
Integrated Proteogenomic Analysis Reveals Distinct Potentially Actionable Therapeutic Vulnerabilities in Triple-Negative Breast Cancer Subtypes

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Data Retrieval and Ethical Approval
2.2. Bioinformatic Analysis Pipeline for Multi-Omics Data
2.3. Validation of the Proteogenomic Workflow
2.4. Statistical Analysis
3. Results
3.1. Proteomic Phenotypes and Protein–Protein Interactions in TNBC Subtypes
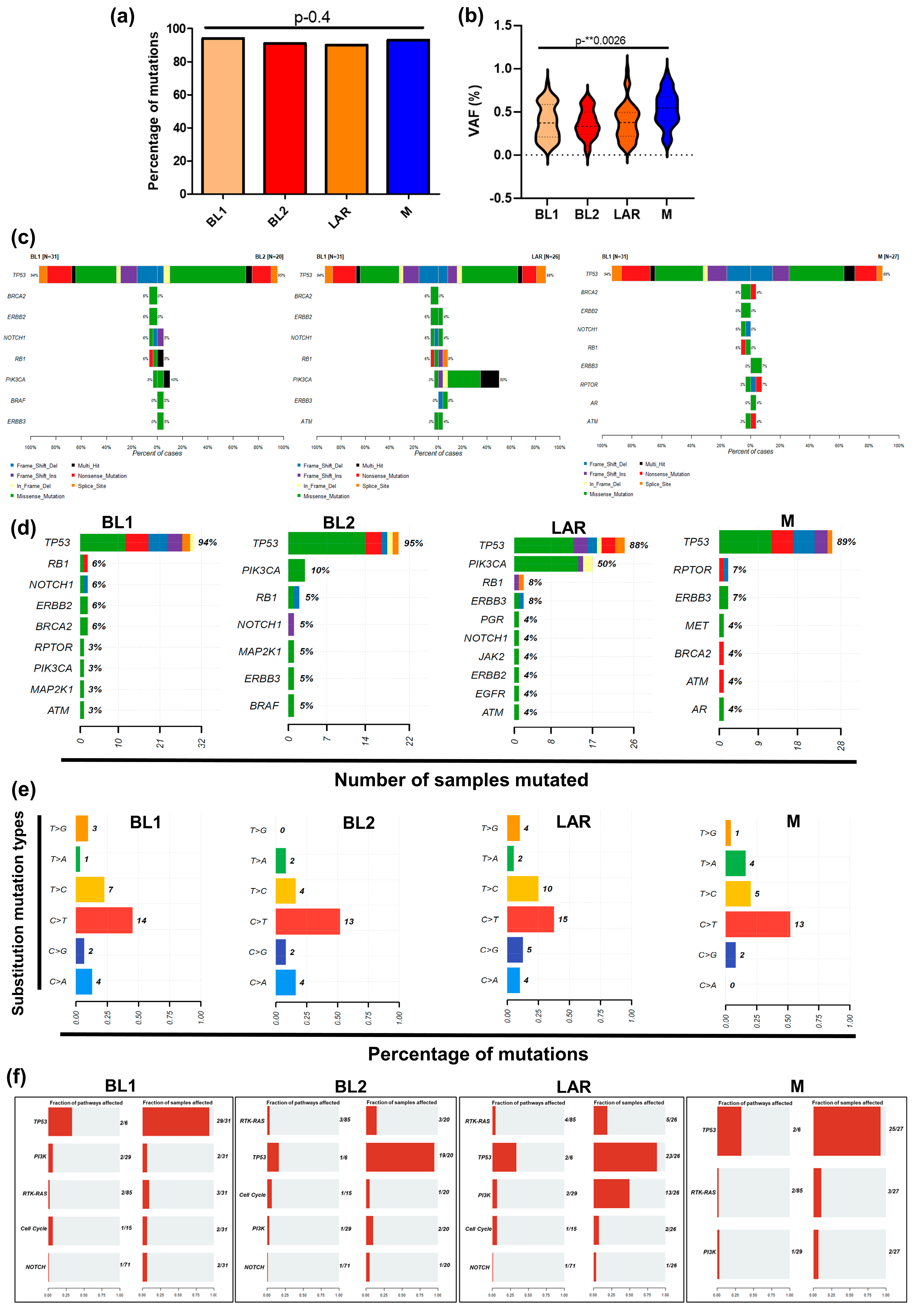
3.2. TP53 and PIK3CA Driver Genomic Alterations
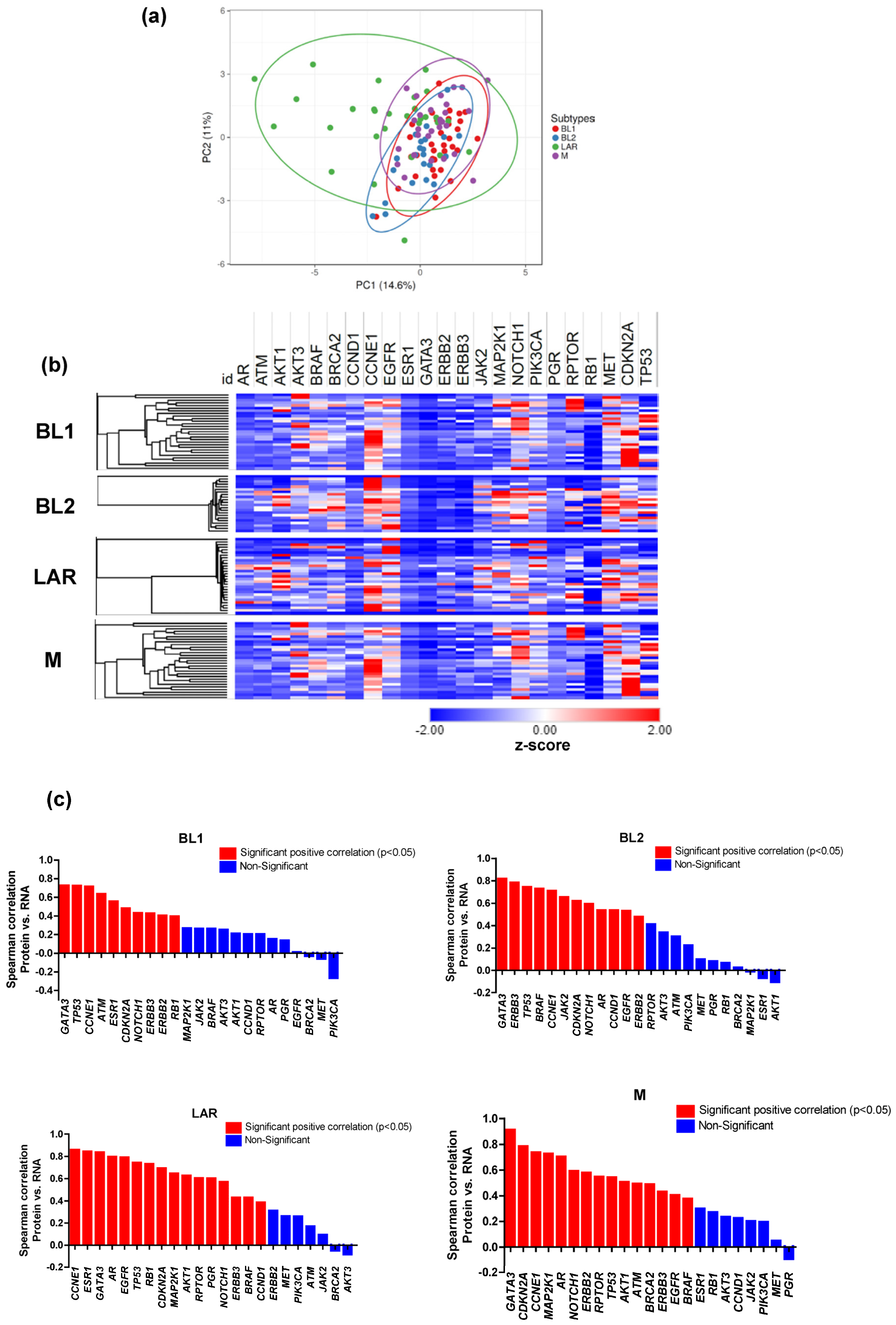
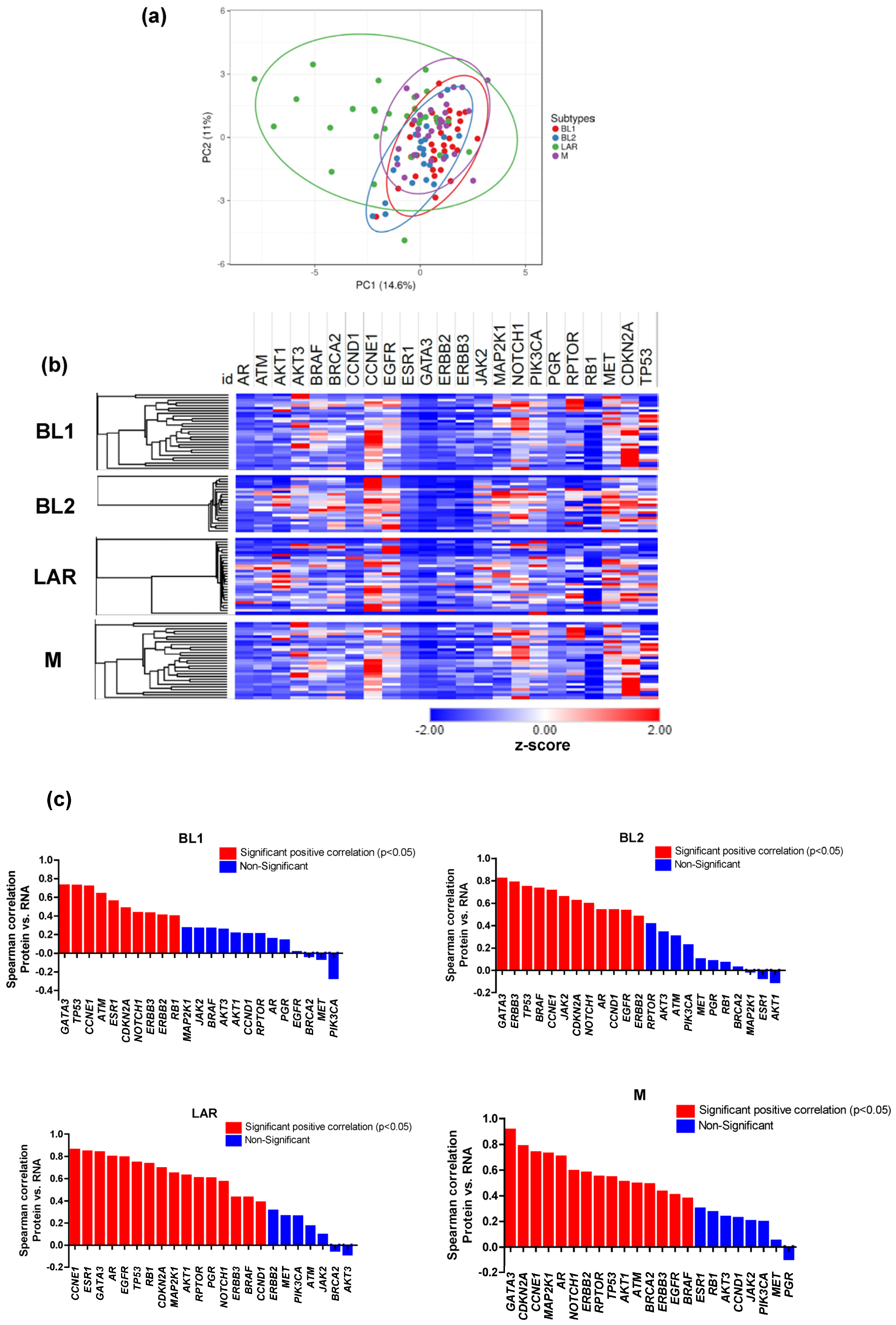
3.3. Proteomic and Transcriptomic Profile of TNBC Subtypes
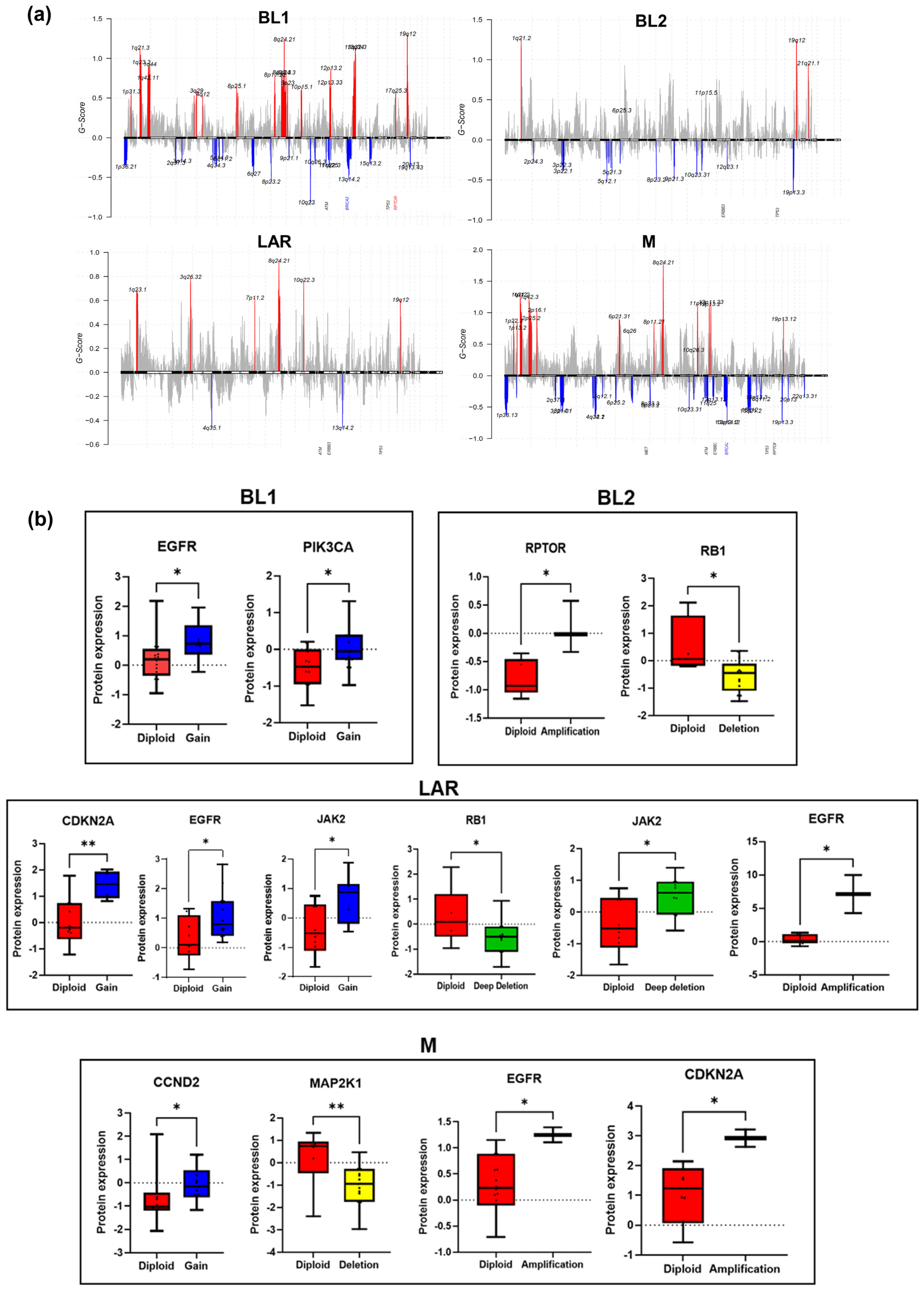
3.4. Effects of CNAs on Transcriptome and Proteome
3.5. Molecular Mechanisms Underlying Drug Sensitivity/Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Horak, C.E.; Pusztai, L.; Xing, G.; Trifan, O.C.; Saura, C.; Tseng, L.M.; Chan, S.; Welcher, R.; Liu, D. Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin. Cancer Res. 2013, 19, 1587–1595. [Google Scholar] [CrossRef]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Usmani, S.S.; Subbarayan, R.; Saini, R.; Pandey, P.K. Hypoxia: Syndicating triple negative breast cancer against various therapeutic regimens. Front. Oncol. 