
Insights into Metabolic Reprogramming in Tumor Evolution and Therapy

 ,
,  , , , , , , and
, , , , , , and 
Simple Summary
Abstract
1. Introduction
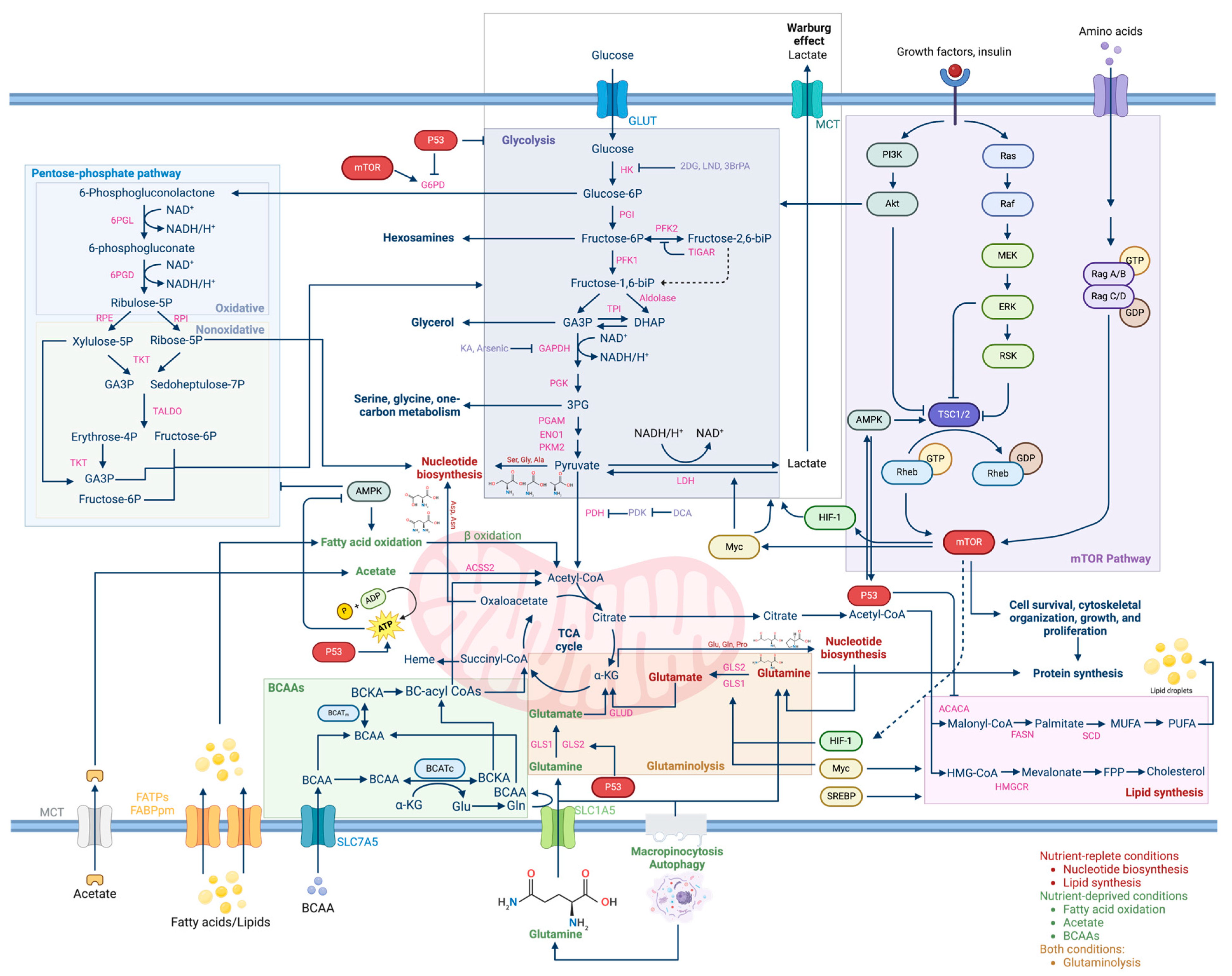
2. Metabolic Reprogramming in Cancer Cells
3. Metabolic Pathways and Genetic Dysfunctions in Cancer

{kind=link}
{kind=link}
| Metabolism | Oncogenic Protein | Metabolic Enzyme Targets | Mechanisms and Phenotype |
|---|---|---|---|
| Glycolysis, glutaminolysis, and amino acid synthesis | Myc | GLUT, HK2, and PFK [40,41]; LDH and MCT1 [42,43]; SLC1A5 and SLC38A5 [44]; GLS [45]; GLUD and transaminase [46,47]; G6PD and TKT [48] | Gain-of-function mutation enhances cell cycle progression and metabolism in cancer by upregulating the expression of glucose transporters and the majority of glycolytic enzymes, promoting glycolysis and glutaminolysis. |
| Glycolysis, tricarboxylic acid cycle, and fatty acid oxidation | p53 | GLUT1/4 [49]; TIGAR [50]; LDH and PDH [51]; CPT1 and LPIN1 [52,53] | A loss of p53 alters the metabolism in cancer cells by downregulating several enzymes and transporters inhibiting mitochondrial respiration, glycolysis, and apoptosis. |
| Fatty acid synthesis and glycolysis | PTEN | PI3K/AKT [8] | Mutations or a loss of PTEN result in negative regulation of the PI3K/AKT signaling pathway and in turn intracellular metabolic reprogramming, promoting the growth and proliferation of cancer cells. |
| Glycolysis, glutaminolysis, and amino acid synthesis | Ras | PI3K/AKT/mTOR [8]; RAF/MEK/ERK [8]; Myc [8]; | Oncogenic mutations lead to the upregulation of enzymes, resulting in tumor metabolic reprogramming and promotion of cell proliferation and survival. |
| Glycolysis and fatty acid synthesis | PIK3CA | PI3K/AKT [8] | Mutations lead to the activation of the PI3K/AKT pathway and enhance intracellular signal transduction, which leads to subsequent metabolic reprogramming of cancer cells. |
| Glycolysis and glutaminolysis | EGFR | PI3K/AKT; RAF/MEK/ERK [54] | EGFR signaling pathways activate lipogenesis through PI3K/AKT and MAPK pathways, leading to increased de novo lipid synthesis and alterations in lipid metabolism that support cancer cell growth and proliferation. |
| Glycolysis | PDK1 | PDHC [55] | Activation promotes a shift from oxidative phosphorylation to glycolysis by inhibiting the pyruvate dehydrogenase complex, thereby redirecting cellular metabolism to support tumorigenesis and metastasis. |
| Glycolysis, de novo lipogenesis, and protein synthesis | NF1 | Neurofibromin [56] | Loss-of-function mutations alter neurofibromin expression, increase RAS and PI3K/AKT pathway signaling, constraining oxidative ATP production, restrict energetic flexibility, and increase glutamine influx into TCA intermediates, expanding lipid pools (especially triglycerides) and altering the synergy between metabolic inhibitors and traditional targeted inhibitors. |
| Glycolysis, tricarboxylic acid cycle, and fatty acid synthesis | HIF-1α | HK2, PDK1, LDHA [57,58] | In response to hypoxia, HIF-1α upregulates the activation of genes involved in glycolysis and metabolism, cell proliferation, angiogenesis, invasion, and metastasis. |
| Glycolysis, protein synthesis, and lipid metabolism | TSC2 | Rheb [59] | Loss-of-function mutations lead to abnormal activation of the mTOR pathway through increased Rheb activity. This results in altered protein synthesis, lipid metabolism, and glucose metabolism. |
| Glycolysis and fatty acid oxidation | SIRT1 | β-catenin [60] | When upregulated in response to glucose deficiency and oxidative stress, SIRT1 deacetylates β-catenin, causing its translocation from the nucleus to the cytoplasm, attenuates glycolysis, and positively correlates with fatty acid oxidation. This promotes the shift in glycolipid metabolism, facilitating tumor development in colorectal carcinoma. |
| Glycolysis, amino acid metabolism, lipid metabolism, and bile acid metabolism | YAP/TAZ | GLUT3 [61]; HK2 [62]; PFKFB3 [63]; SLC1A5 and SLC7A5 [64,65]; GOT1 and PSAT1 [66,67] | Overactivation promotes glycolysis by increasing GLUT3, HK2, and PFKFB3 expression, enhancing glutamine metabolism by upregulating transporters and enzymes. It modulates lipid and bile acid accumulation, aiding cancer metastasis. |
| Glycolysis and fatty acid oxidation | LKB1 | AMPK 1/2, MARK 1/2/3/4, SIK 1/2/3, NUAK 1/2, and SNRK [68] | LKB1 deficiency leads to the dysregulation of cellular energy homeostasis and contributes to the metabolic reprogramming of cancer cells, which induces excess glycolysis, the primary energy supply for cancer cells, enhancing their cellular growth and proliferation. |
| Glycolysis and tricarboxylic acid cycle | FH | PDHA1 [69] | Mutations lead to metabolic reprogramming characterized by increased glycolytic flux, a shift to glutamine as the primary carbon source, the induction of pseudohypoxia, alterations in lipid biosynthesis, and enhanced arginine metabolism, collectively promoting a favorable environment for cancer progression. |
