
RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer

 , ,
, ,  and
and 
Simple Summary
Abstract
1. Introduction
2. Lin28
2.1. (Patho)physiological Function
2.2. Inhibitors

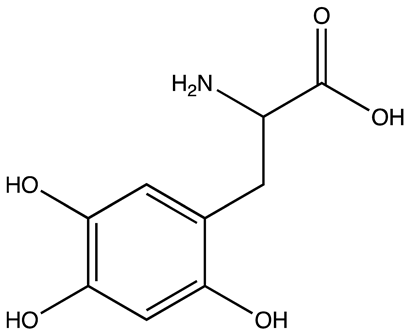
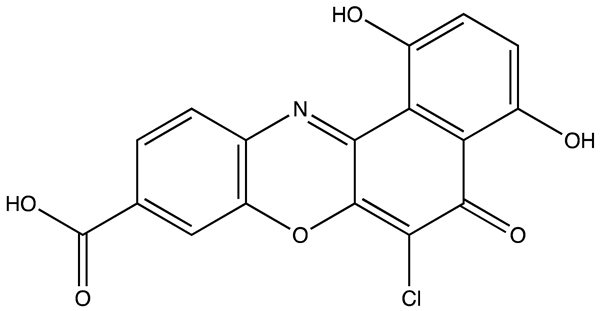
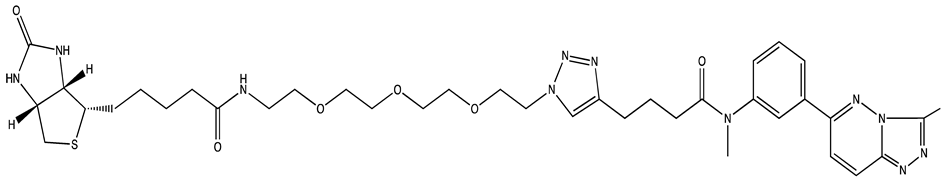

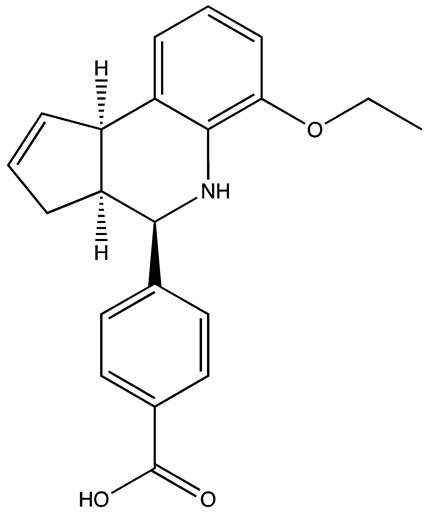
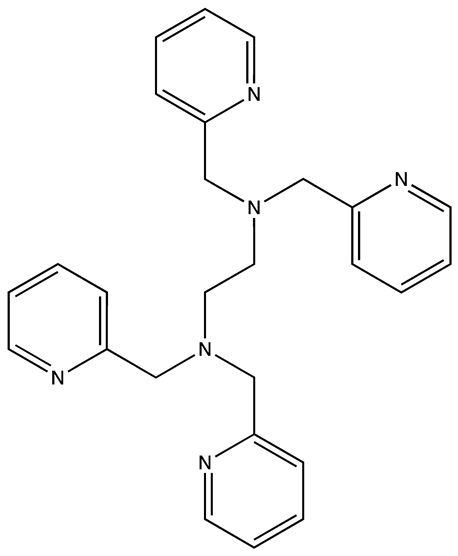

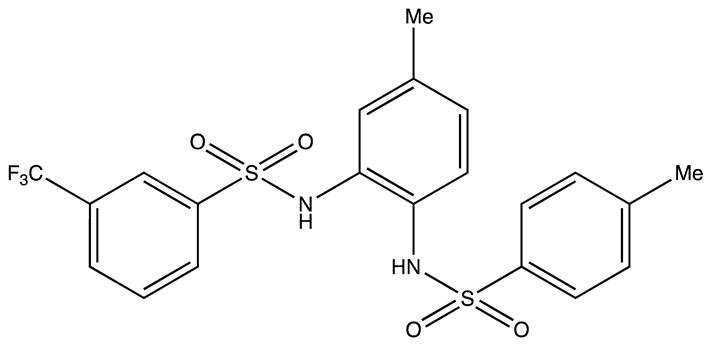
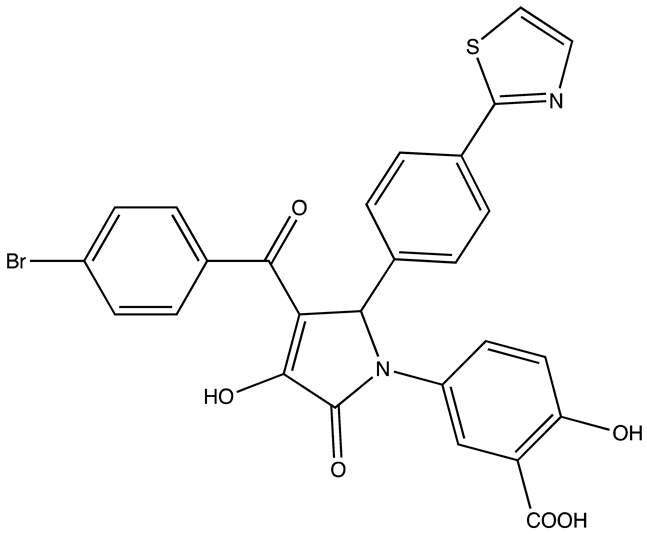
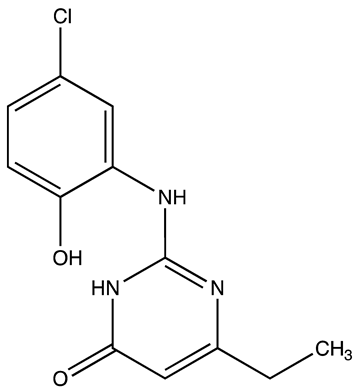
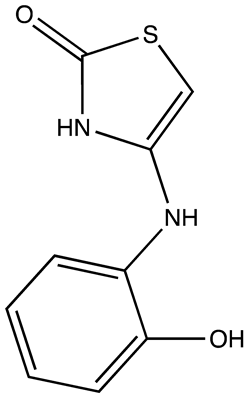
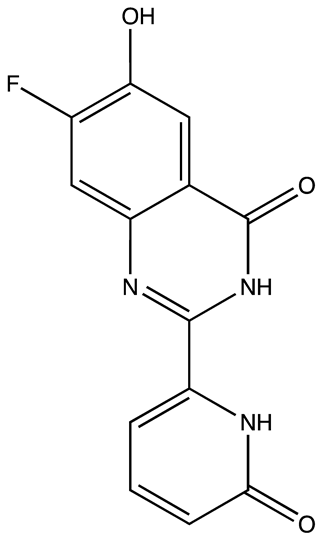
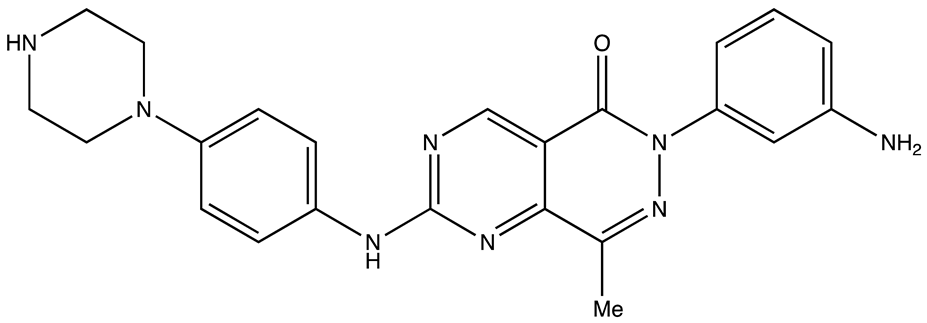
3. IGF2BP’s
3.1. (Patho)physiological Function
3.2. Inhibitors
4. Musashi
4.1. (Patho)physiological Function
