
Emerging Paradigms in Cancer Metastasis: Ghost Mitochondria, Vasculogenic Mimicry, and Polyploid Giant Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
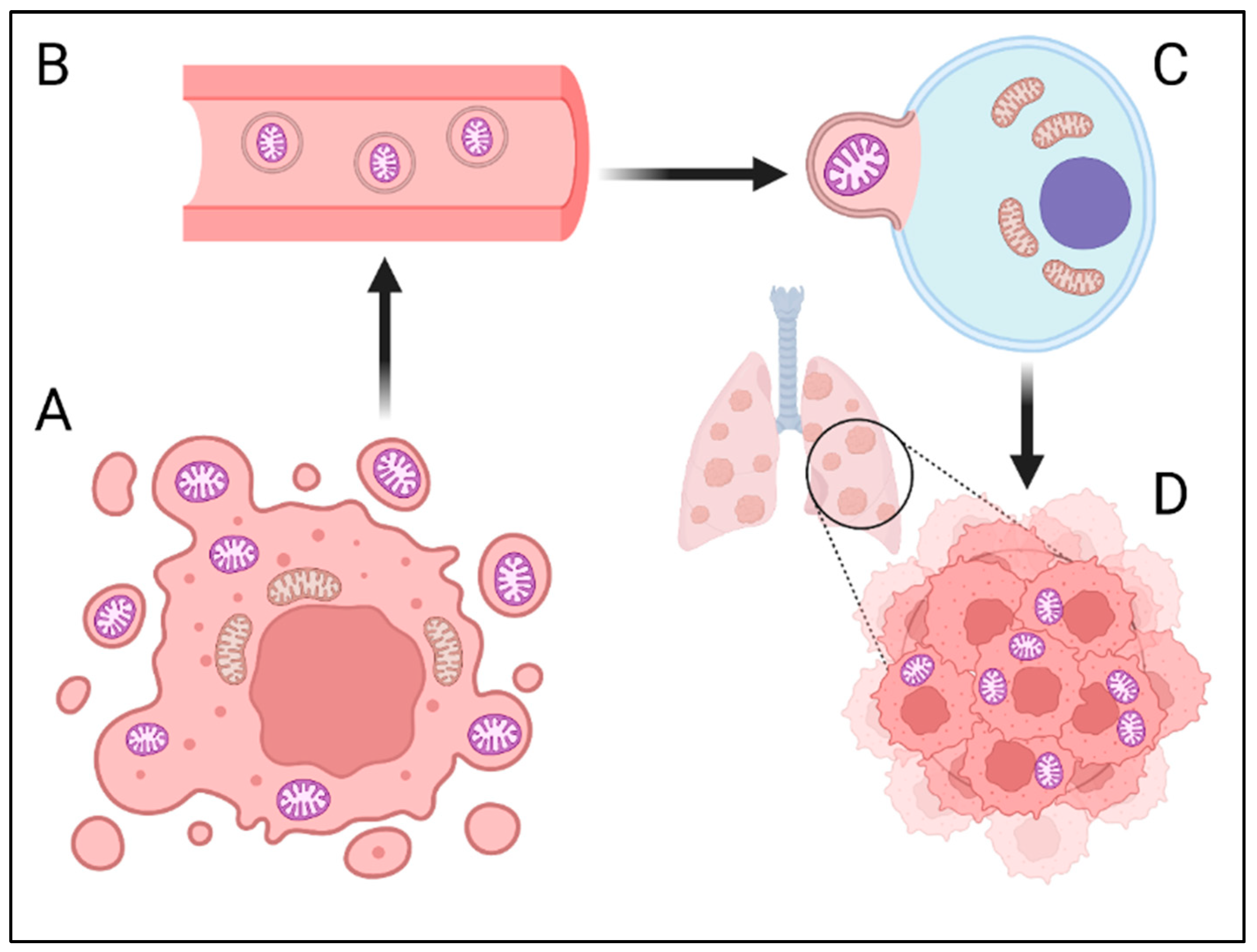
2. “Ghost” Mitochondria (GM)—Cancer Cell Communication or Independent Metastatic Route?
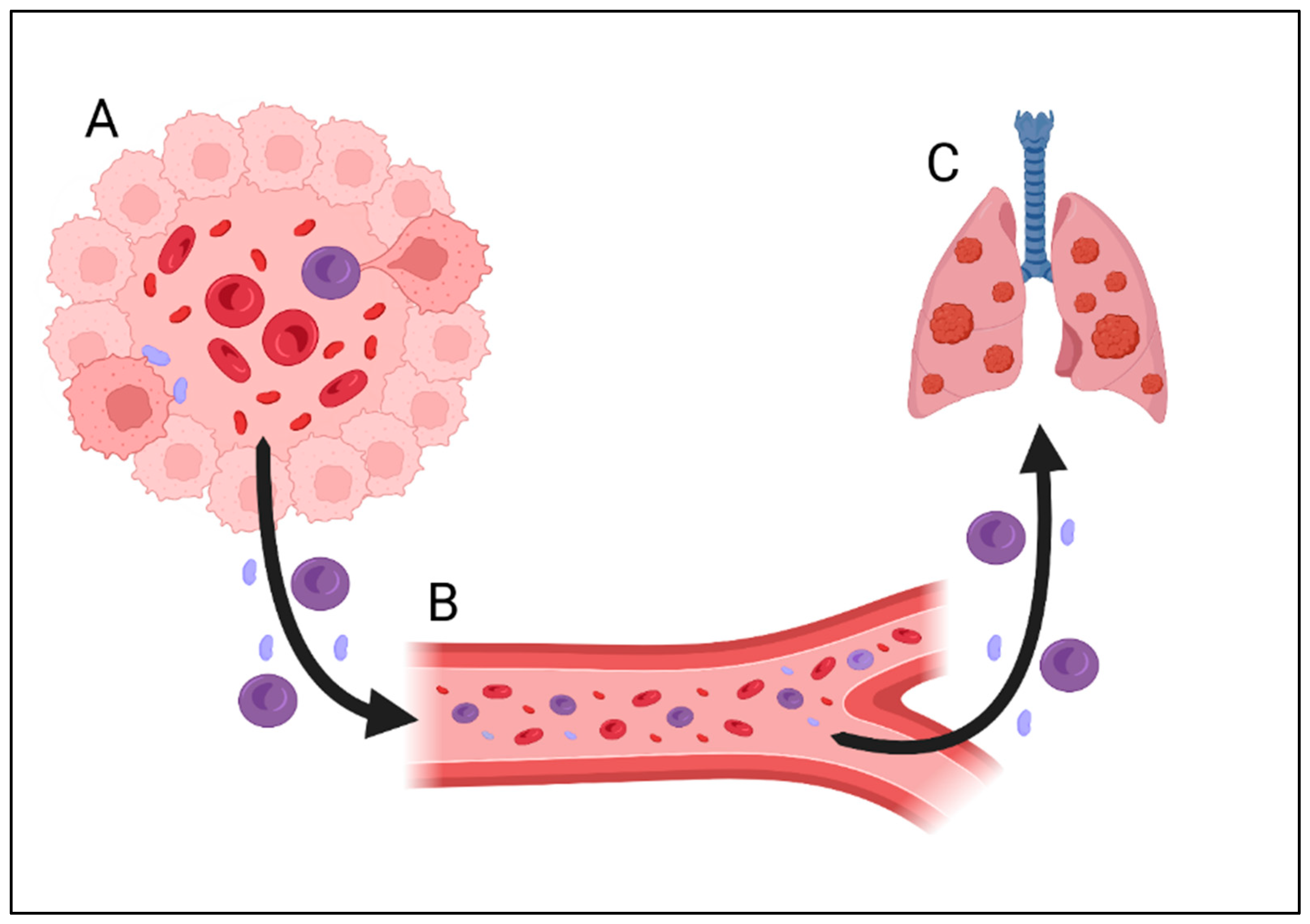
3. Vasculogenic Mimicry (VM)—Need for Nutrients or Method of Metastasis on the Basis of Interactions with Blood Cells?
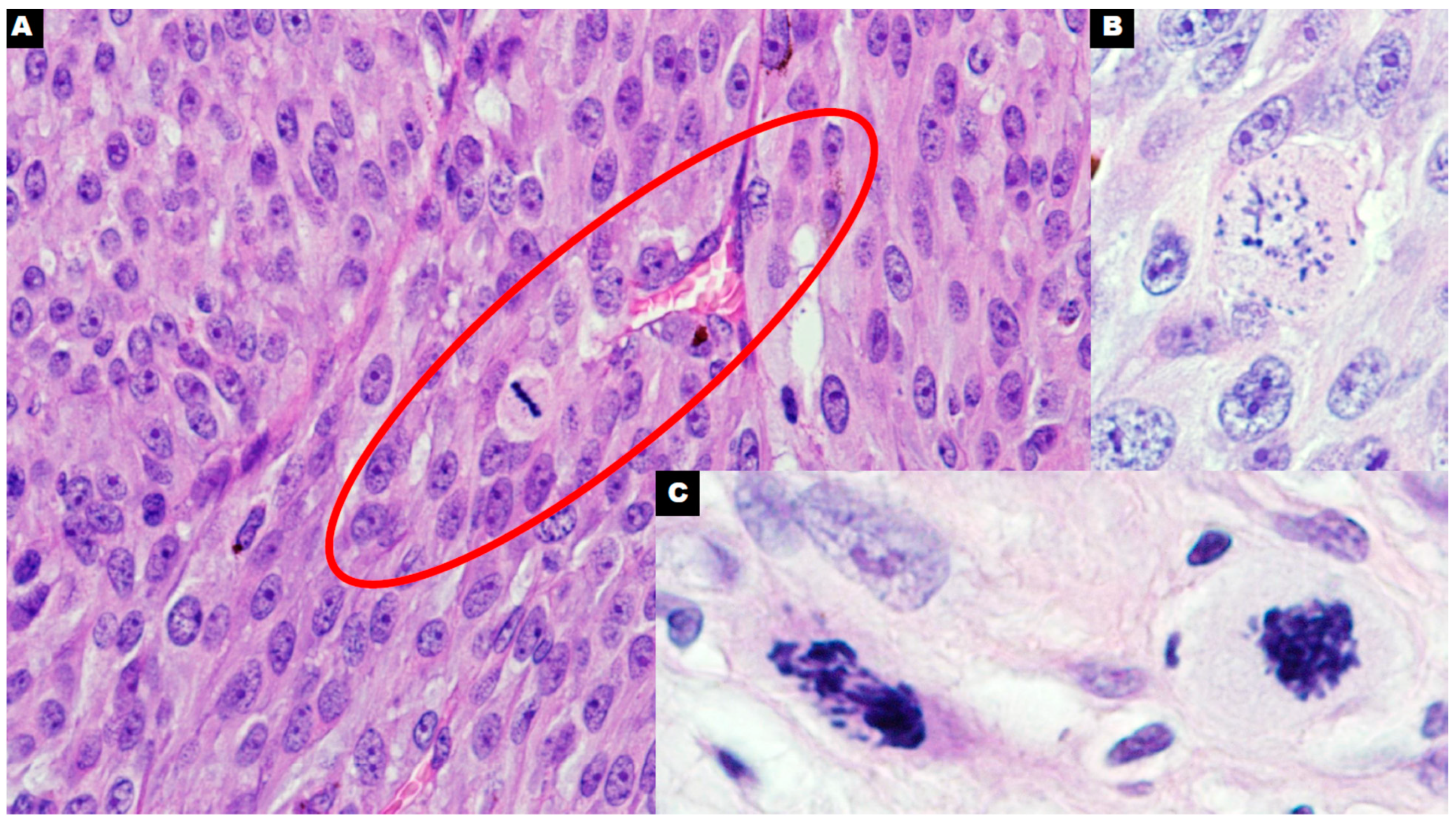
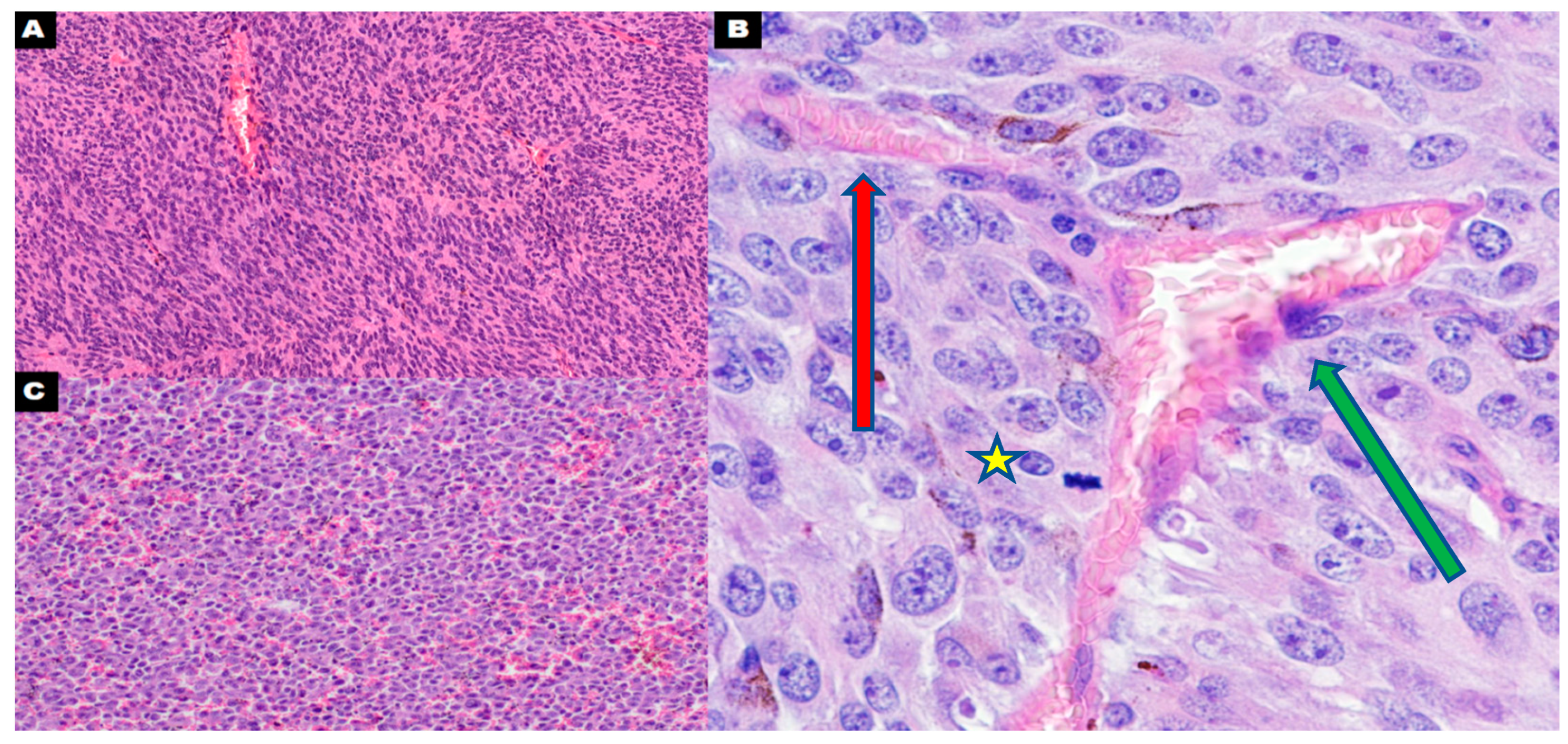
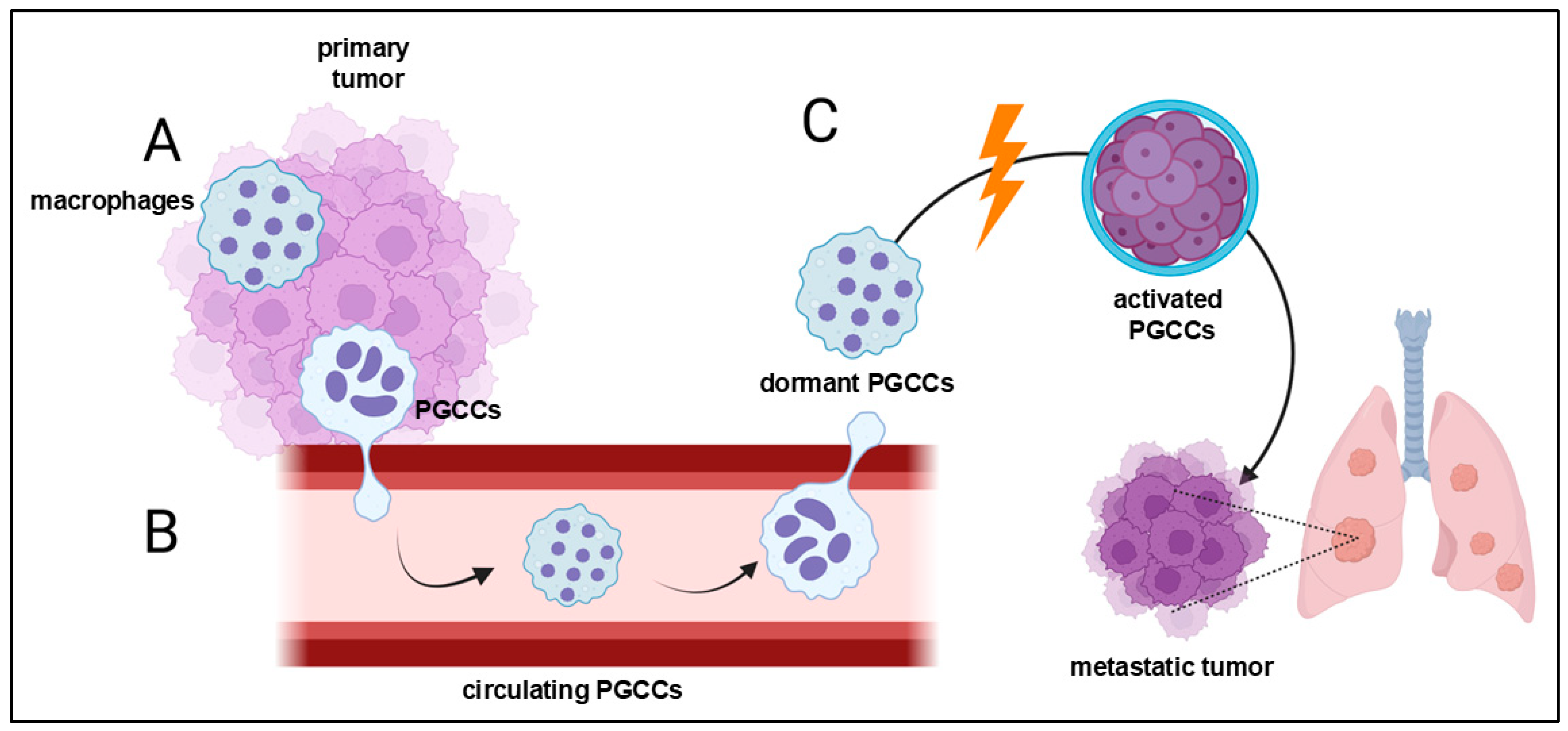
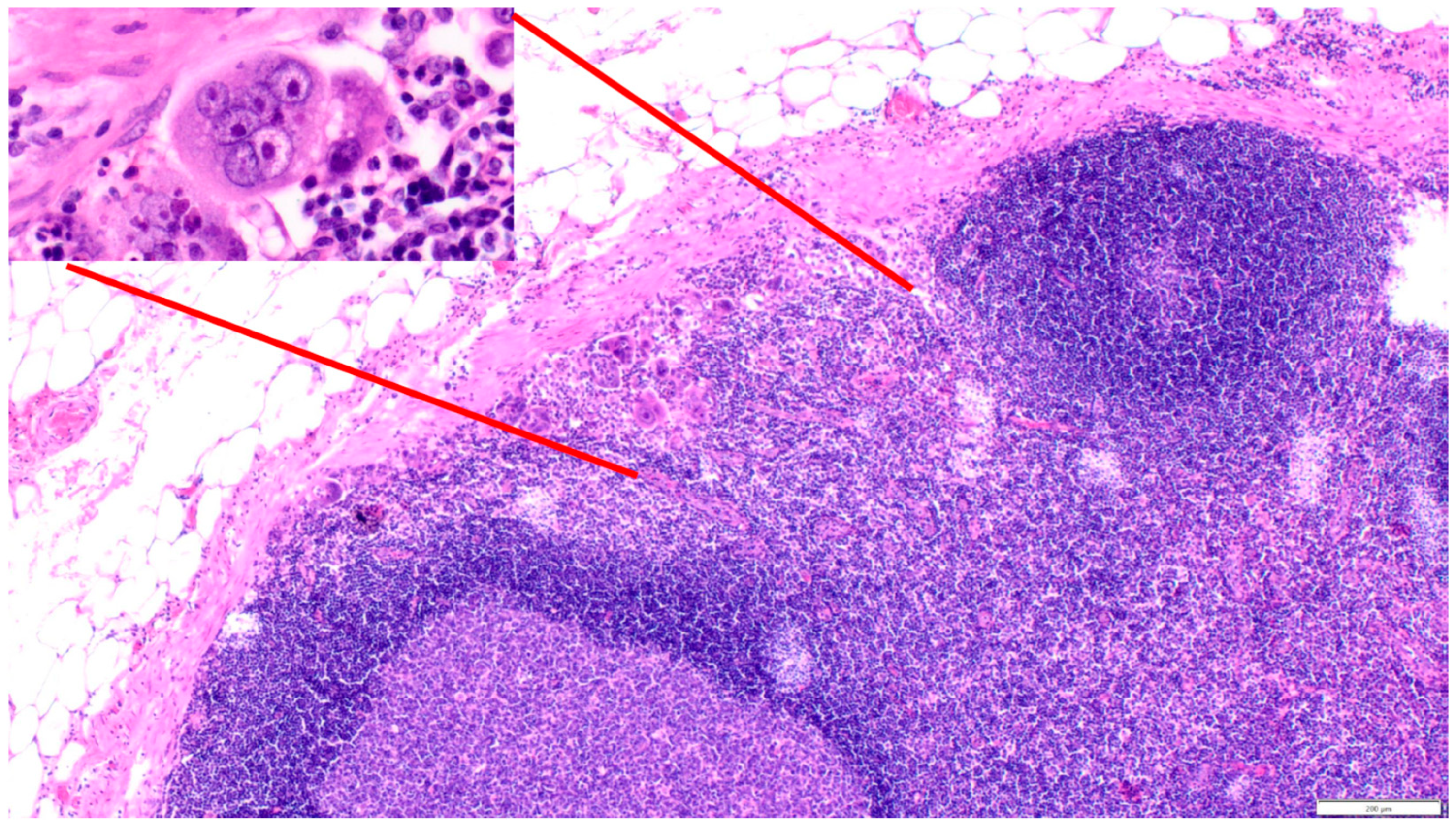
4. Polyploid Giant Cancer Cells—Dormant Locators or Migrating Pioneers
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of Deaths from Cancer Caused by Metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Gennari, A.; Conte, P.; Rosso, R.; Orlandini, C.; Bruzzi, P. Survival of Metastatic Breast Carcinoma Patients over a 20-year Period. Cancer 2005, 104, 1742–1750. [Google Scholar] [CrossRef]
- Mani, K.; Deng, D.; Lin, C.; Wang, M.; Hsu, M.L.; Zaorsky, N.G. Causes of Death among People Living with Metastatic Cancer. Nat. Commun. 2024, 15, 1519. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep Nature of Metastatic Inefficiency: Dormancy of Solitary Cells after Successful Extravasation and Limited Survival of Early Micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Martin, O.A.; Anderson, R.L.; Narayan, K.; MacManus, M.P. Does the Mobilization of Circulating Tumour Cells during Cancer Therapy Cause Metastasis? Nat. Rev. Clin. Oncol. 2017, 14, 32–44. [Google Scholar] [CrossRef]
- Su, J.-X.; Li, S.-J.; Zhou, X.-F.; Zhang, Z.-J.; Yan, Y.; Liu, S.-L.; Qi, Q. Chemotherapy-Induced Metastasis: Molecular Mechanisms and Clinical Therapies. Acta Pharmacol. Sin. 2023, 44, 1725–1736. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Seyfried, T.N. Cancer as a Mitochondrial Metabolic Disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J.C. Crosstalk from Non-Cancerous Mitochondria Can Inhibit Tumor Properties of Metastatic Cells by Suppressing Oncogenic Pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria Organelle Transplantation: Introduction of Normal Epithelial Mitochondria into Human Cancer Cells Inhibits Proliferation and Increases Drug Sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef]
- Shay, J.W. Cytoplasmic Suppression of Tumorigenicity in Reconstructed Mouse Cells. Cancer Res. 1988, 48, 830–833. [Google Scholar]
- Mckinnell, R.G.; Deggins, B.A.; Labat, D.D. Transplantation of Pluripotential Nuclei from Triploid Frog Tumors. Science 1969, 165, 394–396. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Garimella, S.V.; Gampa, S.C.; Chaturvedi, P. Mitochondria in Cancer Stem Cells: From an Innocent Bystander to a Central Player in Therapy Resistance. Stem Cells Cloning 2023, 16, 19. [Google Scholar] [CrossRef]
