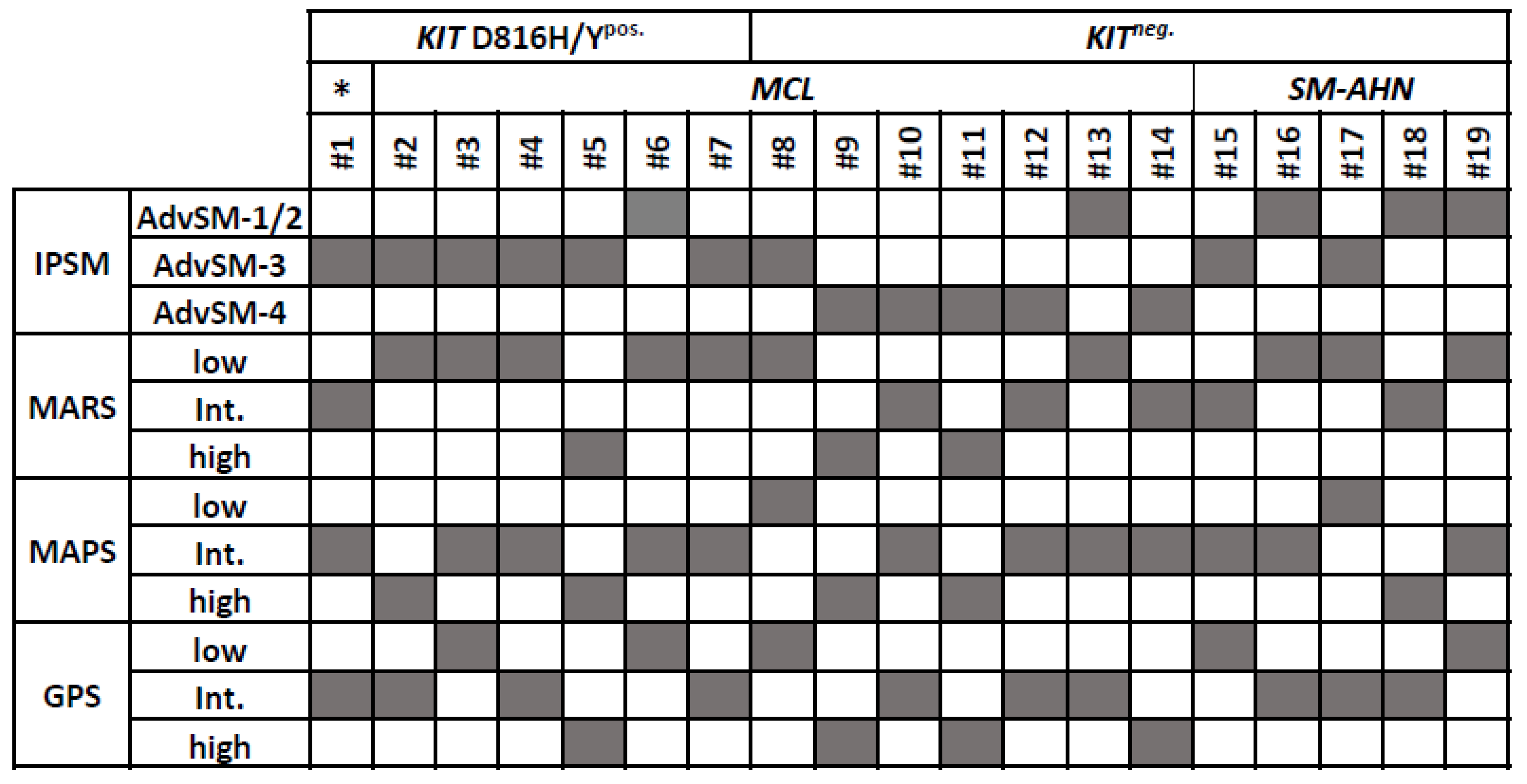
3.3. Commonalities and Differences between the Two Cohorts of KITneg. AdvSM
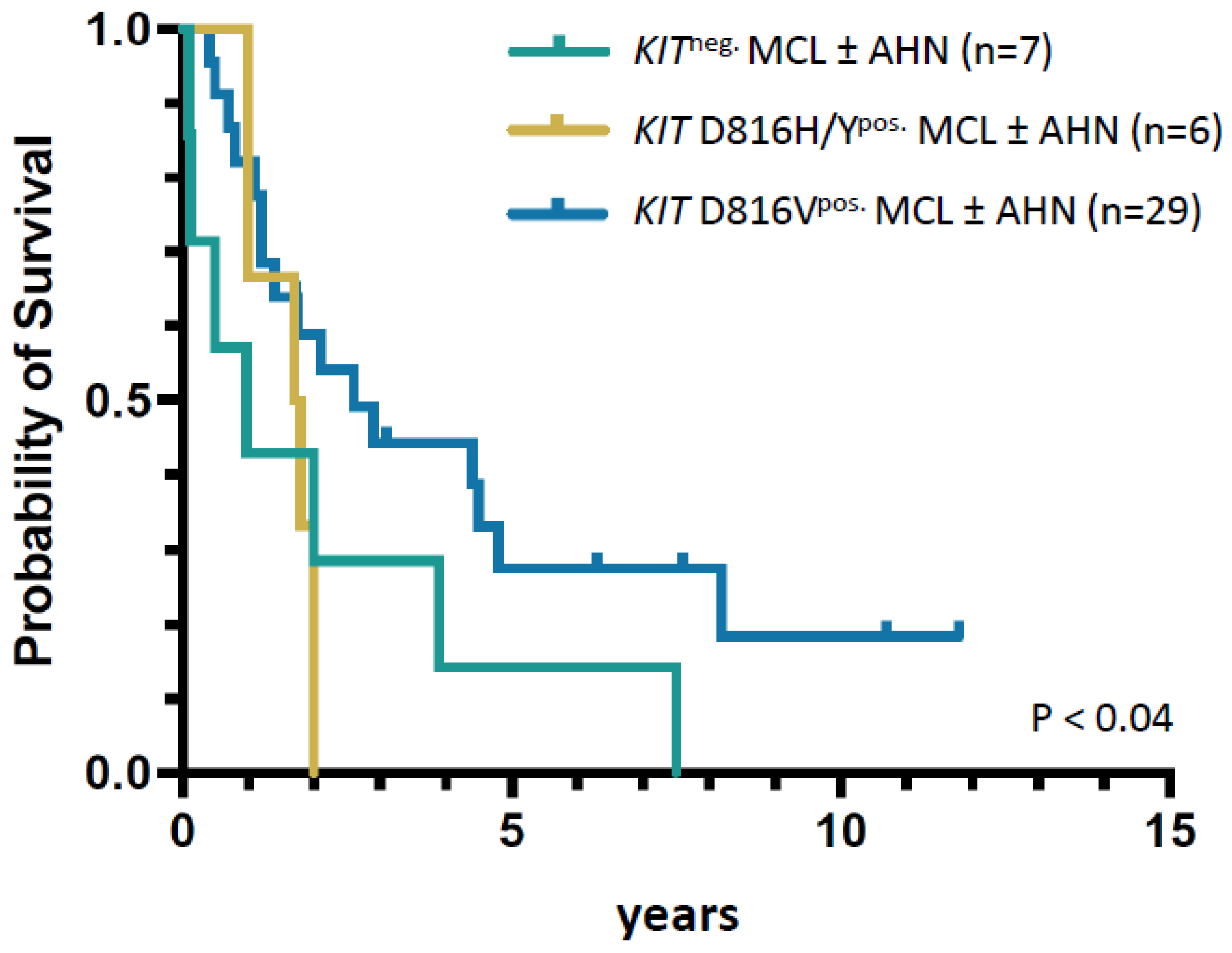
3.4. Analysis of Three MCL Cohorts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #Pat. | Diagnosis | Age (Years) | Sex | Date of Dx | KIT Status | Additional Mutations | Tryptase (µg/L) | BM Infiltration (%) | A/T | M/E | AP (U/L) | Alb (g/L) | Death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | SM-B-NHL | 66 | M | 2017 | D816H | None | 90 | 20 | +/− | −/− | 48 | nk | Alive |
| #2 | MCL | 61 | F | 2011 | D816Y | None | 1300 | 50 | −/− | −/+ | 162 | 36 | Death |
| #3 | MCL | 58 | M | 2013 | D816H | None | 193 | 50 | −/+ | −/− | 83 | 45 | Death |
| #4 | MCL-CEL | 45 | F | 2015 | D816H | NRAS | 157 | 75 | +/− | −/+ | 122 | 32 | Death |
| #5 | MCL-CMML | 69 | F | 2011 | D816Y | TET2, CBL, SRSF2 | 354 | 50 | +/− | +/− | 246 | 39 | Death |
| #6 | MCL-CEL | 51 | F | 2015 | D816H | JAK2 | 135 | 50 | −/− | −/+ | 117 | 35 | Death |
| #7 | MCL | 37 | M | 2018 | D816Y | None | 225 | 60 | +/− | −/− | 106 | 36 | Death |
| #8 | MCL-CLL | 54 | M | 2018 | Negative | None | 885 | 90 | −/− | −/− | 81 | 41 | Death |
| #9 | MCL | 71 | M | 2015 | Negative | None | 1150 | 50 | +/+ | −/− | 95 | 36 | Death |
| #10 | MCL-MM | 79 | M | 2013 | Negative | TET2 | 180 | 50 | −/+ | −/− | 59 | 39 | Death |
| #11 | MCL | 63 | M | 2014 | Negative | None | 451 | 80 | +/+ | −/− | 159 | 35 | Death |
| #12 | MCL | 28 | F | 2018 | Negative | None | 202 | 40 | +/+ | −/− | 131 | 20 | Death |
| #13 | MCL | 27 | M | 2021 | Negative | None | 113 | 80 | −/− | −/− | 437 | 33 | Death |
| #14 | MCL-MDS/MPN | 56 | F | 2021 | Negative | JAK2, DNMT3A, CSF3R | 631 | 90 | +/+ | −/+ | 210 | 31 | Death |
| #15 | SM-MDS | 64 | F | 2017 | Negative | TET2 | 90 | 10 | −/+ | −/− | 69 | 41 | Alive |
| #16 | SM-AML | 57 | F | 2013 | Negative | RUNX1 | 96 | 20 | −/− | −/− | 62 | 39 | Death |
| #17 | SM-MF | 59 | M | 2015 | Negative | JAK2, KRAS, TET2 | 34 | 20 | −/− | +/− | 377 | 43 | Alive |
| #18 | SM-MDS | 62 | M | 2022 | Negative | ASXL1, CSF3R, SRSF2 | 31 | 35 | −/− | −/− | 107 | 38 | Alive |
| #19 | SM-MDS/MPN | 82 | M | 2022 | Negative | FLT3-ITD, U2AF1, CBL, TET2 | 18 | 10 | −/− | +/− | 45 | 36 | Alive |
| #Patient | Gene | Reference Sequence | cDNA Variant | Protein Variant |
|---|---|---|---|---|
| #4 | NRAS | NM_002524.5 | codon 61 | codon 61 |
| #5 | TET2 | NM_001127208.3 | c.4210C>T | p.Arg1404* |
| CBL | NM_005188.4 | c.1169A>T | p.Asp390Val | |