| Glycolysis | PGAM1 | Wnt/β-catenin [70]; BCL-2, BAX, and caspase-3 [71]; ACTA2 [72,73] | Overexpression results in dysregulated glycolysis, leading to altered bioenergetics characterized by increased aerobic glycolysis (Warburg effect), thereby promoting cancer cell growth, proliferation, and invasion. |
| Tricarboxylic acid cycle | IDH1/2 | TET2 [74]; JMJD2A [75] | Mutations lead to altered enzyme function, promoting the production of 2-hydroxyglutarate (2HG) which inhibits enzymes that cause differentiation in hematopoietic cells and histone methylation. |
4. The Tumor Microenvironment
5. Mitochondria
6. Impact of Metabolic Reprogramming on Cancer Progression
7. Interdisciplinary Approaches in Cancer Metabolism Research
8. Therapeutic Implications and Future Directions
9. Challenges and Limitations in Understanding and Targeting Cancer Metabolism
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaidai, O.; Yan, P.; Xing, Y. Future world cancer death rate prediction. Sci. Rep. 2023, 13, 303. [Google Scholar] [CrossRef]
- Pineda, E.; Benavente, R.; Gimmen, M.Y.; DeVille, N.V.; Taparra, K. Cancer Disparities among Pacific Islanders: A Review of Sociocultural Determinants of Health in the Micronesian Region. Cancers 2023, 15, 1392. [Google Scholar] [CrossRef]
- Syrnioti, G.; Eden, C.M.; Johnson, J.A.; Alston, C.; Syrnioti, A.; Newman, L.A. Social Determinants of Cancer Disparities. Ann. Surg. Oncol. 2023, 30, 8094–8104. [Google Scholar] [CrossRef]
- Aguadé-Gorgorió, G.; Costa, J.; Solé, R. An oncospace for human cancers. BioEssays 2023, 45, 2200215. [Google Scholar] [CrossRef]
- Imodoye, S.O.; Adedokun, K.A.; Bello, I.O. From complexity to clarity: Unravelling tumor heterogeneity through the lens of tumor microenvironment for innovative cancer therapy. Histochem. Cell Biol. 2024, 161, 299–323. [Google Scholar] [CrossRef]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Chen, Y.; Oyang, L.; Lin, J.; et al. Metabolic reprogramming and epigenetic modifications in cancer: From the impacts and mechanisms to the treatment potential. Exp. Mol. Med. 2023, 55, 1357–1370. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm 2023, 4, e218. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Ferro, M.; Terracciano, D.; Tataru, O.S.; Battaglia, M.; Ditonno, P.; Lacerelli, G. Emerging hallmarks of metabolic reprogramming in prostate cancer. Int. J. Mol. Sci. 2023, 24, 910. [Google Scholar] [CrossRef]
- Lee, J.; Shin, D.; Roh, J.L. Lipid metabolism alterations and ferroptosis in cancer: Paving the way for solving cancer resistance. Eur. J. Pharmacol. 2023, 941, 175497. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, C.; Martincuks, A.; Herrmann, A.; Yu, H. STAT proteins in cancer: Orchestration of metabolism. Nat. Rev. Cancer 2023, 23, 115–134. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Neugent, M.L.; Goodwin, J.; Sankaranarayanan, I.; Yetkin, C.E.; Hsieh, M.H.; Kim, J.W. A new perspective on the heterogeneity of cancer glycolysis. Biomol. Ther. 2018, 26, 10. [Google Scholar] [CrossRef]
- Abbaszadeh, Z.; Çeşmeli, S.; Avcı, Ç.B. Crucial players in glycolysis: Cancer progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Sun, L.; Yuan, Y.; Zhu, Y. Potential mechanisms of cancer-associated fibroblasts in therapeutic resistance. Biomed. Pharmacother. 2023, 166, 115425. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.a.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef]
- Tsai, C.H.; Chuang, Y.M.; Li, X.; Yu, Y.R.; Tzeng, S.F.; Teoh, S.T.; Lindblad, K.E.; Matteo, M.D.; Cheng, W.-C.; Hsueh, P.-C.; et al. Immunoediting instructs tumor metabolic reprogramming to support immune evasion. Cell Metab. 2023, 35, 118–133. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; ven der Windt, G.J.W.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Chen, J.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Et Biophys. Acta (BBA)–Rev. Cancer 2012, 1826, 370–384. [Google Scholar] [CrossRef]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef]
- Schmidt, C.; Sciacovelli, M.; Frezza, C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin. Cell Dev. Biol. 2020, 98, 15–25. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting glutaminolysis: New perspectives to understand cancer development and novel strategies for potential target therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Shah, R.; Chen, S. Metabolic signaling cascades prompted by glutaminolysis in cancer. Cancers 2020, 12, 2624. [Google Scholar] [CrossRef]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid β–oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty acid oxidation mediated by Acyl–CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism–related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Roy, D.; Sheng, G.Y.; Herve, S.; Carvalho, E.; Mahanty, A.; Yuan, S.; Sun, L. Interplay between cancer cell cycle and metabolism: Challenges, targets and therapeutic opportunities. Biomed. Pharmacother. 2017, 89, 288–296. [Google Scholar] [CrossRef]
- Benfeitas, R.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. New challenges to study heterogeneity in cancer redox metabolism. Front. Cell Dev. Biol. 2017, 5, 65. [Google Scholar] [CrossRef]