- Cheng, X.; Henick, B.S.; Cheng, K. Anticancer Therapy Targeting Cancer-Derived Extracellular Vesicles. ACS Nano 2024, 18, 6748–6765. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-Associated Fibroblasts Promote Prostate Cancer Malignancy via Metabolic Rewiring and Mitochondrial Transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Buller, B.; Chopp, M. Exosomes—Beyond Stem Cells for Restorative Therapy in Stroke and Neurological Injury. Nat. Rev. Neurol. 2019, 15, 193–203. [Google Scholar] [CrossRef]
- Choi, D.; Montermini, L.; Jeong, H.; Sharma, S.; Meehan, B.; Rak, J. Mapping Subpopulations of Cancer Cell-Derived Extracellular Vesicles and Particles by Nano-Flow Cytometry. ACS Nano 2019, 13, 10499–10511. [Google Scholar] [CrossRef]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The Emerging Role of Extracellular Vesicles as Biomarkers for Urogenital Cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef]
- Stier, A. Human Blood Contains Circulating Cell-Free Mitochondria, but Are They Really Functional? Am. J. Physiol. Endocrinol. Metab. 2021, 320, E859–E863. [Google Scholar] [CrossRef]
- Stephens, O.R.; Grant, D.; Frimel, M.; Wanner, N.; Yin, M.; Willard, B.; Erzurum, S.C.; Asosingh, K. Characterization and Origins of Cell-Free Mitochondria in Healthy Murine and Human Blood. Mitochondrion 2020, 54, 102–112. [Google Scholar] [CrossRef]
- Mohd Khair, S.Z.N.; Abd Radzak, S.M.; Mohamed Yusoff, A.A. The Uprising of Mitochondrial DNA Biomarker in Cancer. Dis. Markers 2021, 2021, 7675269. [Google Scholar] [CrossRef]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Olofsson Bagge, R.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lötvall, J. Mitochondrial Protein Enriched Extracellular Vesicles Discovered in Human Melanoma Tissues Can Be Detected in Patient Plasma. J. Extracell. Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets Release Mitochondria Serving as Substrate for Bactericidal Group IIA-Secreted Phospholipase a to Promote Inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Lin, L.; Xu, H.; Bishawi, M.; Feng, F.F.; Samy, K.; Truskey, G.; Barbas, A.S.; Kirk, A.D.; Brennan, T.V. Circulating Mitochondria in Organ Donors Promote Allograft Rejection. Am. J. Transplant. 2019, 19, 1917–1929. [Google Scholar] [CrossRef]
- Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers 2020, 12, 1787. [Google Scholar] [CrossRef]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Kuo, S.J.; Chen, S.T.; Liu, C.S. Mitochondrial Transplantation Regulates Antitumour Activity, Chemoresistance and Mitochondrial Dynamics in Breast Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [Google Scholar] [CrossRef]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Guo, X.; Can, C.; Liu, W.; Wei, Y.; Yang, X.; Liu, J.; Jia, H.; Jia, W.; Wu, H.; Ma, D. Mitochondrial Transfer in Hematological Malignancies. Biomark. Res. 2023, 11, 89. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Ii, M.B.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate Carboxylase Is Critical for Non-Small-Cell Lung Cancer Proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef]
