
BNIP3 Downregulation Ameliorates Muscle Atrophy in Cancer Cachexia

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
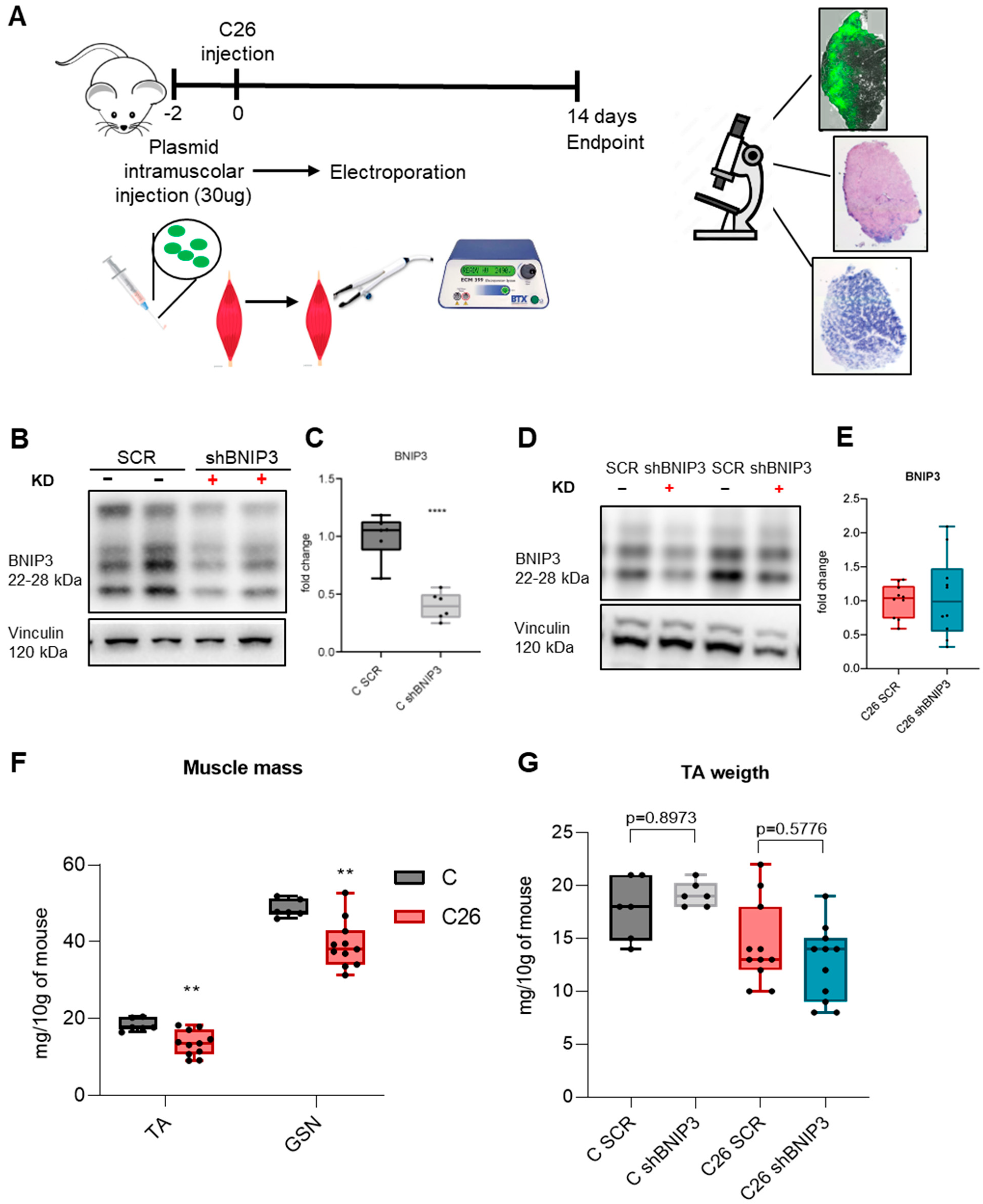
2.1. Animal Model and Experimental Design
2.2. Plasmid Generation and Isolation
2.3. Gene Delivery
2.3.1. Electroporation
2.3.2. Adenovirus
2.4. Histology
2.4.1. Hematoxylin and Eosin Staining and Cross-Sectional Area (CSA)
2.4.2. Succinate Dehydrogenase Staining
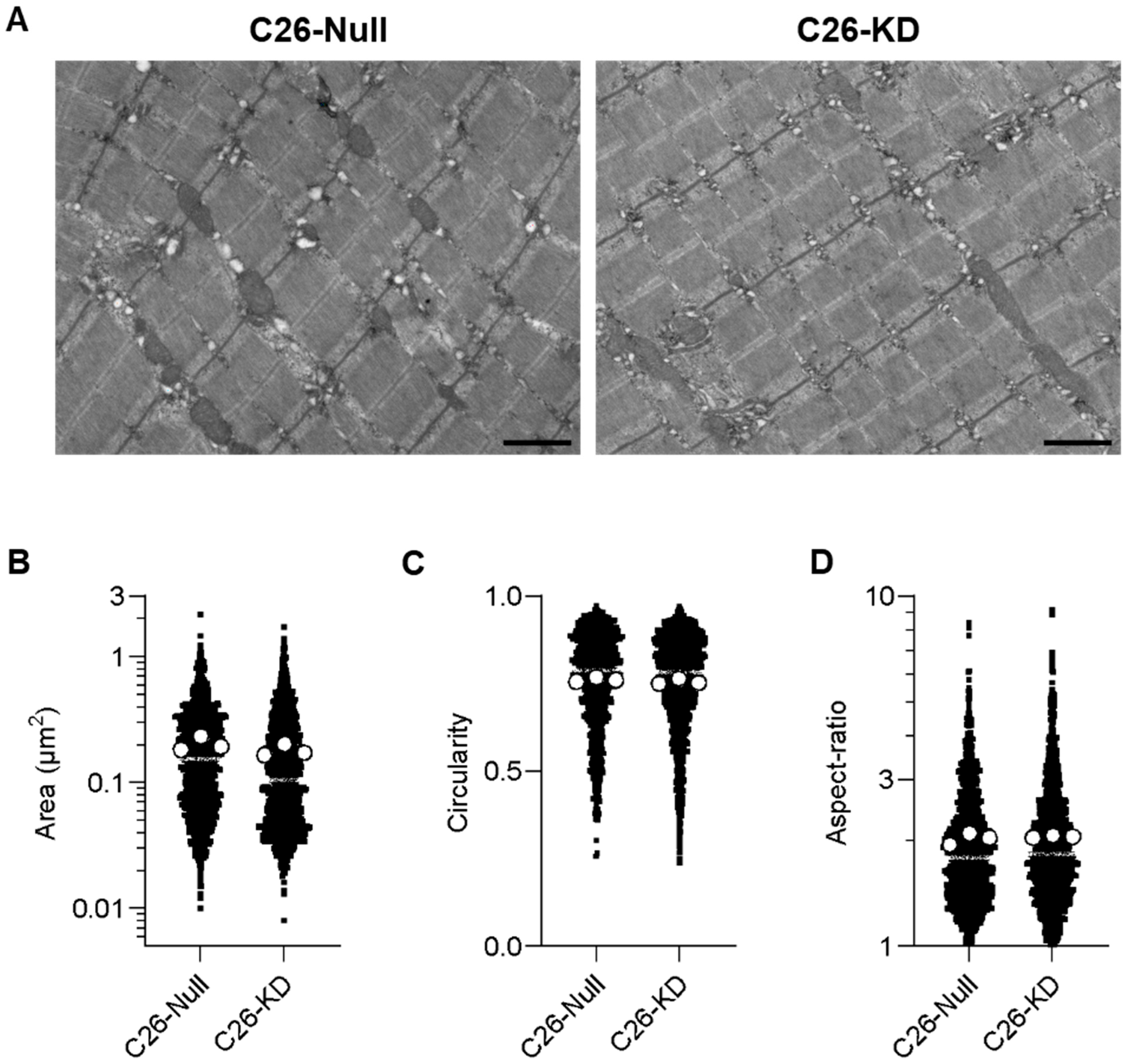
2.5. Transmission Electron Microscopy (TEM)
2.6. Protein Expression Analyses
2.6.1. Protein Extraction
2.6.2. Western Blotting
2.6.3. Amplex™ Red Hydrogen Peroxide/Peroxidase Assay
3. Results
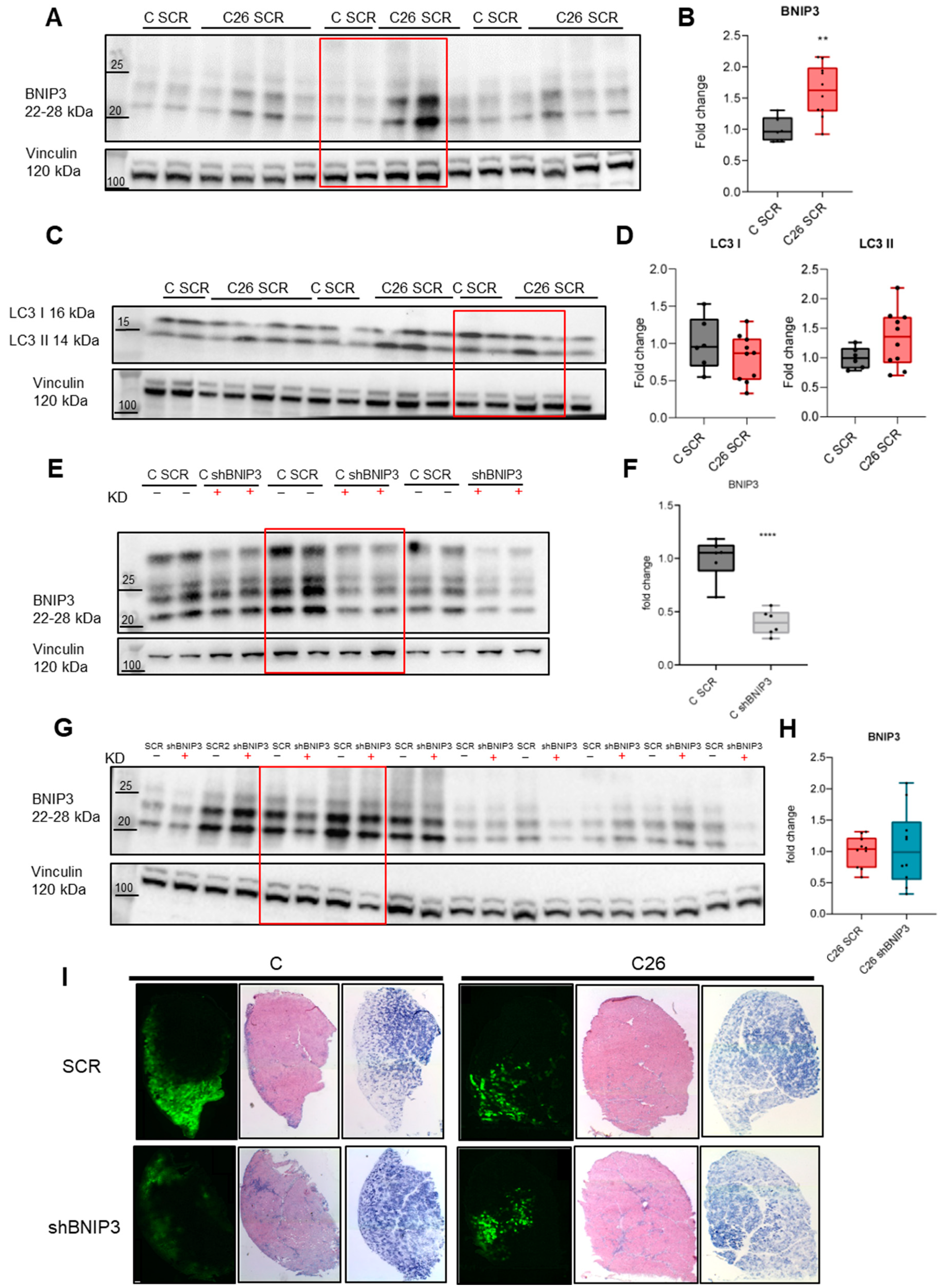
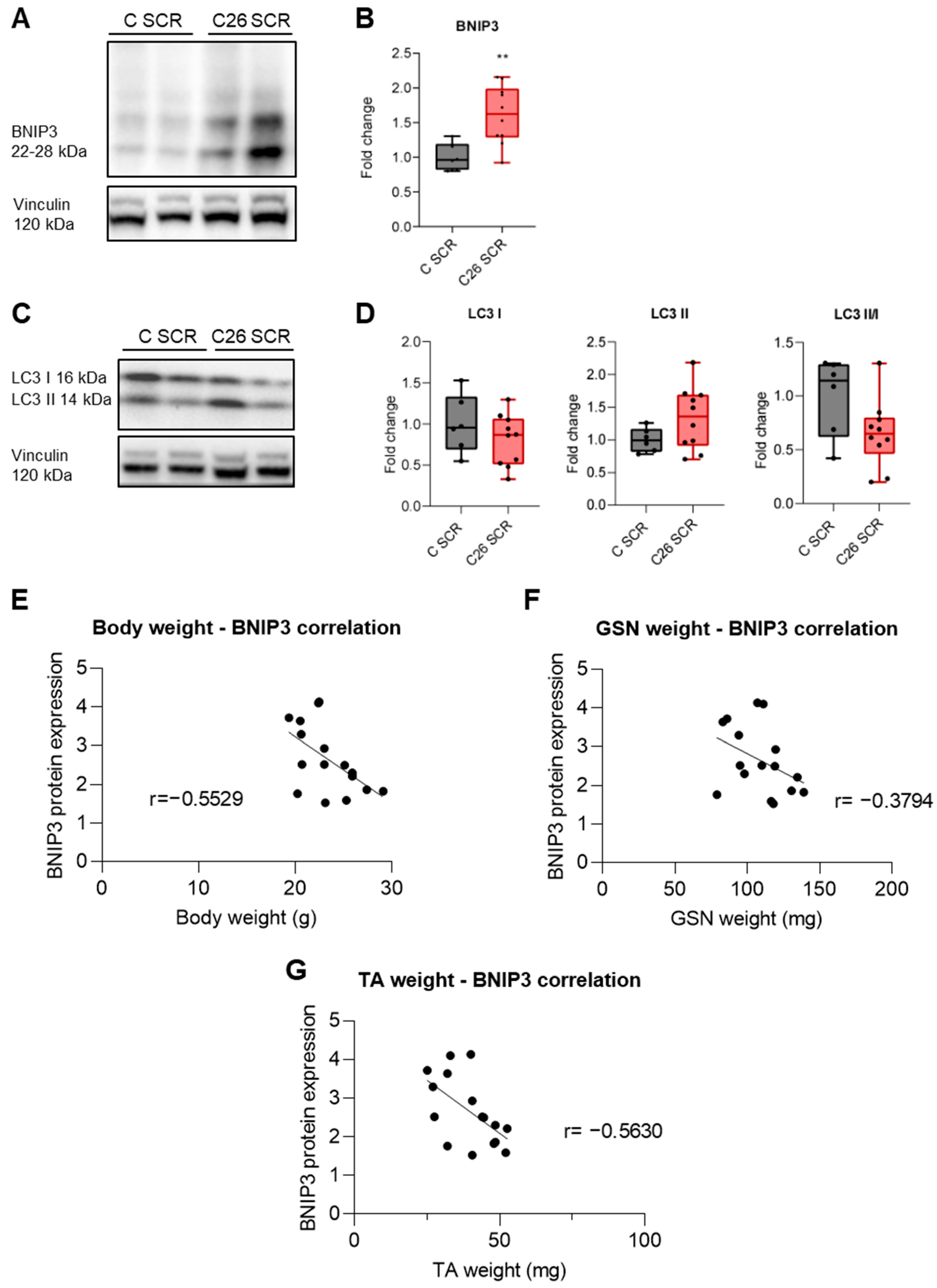
3.1. BNIP3 Is Increased in the Skeletal Muscle of Tumor-Bearing Mice
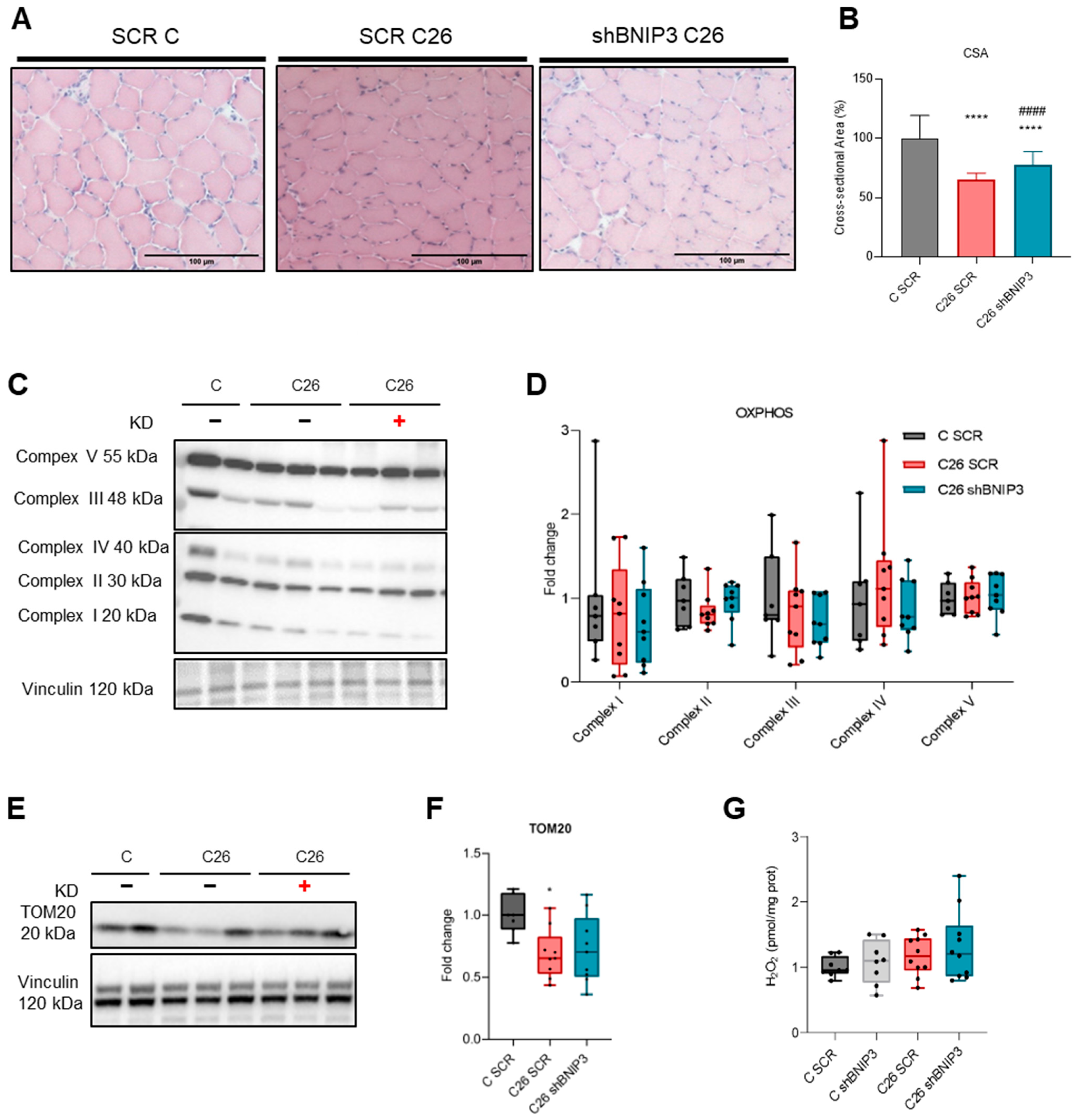