| SRSF2 | NM_001195427.2 | c.284C>A | p.Pro95His | |
| #6 | JAK2 | NM_004972.4 | c.1849G>T | p.Val617Phe |
| #10 | TET2 | NM_001127208.3 | c.2681_2687del | p.Ser894Tyrfs*25 |
| #14 | JAK2 | NM_004972.4 | c.1849G>T | p.Val617Phe |
| DNMT3A | NM_022552.4 | c.2645G>A | p.Arg882His | |
| CSF3R | NM_000760.4 | not available | not available | |
| #15 | TET2 | NM_001127208.3 | c.1795C>T | p.Gln599* |
| TET2 | NM_001127208.3 | c.2428C>T | p.Gln810* | |
| #16 | RUNX1 | ENST00000344691 | c.877C>T | p.Arg293* |
| #17 | JAK2 | NM_004972.4 | c.1849G>T | p.Val617Phe |
| TET2 | NM_001127208.3 | c.4546C>T | p.Arg1516* | |
| TET2 | NM_001127208.3 | c.4621C>T | p.Gln1541* | |
| KRAS | NM_004985.5 | c.108A>G | p.Ile36Met | |
| #18 | SRSF2 | NM_001195427.2 | c.284C>T | p.Pro95Leu |
| ASXL1 | NM_015338.6 | c.3514del | p.Ala1172Leufs*2 | |
| CSF3R | NM_000760.4 | c.2221C>T | p.Gln741* | |
| #19 | TET2 | NM_001127208.3 | c.1526C>G | p.Ser509* |
| TET2 | NM_001127208.3 | c.5688G>T | p.Arg1896Ser | |
| CBL | NM_005188.4 | c.1259G>T | p.Arg420Leu | |
| U2AF1 | NM_006758.3 | c.101C>T | p.Ser34Phe |
References
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef]
- Bellomo, R.G. Stress, Depression, and Dementia Contribute to Neurodegeneration. Eur. J. Neurodegener. Dis. 2023, 12, 15–20. [Google Scholar]
- Pandolfi, F. The Impact of Mast Cells in Neuroimmunology and Cancer. Eur. J. Neurodegener. Dis. 2023, 12, 62–70. [Google Scholar]
- Munoz-Gonzalez, J.I.; Alvarez-Twose, I.; Jara-Acevedo, M.; Zanotti, R.; Perkins, C.; Jawhar, M.; Sperr, W.R.; Shoumariyeh, K.; Schwaab, J.; Greiner, G.; et al. Proposed global prognostic score for systemic mastocytosis: A retrospective prognostic modelling study. Lancet Haematol. 2021, 8, e194–e204. [Google Scholar] [CrossRef]
- Pardanani, A.; Shah, S.; Mannelli, F.; Elala, Y.C.; Guglielmelli, P.; Lasho, T.L.; Patnaik, M.M.; Gangat, N.; Ketterling, R.P.; Reichard, K.K.; et al. Mayo alliance prognostic system for mastocytosis: Clinical and hybrid clinical-molecular models. Blood Adv 2018, 2, 2964–2972. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Hausmann, D.; Clemens, J.; Naumann, N.; Henzler, T.; Horny, H.P.; Sotlar, K.; Schoenberg, S.O.; Cross, N.C.; et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia 2016, 30, 2342–2350. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Alvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lubke, J.; Perkins, C.; Munoz-Gonzalez, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846. [Google Scholar] [CrossRef]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Oude Elberink, J.N.G.; Gorska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef] [PubMed]