- Anderson, R.G.; Ghiraldeli, L.P.; Pardee, T.S. Mitochondria in Cancer Metabolism, an Organelle Whose Time Has Come? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 96–102. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated Stromal Cells Transfer Mitochondria to Rescue Acute Lymphoblastic Leukemia Cells from Oxidative Stress. Blood 2019, 134, 1415. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective Mitochondrial Transfer from Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells during Chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular Nanotubes Mediate Mitochondrial Trafficking between Cancer and Immune Cells. Nat. Nanotechnol. 2022, 17, 98. [Google Scholar] [CrossRef]
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 2021, 33, 283–299. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, H.; Li, H.; Mou, H.; Yinwang, E.; Xue, Y.; Wang, S.; Zhang, Y.; Wang, Z.; Chen, T.; et al. Cancer Cells Reprogram to Metastatic State through the Acquisition of Platelet Mitochondria. Cell Rep. 2023, 42, 113147. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Perego, M.; Agarwal, E.; Bertolini, I.; Wang, Y.; Goldman, A.R.; Tang, H.Y.; Kossenkov, A.V.; Libby, C.J.; Languino, L.R.; et al. Ghost Mitochondria Drive Metastasis through Adaptive GCN2/Akt Therapeutic Vulnerability. Proc. Natl. Acad. Sci. USA 2022, 119, e2115624119. [Google Scholar] [CrossRef]
- Altieri, D.C. Mitochondria in Cancer: Clean Windmills or Stressed Tinkerers? Trends Cell Biol. 2023, 33, 293–299. [Google Scholar] [CrossRef]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin Determines MICOS Assembly Essential for Mitochondrial Dynamics and MtDNA Nucleoid Organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef]
- Kossenkov, A.V.; Milcarek, A.; Notta, F.; Jang, G.H.; Wilson, J.M.; Gallinger, S.; Zhou, D.C.; Ding, L.; Ghosh, J.C.; Perego, M.; et al. Mitochondrial Fitness and Cancer Risk. PLoS ONE 2022, 17, e0273520. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic Mimicry in Carcinogenesis and Clinical Applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of Vasculogenic Mimicry in Hypoxic Tumor Microenvironments. Mol. Cancer 2021, 20, 19. [Google Scholar] [CrossRef]
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem Cells Int. 2022, 2022, 9653244. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-Endothelial Transition and Cancer Stem Cells: Two Cornerstones of Vasculogenic Mimicry in Malignant Tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef]