- Biswas, D.; Khan, M.W. New techniques in understanding cancer biology and metabolism. Technol. Cancer Res. Treat. 2020, 19, 1533033820943248. [Google Scholar] [CrossRef]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef]
- Lewis, B.C.; Prescott, J.E.; Campbell, S.E.; Shim, H.; Orlowski, R.Z.; Dang, C.V. Tumor induction by the c-Myc target genes rcl and lactate dehydrogenase A. Cancer Res. 2000, 60, 6178–6183. [Google Scholar]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F. Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissin, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A.; et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Munger, J.; Green, D.R. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousdan, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S.; et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef]
- Eltayeb, K.; La Monica, S.; Tiseo, M.; Alfieri, R.; Fumarola, C. Reprogramming of lipid metabolism in lung cancer: An overview with focus on EGFR–mutated non–small cell lung cancer. Cells 2022, 11, 413. [Google Scholar] [CrossRef]
- Padder, R.A.; Bhat, Z.I.; Ahmad, Z.; Singh, N.; Husain, M. DRP1 promotes BRAFV600E–driven tumor progression and metabolic reprogramming in colorectal cancer. Front. Oncol. 2021, 10, 592130. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1α. Cancer Res. 2010, 70, 10351–10361. [Google Scholar] [CrossRef]
- Cho, J.; Lee, J.; Kim, J.; Kim, S.T.; Lee, S.; Kim, S.Y.; Ha, S.Y.; Park, C.-K.; Lim, H.Y. Loss of tuberous sclerosis complex 2 (TSC2) as a predictive biomarker of response to mTOR inhibitor treatment in patients with hepatocellular carcinoma. Transl. Oncol. 2016, 9, 466–471. [Google Scholar] [CrossRef]
- Wei, Z.; Xia, J.; Li, J.; Cai, J.; Shan, J.; Zhang, C.; Zhang, L.; Wang, T.; Qian, C.; Liu, L. SIRT1 promotes glucolipid metabolic conversion to facilitate tumor development in colorectal carcinoma. Int. J. Biol. Sci. 2023, 19, 1925–1940. [Google Scholar] [CrossRef]
- Cosset, E.; Ilmjärv, S.; Dutoit, V.; Elliott, K.; von Schalscha, T.; Camargo, M.F.; Reiss, A.; Moroishi, T.; Seguin, L.; Gomez, G.; et al. Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell 2017, 32, 856–868. [Google Scholar] [CrossRef]
- Song, L.; Tang, H.; Liao, W.; Luo, X.; Li, Y.; Chen, T.; Zhang, X. FOXC2 positively regulates YAP signaling and promotes the glycolysis of nasopharyngeal carcinoma. Exp. Cell Res. 2017, 357, 17–24. [Google Scholar] [CrossRef]
- Zheng, X.; Han, H.; Liu, G.P.; Ma, Y.X.; Pan, R.L.; Sang, L.J.; Li, R.-H.; Yang, L.-J.; Marks, J.R.; Wang, W.; et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017, 36, 3325–3335. [Google Scholar] [CrossRef]
- Hansen, C.G.; Ng YL, D.; Lam WL, M.; Plouffe, S.W.; Guan, K.L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015, 25, 1299–1313. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Wang, S.; Shiuan, E.; Brantley-Sieders, D.M.; Kim, L.C.; Reynolds, A.B.; Chen, J. The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Sci. Signal. 2017, 10, eaan4667. [Google Scholar] [CrossRef]
- Yang, C.S.; Stampouloglou, E.; Kingston, N.M.; Zhang, L.; Monti, S.; Varelas, X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018, 19, e43577. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, Q.; Sun, Q.; Xu, Z.; Zhou, H.; Wang, Y. LKB1 deficiency–induced metabolic reprogramming in tumorigenesis and non–neoplastic diseases. Mol. Metab. 2021, 44, 101131. [Google Scholar] [CrossRef]
- Gonçalves, E.; Sciacovelli, M.; Costa, A.S.; Tran, M.G.; Johnson, T.I.; Machado, D.; Frezza, C.; Saez-Rodriguez, J. Post–translational regulation of metabolism in fumarate hydratase deficient cancer cells. Metab. Eng. 2018, 45, 149–157. [Google Scholar] [CrossRef]
- Liu, X.; Tan, X.; Liu, P.; Wu, Y.; Qian, S.; Zhang, X. Phosphoglycerate mutase 1 (PGAM1) promotes pancreatic ductal adenocarcinoma (PDAC) metastasis by acting as a novel downstream target of the PI3K/Akt/mTOR pathway. Oncol. Res. 2018, 26, 1123. [Google Scholar] [CrossRef]
- Wen, Y.A.; Zhou, B.W.; Lv, D.J.; Shu, F.P.; Song, X.L.; Huang, B.; Wang, C.; Zhao, S.C. Phosphoglycerate mutase 1 knockdown inhibits prostate cancer cell growth, migration, and invasion. Asian J. Androl. 2018, 20, 178–183. [Google Scholar]
- Zhang, D.; Jin, N.; Sun, W.; Li, X.; Liu, B.; Xie, Z.; Qu, J.; Xu, J.; Yang, X.; Su, Y.; et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 2017, 36, 2900–2909. [Google Scholar] [CrossRef]
- Ishikawa, M.; Inoue, T.; Shirai, T.; Takamatsu, K.; Kunihiro, S.; Ishii, H.; Nishikata, T. Simultaneous expression of cancer stem cell-like properties and cancer-associated fibroblast-like properties in a primary culture of breast cancer cells. Cancers 2014, 6, 1570–1578. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Alam, M.M.; Lal, S.; FitzGerald, K.E.; Zhang, L. A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin. Transl. Med. 2016, 5, 3. [Google Scholar] [CrossRef]
- Xu, J.; Yu, T.; Zois, C.E.; Cheng, J.X.; Tang, Y.; Harris, A.L.; Huang, W.E. Unveiling cancer metabolism through spontaneous and coherent Raman spectroscopy and stable isotope probing. Cancers 2021, 13, 1718. [Google Scholar] [CrossRef]