4.2. Inhibitors
5. HuR
5.1. (Patho)physiological Function
5.2. Inhibitors
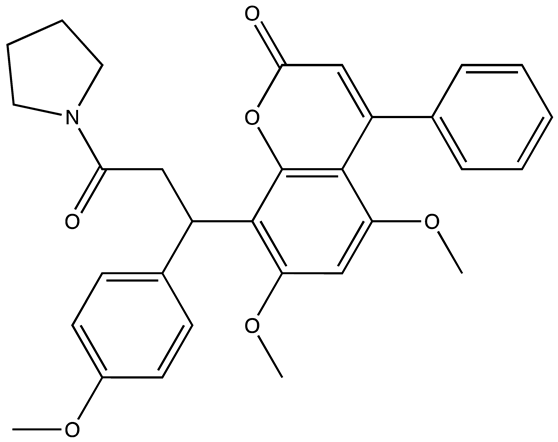


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6. CELF1
6.1. (Patho)physiological Function
6.2. Inhibitor
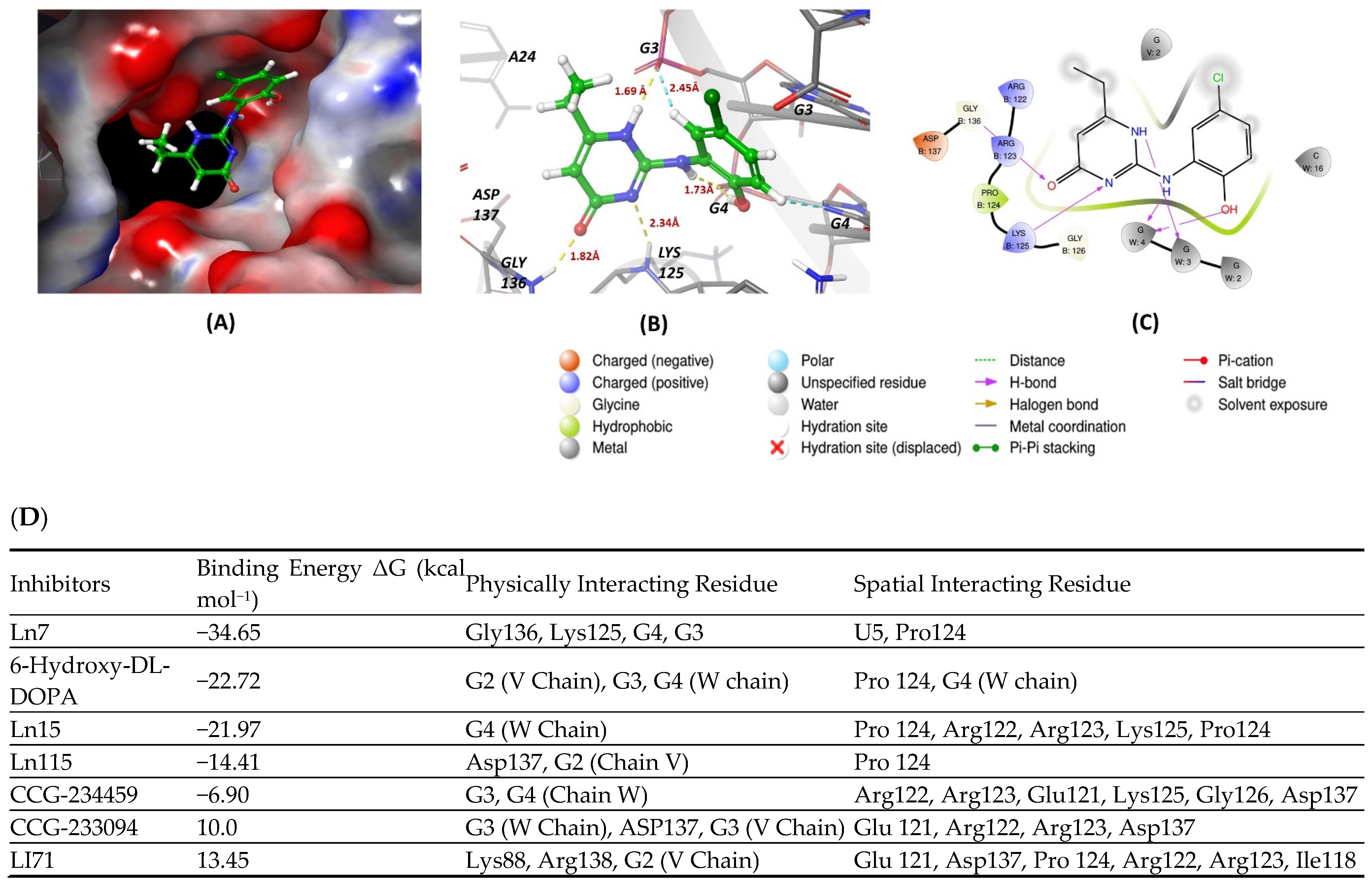
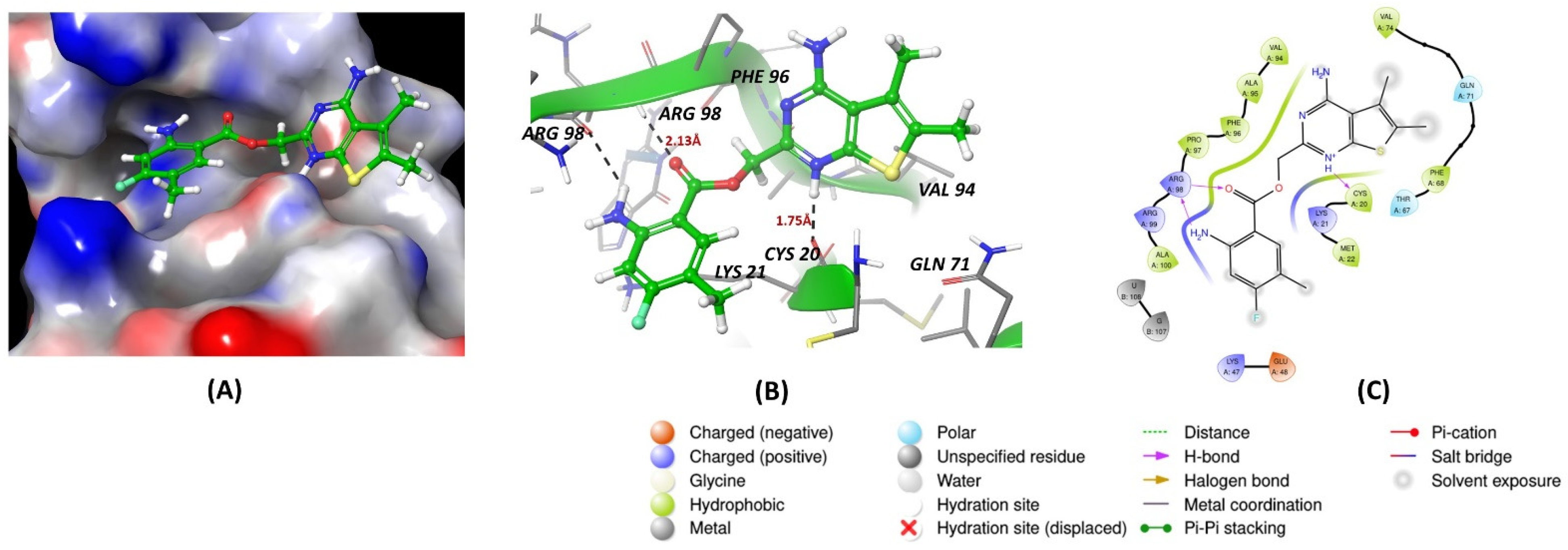
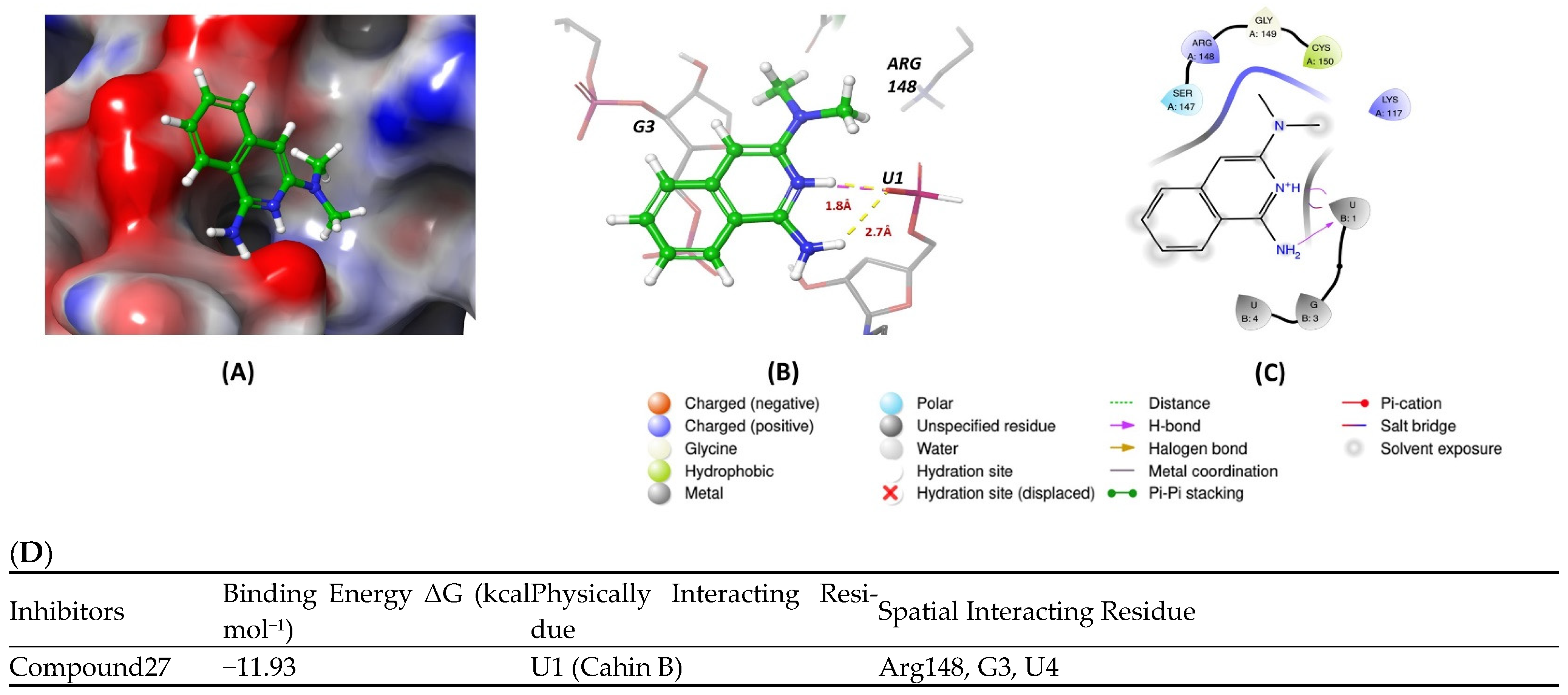
7. Molecular Docking Methods
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Orphanides, G.; Reinberg, D. A unified theory of gene expression. Cell 2002, 108, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Kakumani, P.K. AGO-RBP crosstalk on target mRNAs: Implications in miRNA-guided gene silencing and cancer. Transl. Oncol. 2022, 21, 101434. [Google Scholar] [CrossRef]