- Lapkina, E.Z.; Esimbekova, A.R.; Ruksha, T.G. Vasculogenic Mimicry. Arkh Patol. 2023, 85, 508–525. [Google Scholar] [CrossRef]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Cáceres-Gutiérrez, R.; Caro-Sánchez, C.H.S.; Herrera, L.A.; Díaz-Chávez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef]
- Saini, G.; Joshi, S.; Garlapati, C.; Li, H.; Kong, J.; Krishnamurthy, J.; Reid, M.D.; Aneja, R. Polyploid Giant Cancer Cell Characterization: New Frontiers in Predicting Response to Chemotherapy in Breast Cancer. Semin. Cancer Biol. 2022, 81, 220–231. [Google Scholar] [CrossRef]
- Liu, P.; Wang, L.; Yu, H. Polyploid Giant Cancer Cells: Origin, Possible Pathways of Formation, Characteristics, and Mechanisms of Regulation. Front. Cell Dev. Biol. 2024, 12, 1410637. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, B.; Lin, Y.; Deng, H.; Wang, X.; Xu, C.; Wang, K.; Yu, N.; Liu, R.; Han, M. Lysyl Oxidase Promotes the Formation of Vasculogenic Mimicry in Gastric Cancer through PDGF-PDGFR Pathway. J. Cancer 2024, 15, 1816–1825. [Google Scholar] [CrossRef]
- Han, C.; Sun, B.; Zhao, X.; Zhang, Y.; Gu, Q.; Liu, F.; Zhao, N.; Wu, L. Phosphorylation of STAT3 Promotes Vasculogenic Mimicry by Inducing Epithelial-to-Mesenchymal Transition in Colorectal Cancer. Technol. Cancer Res. Treat. 2017, 16, 1209–1219. [Google Scholar] [CrossRef]
- Lizárraga-Verdugo, E.; Avendaño-Félix, M.; Bermúdez, M.; Ramos-Payán, R.; Pérez-Plasencia, C.; Aguilar-Medina, M. Cancer Stem Cells and Its Role in Angiogenesis and Vasculogenic Mimicry in Gastrointestinal Cancers. Front. Oncol. 2020, 10, 4130. [Google Scholar] [CrossRef]
- Paulis, Y.W.J.; Soetekouw, P.M.M.B.; Verheul, H.M.W.; Tjan-Heijnen, V.C.G.; Griffioen, A.W. Signalling Pathways in Vasculogenic Mimicry. Biochim. Biophys. Acta Rev. Cancer 2010, 1806, 18–28. [Google Scholar] [CrossRef]
- Pereira-Veiga, T.; Schneegans, S.; Pantel, K.; Wikman, H. Circulating Tumor Cell-Blood Cell Crosstalk: Biology and Clinical Relevance. Cell Rep. 2022, 40, 111298. [Google Scholar] [CrossRef]
- Patel, D.; Thankachan, S.; Sreeram, S.; Kavitha, K.P.; Suresh, P.S. The Role of Tumor-Educated Platelets in Ovarian Cancer: A Comprehensive Review and Update. Pathol. Res. Pract. 2023, 241, 154267. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; In ‘t Veld, S.G.J.G.; Vancura, A.; Muller, M.; Niemeijer, A.L.N.; Fejes, A.V.; Tjon Kon Fat, L.A.; Huis In ‘t Veld, A.E.; Leurs, C.; et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell 2017, 32, 238–252.e9. [Google Scholar] [CrossRef]
- Bian, X.; Yin, S.; Yang, S.; Jiang, X.; Wang, J.; Zhang, M.; Zhang, L. Roles of Platelets in Tumor Invasion and Metastasis: A Review. Heliyon 2022, 8, e12072. [Google Scholar] [CrossRef]