- Danzi, F.; Pacchiana, R.; Mafficini, A.; Scupoli, M.T.; Scarpa, A.; Donadelli, M.; Fiore, A. To metabolomics and beyond: A technological portfolio to investigate cancer metabolism. Signal Transduct. Target. Ther. 2023, 8, 137. [Google Scholar] [CrossRef]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting energy metabolism in cancer stem cells: Progress and challenges in leukemia and solid tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef]
- Grima–Reyes, M.; Martinez–Turtos, A.; Abramovich, I.; Gottlieb, E.; Chiche, J.; Ricci, J.E. Physiological impact of in vivo stable isotope tracing on cancer metabolism. Mol. Metab. 2021, 53, 101294. [Google Scholar] [CrossRef]
- de Sá Junior, P.L.; Câmara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The roles of ROS in cancer heterogeneity and therapy. Oxidative Med. Cell. Longev. 2017, 2017, 2467940. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, X.; Chen, H.N.; Wang, K. Amino acid metabolic reprogramming in tumor metastatic colonization. Front. Oncol. 2023, 13, 1123192. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 175. [Google Scholar] [CrossRef]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 735–745. [Google Scholar] [CrossRef]
- Chong SJ, F.; Low IC, C.; Pervaiz, S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion 2014, 19, 39–48. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef]
- Rudin, C.M.; Salgia, R.; Wang, X.; Hodgson, L.D.; Masters, G.A.; Green, M.; Vokes, E.E. Randomized phase II Study of carboplatin and etoposide with or without the bcl-2 antisense oligonucleotide oblimersen for extensive-stage small-cell lung cancer: CALGB 30103. J. Clin. Oncol. 2008, 26, 870–876. [Google Scholar] [CrossRef]
- Yan, C.; Gu, J.; Zhang, Y.; Ma, K.; Lee, R.J. Efficient delivery of the Bcl-2 antisense oligonucleotide G3139 via nucleus-targeted aCD33-NKSN nanoparticles. Int. J. Pharm. 2022, 625, 122074. [Google Scholar] [CrossRef]
- Ferreira-da-Silva, A.; Valacca, C.; Rios, E.; Pópulo, H.; Soares, P.; Sobrinho-Simoes, M.; Scorrano, L.; Maximo, V.; Campello, S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef]
- Schimmer, A.D.; O’Brien, S.; Kantarjian, H.; Brandwein, J.; Cheson, B.D.; Minden, M.D.; Kamel-Reid, S.; Berger, M.; Viallet, J.; Borthakur, G.; et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 8295–8301. [Google Scholar] [CrossRef]
- Kiany, S.; Harrison, D.; Gordon, N. The histone deacetylase inhibitor Entinostat/Syndax 275 in osteosarcoma. Curr. Adv. Osteosarcoma Clin. Perspect. Past Present Future 2020, 1257, 75–83. [Google Scholar]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Letai, A.; Chyla, B.; Potluri, J.; Pollyea, D.A.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Secrist, J.P.; Clark, E.A.; Wilson, D.M.; Hird, A.W.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Zhou, C.; Yang, Z.F.; Sun, B.Y.; Yi, Y.; Wang, Z.; Zhou, J.; Fan, J.; Gan, W.; Ren, N.; Qiu, S.J. Lenvatinib induces immunogenic cell death and triggers toll-like receptor-3/4 ligands in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2023, 10, 697–712. [Google Scholar] [CrossRef]
- Lin, X.H.; Qiu, B.Q.; Ma, M.; Zhang, R.; Hsu, S.J.; Liu, H.H.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; et al. Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis 2020, 9, 67. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Inoue-Yamauchi, A.; Oda, H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 81–85. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Jayasena, S. The role of reprogrammed glucose metabolism in cancer. Metabolites 2023, 13, 345. [Google Scholar] [CrossRef]
- Lei, P.; Wang, W.; Sheldon, M.; Sun, Y.; Yao, F.; Ma, L. Role of glucose metabolic reprogramming in breast cancer progression and drug resistance. Cancers 2023, 15, 3390. [Google Scholar] [CrossRef]
- Xie, S.Z.; Pan, J.J.; Xu, J.F.; Zhu, W.W.; Qin, L.X. The critical function of metabolic reprogramming in cancer metastasis. Aging Cancer 2022, 3, 20–43. [Google Scholar] [CrossRef]
- Jin, H.R.; Wang, J.; Wang, Z.J.; Xi, M.J.; Xia, B.H.; Deng, K.; Yang, J.L. Lipid metabolic reprogramming in tumor microenvironment: From mechanisms to therapeutics. J. Hematol. Oncol. 2023, 16, 103. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one–carbon pathway in cancer. J. Cell Biol. 2019, 219, e201907022. [Google Scholar] [CrossRef]
- Hipólito, A.; Martins, F.; Mendes, C.; Lopes-Coelho, F.; Serpa, J. Molecular and metabolic reprogramming: Pulling the strings toward tumor metastasis. Front. Oncol. 2021, 11, 656851. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Yu, D. Metabolic reprogramming of chemoresistant cancer cells and the potential significance of metabolic regulation in the reversal of cancer chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef]