2023, 13, 1199105. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhao, Y.; Lee, W.C.; Ong, L.-T.; Lee, P.L.; Jiang, Z.; Oguz, G.; Niu, Z.; Liu, M.; Goh, J.Y.; et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 2022, 13, 4118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, C.; Liu, C.; Li, H.; Wu, J.; Sun, C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022, 156, 113861. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.C.; Lee, D.Y.; Lee, B.; Li, H.; Lim, J.C.T.; Lim, J.X.; Yeong, J.P.S.; Lau, H.Y.; Thike, A.A.; Tan, P.H.; et al. Hypoxia-regulated carbonic anhydrase IX (CAIX) protein is an independent prognostic indicator in triple negative breast cancer. Breast Cancer Res. 2022, 24, 38. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Porras, T.B.; Colombo, A.; Ring, A.; Lu, J.; Kang, I.; Lang, J.E. Identification of putative actionable alterations in clinically relevant genes in breast cancer. Br. J. Cancer 2021, 125, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Porras, T.B.; Ring, A.; Carpten, J.D.; Lang, J.E. Comparison of TCGA and GENIE genomic datasets for the detection of clinically actionable alterations in breast cancer. Sci. Rep. 2019, 9, 1482. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Mayakonda, A.; Koeffler, H.P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv 2016. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; van Oudenaarden, A.; Teichmann, S.A. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BL1 (n = 33), n (%) | BL2 (n = 22), n (%) | LAR (n = 29), n (%) | M (n = 29), n (%) | p-Value | |
|---|---|---|---|---|---|
| Age | 0.92 | ||||
| Mean + SD | 11.0 + 5.1 | 7.3 + 4.3 | 9.6 + 3.4 | 9.4 + 2.6 | |
| 26–40 | 4 (12.1%) | 0 (0.0%) | 4 (13.8%) | 5 (17.2%) | |
| 41–60 | 21 (63.6%) | 15 (68.2%) | 16 (55.2%) | 14 (48.3%) | |
| 61–90 | 8 (24.2%) | 7 (31.8%) | 9 (31%) | 10 (34.5%) | |
| Ethnicity | 0.98 | ||||
| Hispanic or Latino | 0 (0.0%) | 3 (13.6%) | 1 (3.4%) | 1 (3.4%) | |
| Not Hispanic or Latino | 29 (87.9%) | 19 (86.4%) | 25 (86.2%) | 25 (86.2%) | |
| NA | 3 (9.1%) | 0 (0.0%) | 3 (10.3%) | 3 (10.3%) | |
| NE | 1 (3.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Race | 0.97 | ||||
| White | 19 (57.6%) | 15 (68.2%) | 21 (72.4%) | 19 (65.5%) | |
| Black or African American | 8 (24.2%) | 1 (4.5%) | 5 (17.2%) | 8 (27.6%) | |
| American Indian or Alsaka Native | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Asian | 4 (12.1%) | 6 (27.3%) | 3 (10.3%) | 2 (6.9%) | |