- Ruf, W.; Yokota, N.; Schaffner, F. Tissue Factor in Cancer Progression and Angiogenesis. Thromb. Res. 2010, 125, S36–S38. [Google Scholar] [CrossRef]
- Li, S.; Lu, Z.; Wu, S.; Chu, T.; Li, B.; Qi, F.; Zhao, Y.; Nie, G. The Dynamic Role of Platelets in Cancer Progression and Their Therapeutic Implications. Nat. Rev. Cancer 2024, 24, 72–87. [Google Scholar] [CrossRef]
- Cluxton, C.D.; Spillane, C.; O’Toole, S.A.; Sheils, O.; Gardiner, C.M.; O’Leary, J.J. Suppression of Natural Killer Cell NKG2D and CD226 Anti-Tumour Cascades by Platelet Cloaked Cancer Cells: Implications for the Metastatic Cascade. PLoS ONE 2019, 14, e0211538. [Google Scholar] [CrossRef]
- Karsten, E.; Breen, E.; McCracken, S.A.; Clarke, S.; Herbert, B.R. Red Blood Cells Exposed to Cancer Cells in Culture Have Altered Cytokine Profiles and Immune Function. Sci. Rep. 2020, 10, 7727. [Google Scholar] [CrossRef]
- Liang, N.; Jiao, Z.; Zhang, C.; Wu, Y.; Wang, T.; Li, S.; Wang, Y.; Song, T.; Chen, J.Q.; Liang, H.; et al. Mature Red Blood Cells Contain Long DNA Fragments and Could Acquire DNA from Lung Cancer Tissue. Adv. Sci. 2023, 10, e2206361. [Google Scholar] [CrossRef]
- Nilsson, R.J.A.; Balaj, L.; Hulleman, E.; Van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood Platelets Contain Tumor-Derived RNA Biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef]
- Girardot, M.; Pecquet, C.; Boukour, S.; Knoops, L.; Ferrant, A.; Vainchenker, W.; Giraudier, S.; Constantinescu, S.N. MiR-28 Is a Thrombopoietin Receptor Targeting MicroRNA Detected in a Fraction of Myeloproliferative Neoplasm Patient Platelets. Blood 2010, 116, 437–445. [Google Scholar] [CrossRef]
- Nilsson, J.; Skog, J.; Nordstrand, A.; Baranov, V.; Mincheva-Nilsson, L.; Breakefield, X.O.; Widmark, A. Prostate Cancer-Derived Urine Exosomes: A Novel Approach to Biomarkers for Prostate Cancer. Br. J. Cancer 2009, 100, 1603–1607. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, H.; Tang, K.; Huang, B. Tumor-Derived Microparticles in Tumor Immunology and Immunotherapy. Eur. J. Immunol. 2020, 50, 1653–1662. [Google Scholar] [CrossRef]
- Bagnall, J.S.; Byun, S.; Begum, S.; Miyamoto, D.T.; Hecht, V.C.; Maheswaran, S.; Stott, S.L.; Toner, M.; Hynes, R.O.; Manalis, S.R. Deformability of Tumor Cells versus Blood Cells. Sci. Rep. 2015, 5, 18542. [Google Scholar] [CrossRef]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef]
- Foster, B.M.; Shi, L.; Harris, K.S.; Patel, C.; Surratt, V.E.; Langsten, K.L.; Kerr, B.A. Bone Marrow-Derived Stem Cell Factor Regulates Prostate Cancer-Induced Shifts in Pre-Metastatic Niche Composition. Front. Oncol. 2022, 12, 855188. [Google Scholar] [CrossRef]
- Xia, Y.; Cai, X.Y.; Fan, J.Q.; Zhang, L.L.; Ren, J.H.; Chen, J.; Li, Z.Y.; Zhang, R.G.; Zhu, F.; Wu, G. Rho Kinase Inhibitor Fasudil Suppresses the Vasculogenic Mimicry of B16 Mouse Melanoma Cells Both in Vitro and in Vivo. Mol. Cancer Ther. 2015, 14, 1582–1590. [Google Scholar] [CrossRef]