- Erben, P.; Schwaab, J.; Metzgeroth, G.; Horny, H.P.; Jawhar, M.; Sotlar, K.; Fabarius, A.; Teichmann, M.; Schneider, S.; Ernst, T.; et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann. Hematol. 2014, 93, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, T.; Vestergaard, H.; Møller, M.B. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J. Mol. Diagn. 2011, 13, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Orfao, A.; Garcia-Montero, A.C.; Sanchez, L.; Escribano, L. Recent advances in the understanding of mastocytosis: The role of KIT mutations. Br. J. Haematol 2007, 138, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef]
- Valent, P.; Sotlar, K.; Sperr, W.R.; Escribano, L.; Yavuz, S.; Reiter, A.; George, T.I.; Kluin-Nelemans, H.C.; Hermine, O.; Butterfield, J.H.; et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): A consensus proposal. Ann. Oncol. 2014, 25, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Reichard, K.K.; Hanson, C.A.; Tefferi, A. World Health Organization class-independent risk categorization in mastocytosis. Blood Cancer J. 2019, 9, 29. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Metzgeroth, G.; Fabarius, A.; Valent, P.; Hofmann, W.K.; et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood 2017, 130, 137–145. [Google Scholar] [CrossRef] [PubMed]
- González-López, O.; Muñoz-González, J.I.; Orfao, A.; Álvarez-Twose, I.; García-Montero, A.C. Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis. Cancers 2022, 14, 2487. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Meggendorfer, M.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Schmitt, K.; Fabarius, A.; Valent, P.; et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 2017, 102, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; Matito, A.; Morgado, J.M.; Sánchez-Muñoz, L.; Jara-Acevedo, M.; García-Montero, A.; Mayado, A.; Caldas, C.; Teodósio, C.; Muñoz-González, J.I.; et al. Imatinib in systemic mastocytosis: A phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget 2017, 8, 68950–68963. [Google Scholar] [CrossRef]
- Broderick, V.; Waghorn, K.; Langabeer, S.E.; Jeffers, M.; Cross, N.C.P.; Hayden, P.J. Molecular response to imatinib in KIT F522C-mutated systemic mastocytosis. Leuk. Res. 2019, 77, 28–29. [Google Scholar] [CrossRef]