3.2. Electroporation-Mediated BNIP3 Silencing Does Not Prevent BNIP3 Overexpression in the Skeletal Muscle of Tumor-Bearing Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Costelli, P. The Redox Balance: A Target for Interventions against Muscle Wasting in Cancer Cachexia? Antioxid. Redox Signal. 2020, 33, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Penna, F.; Baccino, F.M.; Costelli, P. Coming back: Autophagy in cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 241–246. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Guan, K. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.Y.; et al. Autophagy: Cancer’s Friend or Foe? Adv. Cancer Res. 2013, 118, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Hanif, E.A.M.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2016, 6, 422. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gogvadze, V.; Zhivotovsky, B. Mitophagy in carcinogenesis and cancer treatment. Discov. Oncol. 2021, 12, 58. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2019, 66, 45–58. [Google Scholar] [CrossRef]
- Song, C.; Pan, S.; Zhang, J.; Li, N.; Geng, Q. Mitophagy: A novel perspective for insighting into cancer and cancer treatment. Cell Prolif. 2022, 55, e13327. [Google Scholar] [CrossRef]
- Damrauer, J.S.; Stadler, M.E.; Acharyya, S.; Baldwin, A.S.; Couch, M.E.; Guttridge, D.C. Chemotherapy-induced muscle wasting: Association with NF-κB and cancer cachexia. Eur. J. Transl. Myol. 2018, 28, 158–166. [Google Scholar] [CrossRef]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: Molecular mechanisms and implications for human disease. Cell Death Dis. 2022, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Chourasia, H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef]
- Springer, M.Z.; Macleod, K.F. Mitophagy: Mechanisms and Role in Human Disease Maya. J. Pathol. 2016, 240, 253–255. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Poole, L.P.; Bock-Hughes, A.; Berardi, D.E.; Macleod, K.F. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci. Rep. 2021, 11, 20526. [Google Scholar] [CrossRef]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death. Cell Death Differ. 