- Colucci-D’amato, L.; Pastorino, O.; Teresa Gentile, M.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef]
- Hazra, S.; Kalyan Dinda, S.; Kumar Mondal, N.; Hossain, S.R.; Datta, P.; Yasmin Mondal, A.; Malakar, P.; Manna, D. Giant Cells: Multiple Cells Unite to Survive. Front. Cell Infect. Microbiol. 2023, 13, 1220589. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, M.; Zheng, M.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. Polyploid Giant Cancer Cells and Cancer Progression. Front. Cell Dev. Biol. 2022, 10, 1017588. [Google Scholar] [CrossRef]
- Jiao, Y.; Yu, Y.; Zheng, M.; Yan, M.; Wang, J.; Zhang, Y.; Zhang, S. Dormant Cancer Cells and Polyploid Giant Cancer Cells: The Roots of Cancer Recurrence and Metastasis. Clin. Transl. Med. 2024, 14, e1567. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Zhao, Q.; Zhang, K.; Li, Z.; Fu, F.; Zhang, H.; Du, J.; Li, Y.; Zhang, S. Different P53 Genotypes Regulating Different Phosphorylation Sites and Subcellular Location of CDC25C Associated with the Formation of Polyploid Giant Cancer Cells. J. Exp. Clin. Cancer Res. 2020, 39, 83. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of Cancer Stem-like Cells through the Formation of Polyploid Giant Cancer Cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, M.; Fei, F.; Li, C.; Du, J.; Liu, K.; Li, Y.; Zhang, S. EMT-Related Protein Expression in Polyploid Giant Cancer Cells and Their Daughter Cells with Different Passages after Triptolide Treatment. Med. Oncol. 2019, 36, 82. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y.; Wu, M.; Liu, J.; Wu, H.; Xu, C.; Chen, L. Hypoxia-Induced Polypoid Giant Cancer Cells in Glioma Promote the Transformation of Tumor-Associated Macrophages to a Tumor-Supportive Phenotype. CNS Neurosci. Ther. 2022, 28, 1326–1338. [Google Scholar] [CrossRef]
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernandez, V.; Peñarando, J.; Martinez, A.; Lopez-Pedrera, C.; Muñoz-Castañeda, J.R.; De La Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a Mimic of Hypoxia, Induces Formation of Polyploid Giant Cells with Stem Characteristics in Colon Cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef]
- Fei, F.; Qu, J.; Liu, K.; Li, C.; Wang, X.; Li, Y.; Zhang, S. The Subcellular Location of Cyclin B1 and CDC25 Associated with the Formation of Polyploid Giant Cancer Cells and Their Clinicopathological Significance. Lab. Investig. 2019, 99, 483–498. [Google Scholar] [CrossRef]
- Lu, P.; White-Gilbertson, S.; Beeson, G.; Beeson, C.; Ogretmen, B.; Norris, J.; Voelkel-Johnson, C. Ceramide Synthase 6 Maximizes P53 Function to Prevent Progeny Formation from Polyploid Giant Cancer Cells. Cancers 2021, 13, 2212. [Google Scholar] [CrossRef]