- Jang, W.J.; Choi, B.; Song, S.H.; Lee, N.; Kim, D.J.; Lee, S.; Jeong, C.H. Multi-omics analysis reveals that ornithine decarboxylase contributes to erlotinib resistance in pancreatic cancer cells. Oncotarget 2017, 8, 92727. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Ren, S.; Sun, D.; Huang, H.Y.; Wang, H.; Jin, Y.; Li, F.; Zheng, C.; Yang, L.; et al. Branched-chain amino acid metabolic reprogramming orchestrates drug resistance to EGFR tyrosine kinase inhibitors. Cell Rep. 2019, 28, 512–525. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Siddiqui, S.; Ur Rehman, A.; Siddiqui, F.A.; Singh, P.; Kumar, B.; Saluja, D. Multiomics integrative analysis reveals antagonistic roles of CBX2 and CBX7 in metabolic reprogramming of breast cancer. Mol. Oncol. 2021, 15, 1450–1465. [Google Scholar] [CrossRef]
- Weiss, R.H. Metabolomics and metabolic reprogramming in kidney cancer. Semin. Nephrol. 2018, 38, 175–182. [Google Scholar] [CrossRef]
- Draguet, A.; Tagliatti, V.; Colet, J.M. Targeting Metabolic Reprogramming to Improve Breast Cancer Treatment: An In Vitro Evaluation of Selected Metabolic Inhibitors Using a Metabolomic Approach. Metabolites 2021, 11, 556. [Google Scholar] [CrossRef]
- Li, B.; Sui, L. Metabolic reprogramming in cervical cancer and metabolomics perspectives. Nutr. Metab. 2021, 18, 93. [Google Scholar] [CrossRef]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.M.; Journe, F. Metabolic reprogramming in metastatic melanoma with acquired resistance to targeted therapies: Integrative metabolomic and proteomic analysis. Cancers 2020, 12, 1323. [Google Scholar] [CrossRef]
- Klotz, L.O.; Carlberg, C. Nutrigenomics and redox regulation: Concepts relating to the Special Issue on nutrigenomics. Redox Biol. 2023, 68, 102920. [Google Scholar] [CrossRef]
- Malcomson, F.C.; Mathers, J.C. Translation of nutrigenomic research for personalised and precision nutrition for cancer prevention and for cancer survivors. Redox Biol. 2023, 62, 102710. [Google Scholar] [CrossRef]
- Rahman, A.; Chakraborty, S.; Kabir, Y. Harnessing personalized nutrigenomics for cancer prevention and treatment through diet-gene interaction. In Functional Foods in Cancer Prevention and Therapy; Kabir, Y., Ed.; Academic Press: New York, NY, USA, 2020; pp. 387–403. [Google Scholar]
- Aghakhani, S.; Zerrouk, N.; Niarakis, A. Metabolic reprogramming of fibroblasts as therapeutic target in rheumatoid arthritis and cancer: Deciphering key mechanisms using computational systems biology approaches. Cancers 2020, 13, 35. [Google Scholar] [CrossRef]
- Yizhak, K.; Le Dévédec, S.E.; Rogkoti, V.M.; Baenke, F.; De Boer, V.C.; Frezza, C.; Schulze, A.; van de Water, B.; Ruppin, E. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol. Syst. Biol. 2014, 10, 744. [Google Scholar] [CrossRef]
- Meric–Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Tannir, N.M.; Owonikoko, T.K.; Haas, N.B.; Voss, M.H.; et al. CB–839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. J. Clin. Oncol. 2019, 37 (Suppl. S7), 549. [Google Scholar] [CrossRef]
- Mueller, C.; Al–Batran, S.; Jaeger, E.; Schmidt, B.; Bausch, M.; Unger, C.; Sethuraman, N. A phase IIa study of PEGylated glutaminase (PEG–PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. J. Clin. Oncol. 2008, 26 (Suppl. S15), 2533. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Navarro, C.; Ortega, A.; Santeliz, R.; Garrido, B.; Chacín, M.; Galban, N.; Vera, I.; De Sanctis, J.B.; Bermúdez, V. Metabolic reprogramming in cancer cells: Emerging molecular mechanisms and novel therapeutic approaches. Pharmaceutics 2022, 14, 1303. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1. [Google Scholar]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic rewiring faces tumor heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, T.J.; Xu, Y.; Ding, R.; Wang, Y.P.; Jiang, Y.Z.; Shao, Z.M. Emerging therapies in cancer metabolism. Cell Metab. 2023, 35, 1283–1303. [Google Scholar] [CrossRef]
- Roy, S.; Dukic, T.; Bhandary, B.; Tu, K.J.; Molitoris, J.; Ko, Y.H.; Shukla, H.D. 3-Bromopyruvate inhibits pancreatic tumor growth by stalling glycolysis, and dismantling mitochondria in a syngeneic mouse model. Am. J. Cancer Res. 2022, 12, 497. [Google Scholar]
- Zhang, H.; Shangguan, F.; Zhang, L.; Ma, N.; Song, S.; Ma, L.; Liu, C.; Liu, M.; An, J.; Li, H.; et al. A novel mechanism of 6-methoxydihydroavicine in suppressing ovarian carcinoma by disrupting mitochondrial homeostasis and triggering ROS/MAPK mediated apoptosis. Front. Pharmacol. 2023, 14, 1093650. [Google Scholar] [CrossRef]
- Guan, X.; Morris, M.E. Pharmacokinetics of the monocarboxylate transporter 1 inhibitor AZD3965 in mice: Potential enterohepatic circulation and target-mediated disposition. Pharm. Res. 2020, 37, 5. [Google Scholar] [CrossRef]
- Li, W.; Zheng, M.; Wu, S.; Gao, S.; Yang, M.; Li, Z.; Min, Q.; Sun, W.; Chen, L.; Xiang, G.; et al. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36, 58. [Google Scholar] [CrossRef]
- Harding, J.J.; Telli, M.; Munster, P.; Voss, M.H.; Infante, J.R.; DeMichele, A.; Dunphy, M.; Le, M.H.; Molineaux, C.; Orford, K.; et al. A phase I dose-escalation and expansion study of telaglenastat in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 2021, 27, 4994–5003. [Google Scholar] [CrossRef]