- Chan, E.C.; Bai, Y.; Kirshenbaum, A.S.; Fischer, E.R.; Simakova, O.; Bandara, G.; Scott, L.M.; Wisch, L.B.; Cantave, D.; Carter, M.C.; et al. Mastocytosis associated with a rare germline KIT K509I mutation displays a well-differentiated mast cell phenotype. J. Allergy Clin. Immunol. 2014, 134, 178–187. [Google Scholar] [CrossRef]
- Lanternier, F.; Cohen-Akenine, A.; Palmerini, F.; Feger, F.; Yang, Y.; Zermati, Y.; Barète, S.; Sans, B.; Baude, C.; Ghez, D.; et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PLoS ONE 2008, 3, e1906. [Google Scholar] [CrossRef]
- Wang, H.J.; Lin, Z.M.; Zhang, J.; Yin, J.H.; Yang, Y. A new germline mutation in KIT associated with diffuse cutaneous mastocytosis in a Chinese family. Clin. Exp. Dermatol. 2014, 39, 146–149. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Smith, M.L.; Schultheis, B.; Fitzgibbon, J.; Lister, T.A.; Melo, J.V.; Cross, N.C.; Cavenagh, J.D. A novel K509I mutation of KIT identified in familial mastocytosis-in vitro and in vivo responsiveness to imatinib therapy. Leuk. Res. 2006, 30, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol. Allergy Clin. N. Am. 2014, 34, 239–262. [Google Scholar] [CrossRef]
- Pardanani, A.; Tefferi, A.; Singh, A.; Al-Kali, A.; Patnaik, M.M.; Hogan, W.J.; Begna, K.; Elliott, M.A.; Khera, N.; Palmer, J.M.; et al. Reappraisal of Mast Cell Leukemia Based on a Single Institution Review of 16 Cases: Role of Mast Cell Morphology in Determining Clinical Outcome. Blood 2022, 140, 6872–6873. [Google Scholar] [CrossRef]
- Pardanani, A.; Lim, K.H.; Lasho, T.L.; Finke, C.; McClure, R.F.; Li, C.Y.; Tefferi, A. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood 2009, 114, 3769–3772. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Rangit, V.; Lasho, T.L.; Hoversten, K.P.; Finke, C.M.; Ketterling, R.P.; Hanson, C.A.; Gangat, N.; Tefferi, A.; Pardanani, A. A comparison of clinical and molecular characteristics of patients with systemic mastocytosis with chronic myelomonocytic leukemia to CMML alone. Leukemia 2018, 32, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Naumann, N.; Lübke, J.; Shomali, W.; Reiter, L.; Horny, H.P.; Jawhar, M.; Dangelo, V.; Fabarius, A.; Metzgeroth, G.; Kreil, S.; et al. Clinical and histopathological features of myeloid neoplasms with concurrent Janus kinase 2 (JAK2) V617F and KIT proto-oncogene, receptor tyrosine kinase (KIT) D816V mutations. Br. J. Haematol. 