- Li, X.; Zhong, Y.; Zhang, X.; Sood, A.K.; Liu, J. Spatiotemporal View of Malignant Histogenesis and Macroevolution via Formation of Polyploid Giant Cancer Cells. Oncogene 2023, 42, 665–678. [Google Scholar] [CrossRef]
- Zhao, S.; Xing, S.; Wang, L.; Ouyang, M.; Liu, S.; Sun, L.; Yu, H. IL-1β Is Involved in Docetaxel Chemoresistance by Regulating the Formation of Polyploid Giant Cancer Cells in Non-Small Cell Lung Cancer. Sci. Rep. 2023, 13, 12763. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into Blastomere-like Cancer Stem Cells via Formation of Polyploid Giant Cancer Cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of Mitotic Descendants by Giant Cells from Irradiated Burkitt’s Lymphoma Cell Lines. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A Novel Type of Cell Division in Cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef]
- Zhang, Z.; Feng, X.; Deng, Z.; Cheng, J.; Wang, Y.; Zhao, M.; Zhao, Y.; He, S.; Huang, Q. Irradiation-Induced Polyploid Giant Cancer Cells Are Involved in Tumor Cell Repopulation via Neosis. Mol. Oncol. 2021, 15, 2219–2234. [Google Scholar] [CrossRef]
- Liu, J. The “Life Code”: A Theory That Unifies the Human Life Cycle and the Origin of Human Tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef]
- Adams, D.L.; Martin, S.S.; Alpaugh, R.K.; Charpentier, M.; Tsai, S.; Bergan, R.C.; Ogden, I.M.; Catalona, W.; Chumsri, S.; Tang, C.M.; et al. Circulating Giant Macrophages as a Potential Biomarker of Solid Tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 3514–3519. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as Tools and Targets in Cancer Therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Sutton, T.L.; Patel, R.K.; Anderson, A.N.; Bowden, S.G.; Whalen, R.; Giske, N.R.; Wong, M.H. Circulating Cells with Macrophage-like Characteristics in Cancer: The Importance of Circulating Neoplastic-Immune Hybrid Cells in Cancer. Cancers 2022, 14, 3871. [Google Scholar] [CrossRef]
- You, B.; Xia, T.; Gu, M.; Zhang, Z.; Zhang, Q.; Shen, J.; Fan, Y.; Yao, H.; Pan, S.; Lu, Y.; et al. AMPK-MTOR-Mediated Activation of Autophagy Promotes Formation of Dormant Polyploid Giant Cancer Cells. Cancer Res. 2022, 82, 846–858. [Google Scholar] [CrossRef]
- Shen, L.; Chen, Y.L.; Huang, C.C.; Shyu, Y.C.; Seftor, R.E.B.; Seftor, E.A.; Hendrix, M.J.C.; Chien, D.S.; Chu, Y.W. CVM-1118 (Foslinanib), a 2-Phenyl-4-Quinolone Derivative, Promotes Apoptosis and Inhibits Vasculogenic Mimicry via Targeting TRAP1. Pathol. Oncol. Res. 2023, 29, 1611038. [Google Scholar] [CrossRef]