| NA | 1 (3.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| NE | 1 (3.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Tumor Stage | 0.99 | ||||
| Early (Stage I–III) | 32 (97.0%) | 21 (95.5%) | 28 (96.6%) | 28 (96.6%) | |
| Metastatic | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (3.4%) | |
| NA | 1 (3.0%) | 1 (4.5%) | 1 (3.4%) | 0 (0.0%) | |
| Neoadjuvant treatment | 0.99 | ||||
| Yes | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| No | 33 (100%) | 22 (100%) | 29 (100%) | 29 (100%) | |
| NA | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| TNM Stage | |||||
| T Stage | 0.97 | ||||
| T1 | 4 (12.1%) | 4 (18.2%) | 7 (24.1%) | 6 (20.7%) | |
| T2 | 26 (78.8%) | 14 (63.6%) | 18 (62.1%) | 18 (62.1%) | |
| T3 | 2 (6.1%) | 2 (9.1%) | 2 (6.9%) | 5 (17.2%) | |
| T4 | 1 (3.0%) | 2 (9.1%) | 2 (6.9%) | 0 (0.0%) | |
| TX | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| N Stage | 0.99 | ||||
| N0 | 23 (69.7%) | 15 (68.2%) | 14 (48.3%) | 20 (69.0%) | |
| N1 | 9 (27.3%) | 3 (13.6%) | 8 (27.6%) | 6 (20.7%) | |
| N2 | 0 (0.0%) | 3 (13.6%) | 4 (13.8%) | 2 (6.9%) | |
| N3 | 1 (3.0%) | 1 (4.5%) | 3 (10.3%) | 1 (3.4%) | |
| NX | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| M Stage | 0.96 | ||||