- Saddiq, A.A.; El-Far, A.H.; Mohamed, S.A.; Almaghrabi, O.A.; Mousa, S.A. Curcumin and thymoquinone combination attenuates breast cancer cell lines’ progression. Integr. Cancer Ther. 2022, 21, 15347354221099537. [Google Scholar] [CrossRef]
- Pan, L.; Feng, F.; Wu, J.; Fan, S.; Han, J.; Wang, S.; Yang, L.; Liu, W.; Wang, C.; Xu, K. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 2022, 181, 106270. [Google Scholar] [CrossRef]
- Dogra, N.; Kumar, A.; Mukhopadhyay, T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, 1–5. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 433–444. [Google Scholar] [CrossRef]
- Furuse, J.; Ikeda, M.; Ueno, M.; Furukawa, M.; Morizane, C.; Takehara, T.; Nishina, T.; Todaka, A.; Okano, N.; Hara, K.; et al. A Phase II Placebo-Controlled Study of the Effect and Safety of Nanvuranlat in Patients with Advanced Biliary Tract Cancers Previously Treated by Systemic Chemotherapy. Clin. Cancer Res. 2024, 30, 3990–3995. [Google Scholar] [CrossRef]
- Furuse, J.; Ikeda, M.; Ueno, M.; Furukawa, M.; Morizane, C.; Takehara, T.; Nishina, T.; Todaka, A.; Okano, N.; Hara, K.; et al. Nanvuranlat, an L-type amino acid transporter (LAT1) inhibitor for patients with pretreated advanced refractory biliary tract cancer (BTC): Primary endpoint results of a randomized, double-blind, placebo-controlled phase 2 study. J. Clin. Oncol. 2023, 41 (Suppl. S4), 494. [Google Scholar] [CrossRef]
- Coulter, D.W.; Chhonker, Y.S.; Kumar, D.; Kesherwani, V.; Aldhafiri, W.N.; McIntyre, E.M.; Alexander, G.; Ray, S.; Joshi, S.S.; Li, R.; et al. Marinopyrrole derivative MP1 as a novel anti–cancer agent in group 3 MYC–amplified Medulloblastoma. J. Exp. Clin. Cancer Res. 2024, 43, 18. [Google Scholar] [CrossRef]
- McGuire, T.R.; Coulter, D.W.; Bai, D.; Sughroue, J.A.; Li, J.; Yang, Z.; Qiao, Z.; Liu, Y.; Murry, D.J.; Chhonker, Y.S.; et al. Effects of novel pyrrolomycin MP1 in MYCN amplified chemoresistant neuroblastoma cell lines alone and combined with temsirolimus. BMC Cancer 2019, 19, 837. [Google Scholar] [CrossRef]
- Seliger, C.; Meyer, A.L.; Leidgens, V.; Rauer, L.; Moeckel, S.; Jachnik, B.; Proske, J.; Dettmer, K.; Rothhammer-Hampl, T.; Kaulen, L.D.; et al. Metabolic heterogeneity of brain tumor cells of proneural and mesenchymal origin. Int. J. Mol. Sci. 2022, 23, 11629. [Google Scholar] [CrossRef]
- Li, J.; Li, S.; Guo, J.; Li, Q.; Long, J.; Ma, C.; Ding, Y.; Yan, C.; Li, L.; Wu, Z.; et al. Natural product Micheliolide (MCL) irreversibly activates pyruvate kinase M2 and suppresses leukemia. J. Med. Chem. 2018, 61, 4155–4164. [Google Scholar] [CrossRef]
- Sardesai, S.D.; Thomas, A.; Gallagher, C.; Lynce, F.; Ottaviano, Y.L.; Ballinger, T.J.; Schneider, B.P.; Storniolo, A.M.; Bauchle, A.; Althouse, S.K.; et al. Inhibiting fatty acid synthase with omeprazole to improve efficacy of neoadjuvant chemotherapy in patients with operable TNBC. Clin. Cancer Res. 2021, 27, 5810–5817. [Google Scholar] [CrossRef]
- An, J.; Zhang, Y.; He, J.; Zang, Z.; Zhou, Z.; Pei, X.; Zheng, X.; Zhang, W.; Yang, H.; Li, S. Lactate dehydrogenase A promotes the invasion and proliferation of pituitary adenoma. Sci. Rep. 2017, 7, 4734. [Google Scholar] [CrossRef]
- Ruszkowski, M.; Sekula, B.; Ruszkowska, A.; Contestabile, R.; Nogues, I.; Angelaccio, S.; Szczpaniak, A.; Dauter, Z. Structural basis of methotrexate and pemetrexed action on serine hydroxymethyltransferases revealed using plant models. Sci. Rep. 2019, 9, 19614. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin inhibits tumor growth in mice by suppressing pyruvate kinase M2–mediated aerobic glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef]
- Kelly, W.; Diaz Duque, A.E.; Michalek, J.; Konkel, B.; Caflisch, L.; Chen, Y.; Pathuri, S.C.; Madhusudanannair-Kunnuparampil, V.; Floyd, J., II; Brenner, A. Phase II investigation of TVB-2640 (denifanstat) with bevacizumab in patients with first relapse high-grade astrocytoma. Clin. Cancer Res. 2023, 29, 2419–2425. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185. [Google Scholar] [CrossRef]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, D.S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef]
- Spinello, I.; Labbaye, C.; Saulle, E. Metabolic function and Therapeutic Potential of CD147 for Hematological Malignancies: An overview. Int. J. Mol. Sci. 2024, 24, 9178. [Google Scholar] [CrossRef]
- Zuo, F.; Yu, J.; He, X. Single-cell metabolomics in hematopoiesis and hematological malignancies. Front. Oncol. 2022, 12, 931393. [Google Scholar] [CrossRef]

| Compound or Drug | Mechanism | Target | Specific Cancer | Findings | Clinical Pipeline | Reference |
|---|---|---|---|---|---|---|
| Entinostat | Inhibits the function of histone deacetylases leading to more relaxed chromatin and gene expression | HDAC1/3 | Osteosarcoma | Shown to upregulate the expression of Fas, leading to decreased pulmonary metastasis and improved outcomes | Preclinical | [93] |
| Venetoclax | Selective BCL-2 inhibitor that releases pro-apoptotic proteins | BCL-2 | Acute myeloid leukemia | Overall response rate of 67% when combined with hypomethylating agents in elderly AML patients | FDA-approved, Phase III | [94] |