2011, 16, 515–523. [Google Scholar] [CrossRef]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Fanelli, F.R.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Irazoki, A.; Martinez-Vicente, M.; Aparicio, P.; Aris, C.; Alibakhshi, E.; Rubio-Valera, M.; Castellanos, J.; Lores, L.; Palacín, M.; Gumà, A.; et al. Coordination of mitochondrial and lysosomal homeostasis mitigates inflammation and muscle atrophy during aging. Aging Cell 2022, 21, e13583. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.C.; Hardee, J.P.; Waddell, D.S.; Goodman, C.A. CORP: Gene delivery into murine skeletal muscle using in vivo electroporation. J. Appl. Physiol. 2022, 133, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Deaver, J.W.; Rosa-Caldwell, M.E.; Haynie, W.S.; da Silva, F.M.; Cabrera, A.R.; Schrems, E.R.; Saling, L.W.; Jansen, L.T.; Dunlap, K.R.; et al. Development of metabolic and contractile alterations in development of cancer cachexia in female tumor-bearing mice. J. Appl. Physiol. 2022, 132, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Beltrà, M.; Pöllänen, N.; Fornelli, C.; Tonttila, K.; Hsu, M.Y.; Zampieri, S.; Moletta, L.; Corrà, S.; Porporato, P.E.; Kivelä, R.; et al. NAD+ repletion with niacin counteracts cancer cachexia. Nat. Commun. 2023, 14, 1849. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.; Ma, Y.; Yan, X.; Lai, N.; Wang, H.; Gao, B.; Gu, A.M.; Han, Q.; Zhang, Q.; et al. PGAM5 interacts with and maintains BNIP3 to license cancer-associated muscle wasting. Autophagy 2024, 20, 2205–2220. [Google Scholar] [CrossRef]
- Sartori, R.; Hagg, A.; Zampieri, S.; Armani, A.; Winbanks, C.E.; Viana, L.R.; Haidar, M.; Watt, K.I.; Qian, H.; Pezzini, C.; et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci. Transl. Med. 2021, 13, eaay9592. [Google Scholar] [CrossRef]
- Huot, J.R.; Baumfalk, D.; Resendiz, A.; Bonetto, A.; Smuder, A.J.; Penna, F. Targeting Mitochondria and Oxidative Stress in Cancer-and Chemotherapy-Induced Muscle Wasting. Antioxid. Redox Signal. 2023, 38, 352–370. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fornelli, C.; Beltrà, M.; Zorzano, A.; Costelli, P.; Sebastian, D.; Penna, F. BNIP3 Downregulation Ameliorates Muscle Atrophy in Cancer Cachexia. Cancers 2024, 16, 4133. https://doi.org/10.3390/cancers16244133
Fornelli C, Beltrà M, Zorzano A, Costelli P, Sebastian D, Penna F. BNIP3 Downregulation Ameliorates Muscle Atrophy in Cancer Cachexia. Cancers. 2024; 16(24):4133. https://doi.org/10.3390/cancers16244133
Chicago/Turabian StyleFornelli, Claudia, Marc Beltrà, Antonio Zorzano, Paola Costelli, David Sebastian, and Fabio Penna. 2024. "BNIP3 Downregulation Ameliorates Muscle Atrophy in Cancer Cachexia" Cancers 16, no. 24: 4133. https://doi.org/10.3390/cancers16244133
APA StyleFornelli, C., Beltrà, M., Zorzano, A., Costelli, P., Sebastian, D., & Penna, F. (2024). BNIP3 Downregulation Ameliorates Muscle Atrophy in Cancer Cachexia. Cancers, 16(24), 4133. https://doi.org/10.3390/cancers16244133