| M0 | 31 (93.9%) | 20 (90.9%) | 26 (89.7%) | 25 (86.2%) | |
| M1 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 2 (6.9%) | |
| MX | 2 (6.1%) | 2 (9.1%) | 3 (10.3%) | 2 (6.9%) | |
| Hormone Status | |||||
| ER status | 0.99 | ||||
| ER-positive | 0 (0.0%) | 2 (9.1%) | 0 (0.0%) | 2 (6.9%) | |
| ER-negative | 31 (93.9%) | 20 (90.9%) | 28 (96.6%) | 26 (89.7%) | |
| NE | 2 (6.1%) | 0 (0.0%) | 1 (3.4%) | 1 (3.4%) | |
| Indeterminate | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Equivocal | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| PR status | 0.99 | ||||
| PR-positive | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| PR-negative | 31 (93.9%) | 22 (100%) | 28 (96.6%) | 27 (93.1%) | |
| NE | 2 (6.1%) | 0 (0.0%) | 1 (3.4%) | 2 (6.9%) | |
| Indeterminate | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Equivocal | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| HER2 status | 0.96 | ||||
| HER2-positive | 2 (6.1%) | 0 (0.0%) | 3 (10.3%) | 3 (10.3%) | |
| HER2-negative | 19 (57.6%) | 16 (72.7%) | 19 (65.5%) | 15 (51.7%) | |
| NE | 6 (18.2%) | 1 (4.5%) | 3 (10.3%) | 5 (17.2%) | |
| Indeterminate | 0 (0.0%) | 1 (4.5%) | 0 (0.0%) | 0 (0.0%) | |
| Equivocal | 6 (18.2%) | 4 (18.2%) | 4 (13.8%) | 5 (17.2%) | |