2021, 194, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Perkins, C.; Reiter, A.; Jawhar, M.; Lübke, J.; Kluin-Nelemans, H.C.; Shomali, W.; Langford, C.; Abuel, J.; Hermine, O.; et al. Mast Cell Leukemia: Clinical and Molecular Features and Survival Outcomes of Patients in the ECNM Registry. Blood Adv. 2022, 7, 1713–1724. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Lhermitte, L.; Dubreuil, P.; Chandesris, M.-O.; Hermine, O.; Damaj, G. Mast cell leukemia. Blood 2013, 121, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Wang, S.; Patel, K.P.; Sarwari, N.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Mast cell leukemia (MCL): Clinico-pathologic and molecular features and survival outcome. Leuk. Res. 2017, 59, 105–109. [Google Scholar] [CrossRef] [PubMed]


| (a) | ||||||
| Variables | KIT D816H/Ypos. | KITneg. AdvSM | p-Value | KIT D816Vpos. AdvSM | p-Value D816Vpos. vs. D816H/Ypos. | p-Value D816Vpos. vs. KITneg. |
| Number of patients, n | 7 | 12 | - | 118 | - | - |
| Age in years, median (range) | 58 (37–69) | 60 (27–82) | n.s. | 68 (33–87) | 0.0047 | 0.0128 |
| Male, n (%) | 3 (43) | 8 (67) | n.s. | 83 (70) | n.s. | n.s. |
| Diagnosis | ||||||
| SM-AHN, n (%) | 1 (14) | 5 (41) | n.s. | 89 (75) | - | - |
| MCL ± AHN, n (%) | 6 (86) | 7 (58) | n.s. | 29 (25) | - | - |
| AHN-subtypes, n (%) | 4 (57) | 8 (67) | n.s. | 111 (94) | ||
| B-NHL, n (%) | 1 (25) | - | - | |||
| MDS/MPN, n (%) | - | 2 (25) | - | 28 | - | - |
| CLL, n (%) | - | 1 (13) | - | - | - | |
| MM, n (%) | - | 1 (13) | - | 1 (1) | - | - |
| CMML, n (%) | 1 (25) | - | - | 37 | - | - |
| MDS, n (%) | - | 2 (13) | - | 15 | - | - |
| AML, n (%) | - | 1 (25) | - | 5 | - | - |
| CEL, n (%) | 2 (50) | - | - | 10 | - | - |
| MF, n (%) | - | 1 (13) | - | 3 (3) | - | - |
| CNL, n (%) | - | - | - | 1 (1) | - | - |
| ET, n (%) | - | - | - | 2 (2) | - | - |
| PV, n (%) | - | - | - | 1 (1) | - | - |
| MPN, n (%) | - | - | - | 7 (6) | - | - |
| Lymphoma n (%) | - | - | - | 1 (1) | - | - |
| (b) | ||||||
| Variables | KIT D816H/Ypos. | KITneg. AdvSM | p-Value | KIT D816Vpos. AdvSM | p-Value D816Vpos. vs. D816H/Ypos. | p-Value D816Vpos. vs. KITneg. |
| Number of patients, n | 7 | 12 | - | 118 | - | - |
| C-findings | ||||||
| Platelets, ×109/L; median (range) | 149 (100–242) | 95 (24–247) | n.s. | 94 (5–800) | n.s. | n.s. |
| <100 × 109/L, n (%) | 0 (0) | 6 (50) | 0.0228 | 64 (54) | 0.005 | n.s. |
| Alkaline phosphatase, U/L; median (range) | 117 (48–246) | 101 (45–437) | n.s. | 198 (53–1730) | n.s. | n.s. |
| >150 U/L, n (%) | 2 (28) | 4 (33) | n.s. | 73 (62) | n.s. | n.s. |