| MCL-1- specific inhibitor AZD5991 | Binds directly to Mcl-1 and induces rapid apoptosis in cancer cells by activating the Bak-dependent mitochondrial apoptotic pathway | MCL-1 | Myeloma and acute myeloid leukemia | Induces apoptosis in >80% of multiple MCL-1-dependent myeloma cell lines in vitro | Preclinical | [95] |
| Lenvatinib | Induces immunogenic cell death and activates TLR 3/4 ligands, enhancing immune response | TLR 3/4 | Hepatocellular carcinoma | Triggers immunogenic cell death, enhancing anti-tumor immunity; increases T-cell activation and infiltration by 40% in HCC models | Preclinical | [96] |
| Obatoclax | Directly induces apoptosis through the activation of BAX/BAK following their release from the pro-survival BCL-2 members | Pan-BCL -2 family | Hematologic malignancies | Demonstrates tolerability and partial responses in patients with chronic lymphocytic leukemia and Hodkins lymphoma | Phase I clinical trial | [97] |
| Metabolism | Compound or Drug | Target | Mechanism | Findings | Clinical Pipeline | Reference |
|---|---|---|---|---|---|---|
| Glycolysis | 3-Bromopyruvate | Hexokinase II | Irreversibly alkylates HK2, resulting in the disruption of glucose metabolism, leading to cancer cell death | 3-BP (20 mg/kg) reduced tumor size by 75–80% in animals and induced apoptosis and necrosis in drug-treated tumor tissues | Animal Studies | [128] |
| TCA cycle | 6-Methoxydihydroavicine (6-ME) | Oxaloacetic Acid Metabolism | Disrupts OAA metabolism, leading to ROS accumulation and resulting in disrupted mitochondrial homeostasis, ultimately driving apoptosis in ovarian cancer cells | 6-ME significantly reduced tumor growth in a nude mouse model of ovarian cancer without causing physiologically harmful effects on the animal | Animal Studies | [129] |
| Glycolysis | AZD3965 | MCT-1 | Inhibition of MCT1-mediated lactic acid efflux during T-cell lymphocyte proliferation | The drug showed rapid oral absorption, nearly complete bioavailability, nonlinear pharmacokinetics, and potential involvement in enterohepatic circulation (EHC), with evidence of target-mediated drug disposition (TMDD) | Phase I | [130] |
| Glycolysis | Benserazide (Benz) | Hexokinase II | Competitive and noncompetitive binding to selectively inhibit HK2 | In vivo, it suppresses tumor growth in mice without toxicity; when formulated as liposomal nanoparticles, Benz enhances tumor targeting and efficacy at lower doses | Animal Studies | [131] |
| Glutaminolysis | CB839 (Telaglenastat) | Glutamine oxidase | Block glutamine-to-glutamate conversion, reducing the number of immunosuppressive cells and reshaping the tumor microenvironment | The study established a recommended phase II dose (RP2D) for telaglenastat, demonstrating safety, strong GLS inhibition, and early anticancer activity, prompting further investigation | Phase I | [132] |
| Glycolysis | Curcumin (Cur) + Thymoquinone (TQ) | Caspase-3 and PI3K/AKT | Induces apoptosis and cell cycle arrest, and decreases proliferation, colony formation, and migration of MCF7 and MDA-MB-231 cells | Cur and TQ significantly inhibited cancer cell growth and migration, increased apoptosis (73.96% for Cur, 75.76% for Cur + TQ), and reduced S-phase values compared to controls | In vitro | [133] |
| Glycolysis | Demethylzeylasteral (DML) | Lactate |
Dose-dependent decrease in intracellular lactate levels Regulation of histone acetylation via H3K9la and H3K56la modification sites | DML treatment significantly inhibited tumor growth in vivo, as shown by slower tumor growth rates in treated groups compared to controls, and regulated Pan Kla expression, correlating with decreased cancer cell proliferation | In vitro | [134] |
| Glycolysis | Fenbendazole (FZ) |
Microtubules p53 Hexokinase II |
Disruption of microtubule dynamics Increases p53 translocation to mitochondria, which is suggested to induce cell death Inhibition of HK2 activity, leading to apoptosis | FZ administration significantly reduced tumor size and weight in A549 xenografted nude mice | Animal studies | [135] |
| TCA cycle | Ivosidenib | Isocitrate dehydrogenase 1 | Inhibits IDH1 catalysis of the oncometabolite 2HG that disrupts epigenetic regulation, blocks cellular differentiation, and contributes to tumorigenesis | Ivosidenib effectively suppresses plasma 2-HG in IDH1-mutated cholangiocarcinoma and chondrosarcoma, supporting a dose of 500 mg QD for advanced solid tumors | Phase I | [136] |
| Amino acid synthesis | JPH203 (Nanvuranlat) | L-type Amino Acid Transporter 1 | LAT1 inhibition | The drug showed significant improvement in progression-free survival in patients with advanced, refractory biliary tract cancers compared to placebo, with a disease control rate of 25%; the treatment was found to be safe and well tolerated | Phase II | [137,138] |
| Glycolysis | Marinopyrrole derivative MP1 | Myc and mTOR signaling | Modulate global gene expression and inhibit Myc-associated transcriptional targets including translation/mTOR targets. Inhibit tumor growth and Myc expression | MP1 is an orally bioavailable compound with favorable pharmacokinetics and pharmacodynamics, crossing the blood–brain barrier and achieving concentrations above IC50 in tumors, including in the brain, with good tolerability and no significant toxicity | Animal studies | [139,140] |