| NA | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (3.4%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, P.; Ring, A.; Porras, T.B.; Zhou, G.; Lu, J.; Kang, I.; Lang, J.E. Integrated Proteogenomic Analysis Reveals Distinct Potentially Actionable Therapeutic Vulnerabilities in Triple-Negative Breast Cancer Subtypes. Cancers 2024, 16, 516. https://doi.org/10.3390/cancers16030516
Kaur P, Ring A, Porras TB, Zhou G, Lu J, Kang I, Lang JE. Integrated Proteogenomic Analysis Reveals Distinct Potentially Actionable Therapeutic Vulnerabilities in Triple-Negative Breast Cancer Subtypes. Cancers. 2024; 16(3):516. https://doi.org/10.3390/cancers16030516
Chicago/Turabian StyleKaur, Pushpinder, Alexander Ring, Tania B. Porras, Guang Zhou, Janice Lu, Irene Kang, and Julie E. Lang. 2024. "Integrated Proteogenomic Analysis Reveals Distinct Potentially Actionable Therapeutic Vulnerabilities in Triple-Negative Breast Cancer Subtypes" Cancers 16, no. 3: 516. https://doi.org/10.3390/cancers16030516
APA StyleKaur, P., Ring, A., Porras, T. B., Zhou, G., Lu, J., Kang, I., & Lang, J. E. (2024). Integrated Proteogenomic Analysis Reveals Distinct Potentially Actionable Therapeutic Vulnerabilities in Triple-Negative Breast Cancer Subtypes. Cancers, 16(3), 516. https://doi.org/10.3390/cancers16030516