| Albumin, g/L; median (range) | 36 (32–45) | 37 (20–43) | n.s. | 36 (19–48) | n.s. | n.s. |
| <34 g/L, n (%) | 1 (14) | 3 (25) | n.s. | 38 (32) | n.s. | n.s. |
| Ascites, n (%) | 5 (71) | 1 (8) | 0.0023 | 53 (45) | n.s. | 0.0141 |
| B-findings | ||||||
| MC-infiltration in BM histology, %; median (range) | 50 (20–75) | 45 (10–90) | n.s. | 30 (5–100) | n.s. | n.s. |
| Serum tryptase, µg/L; median (range) | 193 (90–1300) | 147 (18–1150) | n.s. | 179 (5–1675) | n.s. | n.s. |
| Splenomegaly, n (%) | 6 (86) | 7 (58) | n.s. | 99 (84) | n.s. | n.s. |
| Hepatomegaly, n (%) | 2 (28) | 4 (33) | n.s. | 57 (48) | n.s. | n.s. |
| Lymphadenopathy, n (%) | 3 (43) | 5 (41) | n.s. | 62 (53) | n.s. | n.s. |
| Additional SM and/or AHN relevant findings | ||||||
| Leukocytes, ×109/L; median (range) | 8.9 (1.6–13.5) | 5 (2–91) | n.s. | 11 (1–130) | n.s. | n.s. |
| Monocytes, %; median (range) | 5 (1–15) | 6 (0–26) | n.s. | 8 (0–42) | n.s. | n.s. |
| Eosinophils, %, median (range) | 11 (0–33) | 1 (0–28) | n.s. | 4 (0–81) | n.s. | n.s. |
| Vitamin B12, ng/L; median (range) | 322 (129–1526) | 406 (303–6000) | n.s. | 1575 (144–6000) | 0.0154 | n.s. |
| >180 ng/L, n (%) | 6 (86) | 8 (100) | n.s. | 94 (100) | n.s. | n.s. |
| S/A/R/D/N mutation(s), n (%) | 2 (29) | 4 (33) | n.s. | 80 (68) | 0.0037 | 0.0042 |
| Other additional mutations, n (%) | 4 (57) | 6 (50) | n.s. | 87 (74) | n.s. | n.s. |
| Anaphylaxis, n (%) | 1 (14) | 1 (8) | n.s. | n.k. | - | - |
| Treatment | ||||||
| Midostaurin, n (%) | 5 (71) | 7 (58) | n.s. | 72 (61) | n.s. | n.s. |
| Cladribine, n (%) | 3 (43) | 6 (50) | n.s. | 47 (40) | n.s. | n.s. |
| Imatinib, n (%) | 0 (0) | 4 (33) | n.s. | 2 (2) | n.s. | <0.0001 |
| Outcome | ||||||
| Follow-up, years, median (range) | 1.8 (1–2) | 1.5 (0.1–7.5) | n.s. | 1.8 (0.04–11.82) | n.s. | n.s. |
| Death, n (%) | 6 (86) | 8 (67) | n.s. | 85 (72) | n.s. | n.s. |
| Overall survival, median, years | 1.8 | 3.9 | n.s. | 2.4 | n.s. | n.s. |
| Variables | KIT D816H/Ypos. MCL | KITneg. MCL | p-Value | KIT D816Vpos. MCL | p-Value D816Vpos. vs. D816H/Ypos. | p-Value D816Vpos. vs. KITneg. |
|---|---|---|---|---|---|---|
| Number of patients, n | 6 | 7 | - | 29 | - | - |
| Age in years, median (range) | 55 (37–69) | 56 (27–78) | n.s. | 68 (45–86) | 0.0104 | 0.0258 |
| Male, n (%) | 2 (33) | 5 (71) | n.s. | 20 (69) | n.s. | n.s. |
| AHN-subtypes, n (%) | 3 (50) | 3 (43) | n.s. | 22 (76) | n.s. | n.s. |
| MDS/MPN, n (%) | 0 (0) | 1 (33) | - | 9 (41) | - | - |
| CLL, n (%) | 0 (0) | 1 (33) | - | 0 (0) | - | - |