| Oxidative phosphorylation | Metformin | NADH | Increased flux of glucose carbons via the pentose phosphate pathway, leading to the inhibition of complex I (NADH:ubiquinone oxidoreductase) | Proneural BTICs respond better to metformin, while mesenchymal BTICs are more glycolytic and less responsive; glycolysis targeting may be more effective for mesenchymal BTICs. | Phase II | [141] |
| Glycolysis | Dimethylaminomicheliolide (DMAMCL), a Micheliolide derivative | Pyruvate kinase | Covalent binding at residue cysteine424 to promote tetramer formation and selectively activate PKM2 | DMAMCL significantly suppresses tumor growth in vivo by activating PKM2, showing potential as a novel anticancer therapeutic drug, with optimal effects observed at 10 μg/mL | Discovery | [142] |
| Fatty acid synthesis | Omeprazole | Fatty acid synthase (FASN), which is a rate-limiting enzyme in synthesizing fatty acids | Proton pump inhibitors selectively inhibit FASN activity and induce apoptosis in Triple-Negative Breast Cancer (TNBC) cell lines via AKT and HIF-1 under hypoxic stress, allowing for adaptations in the tumor microenvironment | Omeprazole, when added to neoadjuvant AC-T, can safely inhibit FASN and shows a promising pCR rate, though further confirmation is needed | Phase II | [143] |
| Glycolysis | Oxamate | Lactate Dehydrogenase A | Induces inhibition of LDHA which suppress glucose uptake, lactate secretion, invasion, and proliferation in GH3 cells via the downregulation of GLUT1 and MMP2 expression and the inhibition of the Akt-GSK-3β-cyclinD1 pathway | Oxamate significantly inhibits the invasion and proliferation of primary pituitary PA cells derived from patients after transsphenoidal resection, confirming its potential as a therapeutic agent against human-invasive PA cells | Discovery | [144] |
| One-carbon metabolism | Methotrexate (MTX), Pemetrexed (PTX) | Serine hydroxymethyltransferases | Inhibit growth of cancer cells by cutting off the supply of 5,10-meTHF (utilized for nucleotide biosynthesis and hyperactivated in cancer) | PTX binds deeper in SHMTs than MTX due to its unique P-moiety structure, making it a more potent inhibitor; polyglutamylation significantly enhances the inhibitory activity of antifolates like PTX and MTX against SHMTs in vivo | Drug repurposing | [145] |
| Glycolysis | Shikonin | Pyruvate kinase | Decreases the PKM2-mediated aerobic glycolysis switch in tumor cells, thereby inhibiting tumor proliferation | Shikonin suppresses tumor growth in a dose-dependent manner in a mouse model of B16 melanoma at concentrations of 1 mg/kg and 10 mg/kg | Animal studies | [146] |
| Fatty acid synthesis | TVB 2640 (Denifanstat) + bevacizumab | Fatty acid synthase | Alternation of fatty acid synthase signaling which can drive phenotypic plasticity and cell fate decisions, mitochondrial regulation of cell death, immune escape, and organ-specific metastatic potential | TVB-2640 combined with bevacizumab significantly improved progression-free survival (PFS) in patients with recurrent high-grade astrocytoma compared to historical bevacizumab monotherapy, demonstrating a favorable safety profile and promising efficacy | Phase II | [147] |
| Amino acid synthesis | Venetoclax with azacitidine | BCL-2 | Inhibition of BCL-2 which leads to the suppression of oxidative phosphorylation | Venetoclax and azacitidine show high response rates in treatment-naive patients, but relapsed patients exhibit reduced sensitivity due to metabolic adaptations in leukemic stem cells, indicating potential for targeting fatty acid metabolism | Phase I | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-F.; Guerrero, J.J.G.; Regalado, R.R.H.; Zhou, J.; Notarte, K.I.; Lu, Y.-W.; Encarnacion, P.C.; Carles, C.D.D.; Octavo, E.M.; Limbaroc, D.C.I.; et al. Insights into Metabolic Reprogramming in Tumor Evolution and Therapy. Cancers 2024, 16, 3513. https://doi.org/10.3390/cancers16203513
Chiu C-F, Guerrero JJG, Regalado RRH, Zhou J, Notarte KI, Lu Y-W, Encarnacion PC, Carles CDD, Octavo EM, Limbaroc DCI, et al. Insights into Metabolic Reprogramming in Tumor Evolution and Therapy. Cancers. 2024; 16(20):3513. https://doi.org/10.3390/cancers16203513
Chicago/Turabian StyleChiu, Ching-Feng, Jonathan Jaime G. Guerrero, Ric Ryan H. Regalado, Jiayan Zhou, Kin Israel Notarte, Yu-Wei Lu, Paolo C. Encarnacion, Cidne Danielle D. Carles, Edrian M. Octavo, Dan Christopher I. Limbaroc, and et al. 2024. "Insights into Metabolic Reprogramming in Tumor Evolution and Therapy" Cancers 16, no. 20: 3513. https://doi.org/10.3390/cancers16203513
APA StyleChiu, C.-F., Guerrero, J. J. G., Regalado, R. R. H., Zhou, J., Notarte, K. I., Lu, Y.-W., Encarnacion, P. C., Carles, C. D. D., Octavo, E. M., Limbaroc, D. C. I., Saengboonmee, C., & Huang, S.-Y. (2024). Insights into Metabolic Reprogramming in Tumor Evolution and Therapy. Cancers, 16(20), 3513. https://doi.org/10.3390/cancers16203513