- Su, W.-C.; Chen, M.H.; Bai, L.-Y.; Chen, J.-S.; Chen, Y.-Y.; Shih, Y.-H.; Wu, I.-C.; Gutheil, J.; Melink, T.J.; Chu, Y.-W.; et al. CVM-1118: A Potent Oral Anti-Vasculogenic Mimicry (VM) Agent in Patients with Advanced Neuroendocrine Tumors (NETs)-A Phase IIa Study. Am. Soc. Clin. Oncol. 2023, 41, e16235. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, L.; Rong, Z.; He, T.; Zhang, S. Number of Glioma Polyploid Giant Cancer Cells (PGCCs) Associated with Vasculogenic Mimicry Formation and Tumor Grade in Human Glioma. J. Exp. Clin. Cancer Res. 2013, 32, 75. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, P.; Lv, H.; Zhang, D.; Liu, G.; Yang, Z.; Li, Y.; Liu, J.; Zhang, S. Number of Polyploid Giant Cancer Cells and Expression of EZH2 Are Associated with VM Formation and Tumor Grade in Human Ovarian Tumor. Biomed. Res. Int. 2014, 2014, 903542. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S.; Qu, J.; Zhang, M.; Li, Y.; Zhang, X.; Zhang, S. Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J. Cancer 2017, 8, 469–478. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Y.; Fei, F.; Wang, X.; Li, C.; Liu, K.; Du, J.; Cao, Y.; Zhang, S. Clinical Characteristics and Preliminary Morphological Observation of 47 Cases of Primary Anorectal Malignant Melanomas. Melanoma Res. 2018, 28, 592–599. [Google Scholar] [CrossRef]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting Mitochondrial Metabolism for Precision Medicine in Cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Piecyk, M.; Ferraro-Peyret, C.; Laville, D.; Perros, F.; Chaveroux, C. Novel Insights into the GCN2 Pathway and Its Targeting. Therapeutic Value in Cancer and Lessons from Lung Fibrosis Development. FEBS J. 2024. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, J.; Li, X.; Niu, N.; Liu, Y.; Hajek, R.A.; Peng, G.; Westin, S.; Sood, A.K.; Liu, J. Targeting Polyploid Giant Cancer Cells Potentiates a Therapeutic Response and Overcomes Resistance to PARP Inhibitors in Ovarian Cancer. Sci. Adv. 2023, 9, eadf7195. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krotofil, M.; Tota, M.; Siednienko, J.; Donizy, P. Emerging Paradigms in Cancer Metastasis: Ghost Mitochondria, Vasculogenic Mimicry, and Polyploid Giant Cancer Cells. Cancers 2024, 16, 3539. https://doi.org/10.3390/cancers16203539
Krotofil M, Tota M, Siednienko J, Donizy P. Emerging Paradigms in Cancer Metastasis: Ghost Mitochondria, Vasculogenic Mimicry, and Polyploid Giant Cancer Cells. Cancers. 2024; 16(20):3539. https://doi.org/10.3390/cancers16203539
Chicago/Turabian StyleKrotofil, Mateusz, Maciej Tota, Jakub Siednienko, and Piotr Donizy. 2024. "Emerging Paradigms in Cancer Metastasis: Ghost Mitochondria, Vasculogenic Mimicry, and Polyploid Giant Cancer Cells" Cancers 16, no. 20: 3539. https://doi.org/10.3390/cancers16203539
APA StyleKrotofil, M., Tota, M., Siednienko, J., & Donizy, P. (2024). Emerging Paradigms in Cancer Metastasis: Ghost Mitochondria, Vasculogenic Mimicry, and Polyploid Giant Cancer Cells. Cancers, 16(20), 3539. https://doi.org/10.3390/cancers16203539