| MM, n (%) | 0 (0) | 1 (33) | - | 0 (0) | - | - |
| CMML, n (%) | 1 (33) | 0 (0) | - | 6 (27) | - | - |
| MDS, n (%) | 0 (0) | 0 (0) | - | 5 (23) | - | - |
| AML, n (%) | 0 (0) | 0 (0) | - | 1 (5) | - | - |
| CEL, n (%) | 2 (66) | 0 (0) | - | 1 (5) | - | - |
| C-findings | ||||||
| Platelets, ×109/L; median (range) | 145 (100–242) | 59 (24–238) | n.s. | 93 (18–795) | n.s. | n.s. |
| <100 × 109/L, n (%) | 0 (0) | 5 (71) | 0.0045 | 19 (66) | 0.0025 | n.s. |
| Alkaline phosphatase, U/L; median (range) | 120 (83–246) | 131 (59–437) | n.s. | 214 (62–548) | n.s. | n.s. |
| >150 U/L, n (%) | 2 (33) | 3 (43) | n.s. | 21 (72) | n.s. | n.s. |
| Albumin, g/L; median (range) | 36 (32–45) | 35 (20–41) | n.s. | 34.4 (23.4–46) | n.s. | n.s. |
| <34 g/L, n (%) | 1 (17) | 3 (43) | n.s. | 11 (39) | n.s. | n.s. |
| Ascites, n (%) | 5 (83) | 1 (14) | 0.009 | 16 (57) | n.s. | n.s. |
| B-findings | ||||||
| MC-infiltration in BM histology, %; median (range) | 50 (50–75) | 80 (40–90) | n.s. | 50 (20–100) | n.s. | n.s. |
| Serum tryptase, µg/L; median (range) | 209 (135–1300) | 451 (113–1150) | n.s. | 371 (41–1675) | n.s. | n.s. |
| Splenomegaly, n (%) | 6 (100) | 4 (57) | n.s. | 25 (93) | n.s. | n.s. |
| Hepatomegaly, n (%) | 2 (33) | 2 (29) | n.s. | 19 (70) | n.s. | n.s. |
| Lymphadenopathy, n (%) | 3 (50) | 3 (43) | n.s. | 20 (74) | n.s. | n.s. |
| Additional SM and/or AHN relevant findings | ||||||
| Leukocytes, ×109/L; median (range) | 9 (3.6–13.5) | 6 (2–32) | n.s. | 6.4 (1.3–66.1) | n.s. | n.s. |
| Monocytes, %; median (range) | 4.8 (1.3–15) | 5.4 (1–26) | n.s. | 7.8 (1–42) | n.s. | n.s. |
| Eosinophils, %, median (range) | 19.6 (1–33) | 1 (0–28) | n.s. | 2 (0–38) | 0.0164 | n.s. |
| Vitamin B12, ng/L; median (range) | 300 (129–1526) | 391 (303–6000) | n.s. | 1597 (288–6000) | 0.0379 | n.s. |
| >180 ng/L, n (%) | 5 (83) | 7 (100) | n.s. | 17 (100) | n.s. | n.s. |
| S/A/R/D/N mutation(s), n (%) | 2 (33) | 2 (29) | n.s. | 19 (66) | 0.0278 | 0.0135 |
| Other additional mutations, n (%) | 4 (67) | 2 (29) | n.s. | 15 (52) | n.s. | n.s. |
| Anaphylaxis, n (%) | 1 (17) | 1 (14) | n.s. | - | - | - |
| Treatment | ||||||
| Midostaurin, n (%) | 5 (83) | 6 (86) | - | 26 (90) | - | - |
| Cladribine, n (%) | 3 (50) | 6 (86) | - | 13 (45) | - | - |
| Imatinib, n (%) | 0 (0) | 3 (43) | - | 0 (0) | - | - |
| Follow-up, years, median (range) | 1.8 (1–2) | 0.9 (0.1–7.5) | n.s. | 3.7 (0.04–11.8) | n.s. | n.s. |
| Death, n (%) | 6 (100) | 7 (100) | n.s. | 18 (62) | n.s. | n.s. |
| Overall survival, median, years | 1.7 | 0.9 | n.s. | 2.6 | 0.043 | 0.046 |
| #pat. | Age | Sex | Diagnosis | SM-Subtype | CD25 | CD2 | CD30 | Differentiation | Tryptase [ng/mL] |
|---|---|---|---|---|---|---|---|---|---|
| 8 | 54 | M | SM-AHN | MCL | - | + | + | Well-differentiated | 885 |
| 9 | 71 | M | SM | MCL | - | + | + | Immature | 1150 |
| 10 | 79 | M | SM-AHN | MCL | - | + | + | Well-differentiated | 180 |
| 11 | 63 | M | SM | MCL | + | + | - | Immature | 451 |
| 12 | 28 | F | SM | MCL | + | n.d. | n.d. | Immature | 202 |
| 13 | 27 | M | SM | MCL | + | + | + | Immature | 113 |
| 14 | 56 | F | SM-AHN | MCL | n.k. | n.k. | n.k. | n.k. | 631 |
| 15 | 64 | F | SM-AHN | ISM | - | - | - | Well-differentiated | 90 |
| 16 | 57 | F | SM-AHN | ISM | + | - | - | Immature | 96 |
| 17 | 59 | M | SM-AHN | ISM | + | + | + | Immature | 34 |
| 18 | 62 | M | SM-AHN | ISM | + | - | - | Immature | 31 |
| 19 | 82 | M | SM-AHN | ISM | + | - | - | Immature | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumann, N.; Rudelius, M.; Lübke, J.; Christen, D.; Bresser, J.; Sotlar, K.; Metzgeroth, G.; Fabarius, A.; Hofmann, W.-K.; Panse, J.; et al. Poor Applicability of Currently Available Prognostic Scoring Systems for Prediction of Outcome in KIT D816V-Negative Advanced Systemic Mastocytosis. Cancers 2024, 16, 593. https://doi.org/10.3390/cancers16030593
Naumann N, Rudelius M, Lübke J, Christen D, Bresser J, Sotlar K, Metzgeroth G, Fabarius A, Hofmann W-K, Panse J, et al. Poor Applicability of Currently Available Prognostic Scoring Systems for Prediction of Outcome in KIT D816V-Negative Advanced Systemic Mastocytosis. Cancers. 2024; 16(3):593. https://doi.org/10.3390/cancers16030593
Chicago/Turabian StyleNaumann, Nicole, Martina Rudelius, Johannes Lübke, Deborah Christen, Jakob Bresser, Karl Sotlar, Georgia Metzgeroth, Alice Fabarius, Wolf-Karsten Hofmann, Jens Panse, and et al. 2024. "Poor Applicability of Currently Available Prognostic Scoring Systems for Prediction of Outcome in KIT D816V-Negative Advanced Systemic Mastocytosis" Cancers 16, no. 3: 593. https://doi.org/10.3390/cancers16030593
APA StyleNaumann, N., Rudelius, M., Lübke, J., Christen, D., Bresser, J., Sotlar, K., Metzgeroth, G., Fabarius, A., Hofmann, W.-K., Panse, J., Horny, H.-P., Cross, N. C. P., Reiter, A., & Schwaab, J. (2024). Poor Applicability of Currently Available Prognostic Scoring Systems for Prediction of Outcome in KIT D816V-Negative Advanced Systemic Mastocytosis. Cancers, 16(3), 593. https://doi.org/10.3390/cancers16030593