- Kechavarzi, B.; Janga, S.C. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014, 15, R14. [Google Scholar] [CrossRef]
- Neelamraju, Y.; Gonzalez-Perez, A.; Bhat-Nakshatri, P.; Nakshatri, H.; Janga, S.C. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol. 2018, 15, 115–129. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Wang, Z.L.; Li, B.; Luo, Y.X.; Lin, Q.; Liu, S.R.; Zhang, X.Q.; Zhou, H.; Yang, J.H.; Qu, L.H. Comprehensive Genomic Characterization of RNA-Binding Proteins across Human Cancers. Cell Rep. 2018, 22, 286–298. [Google Scholar] [CrossRef]
- Byun, W.G.; Lim, D.; Park, S.B. Small-molecule modulators of protein–RNA interactions. Curr. Opin. Chem. Biol. 2022, 68, 102149. [Google Scholar] [CrossRef]
- Ambros, V.; Horvitz, H.R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef]
- Zhu, H.; Shah, S.; Shyh-Chang, N.; Shinoda, G.; Einhorn, W.S.; Viswanathan, S.R.; Takeuchi, A.; Grasemann, C.; Rinn, J.L.; Lopez, M.F.; et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat. Genet. 2010, 42, 626–630. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Zhu, H.; Yvanka de Soysa, T.; Shinoda, G.; Seligson, M.T.; Tsanov, K.M.; Nguyen, L.; Asara, J.M.; Cantley, L.C.; Daley, G.Q. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 2013, 155, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhong, X.; Lin, X.; Guo, J.; Zou, L.; Tanyi, J.L.; Shao, Z.; Liang, S.; Wang, L.P.; Hwang, W.T.; et al. Lin-28 homologue A (LIN28A) promotes cell cycle progression via regulation of cyclin-dependent kinase 2 (CDK2), cyclin D1 (CCND1), and cell division cycle 25 homolog A (CDC25A) expression in cancer. J. Biol. Chem. 2012, 287, 17386–17397. [Google Scholar] [CrossRef] [PubMed]
- Maklad, A.; Sedeeq, M.; Chan, K.M.; Gueven, N.; Azimi, I. Exploring Lin28 proteins: Unravelling structure and functions with emphasis on nervous system malignancies. Life Sci. 2023, 335, 122275. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66–80. [Google Scholar] [CrossRef]
- Tu, H.C.; Schwitalla, S.; Qian, Z.; LaPier, G.S.; Yermalovich, A.; Ku, Y.C.; Chen, S.C.; Viswanathan, S.R.; Zhu, H.; Nishihara, R.; et al. LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev. 2015, 29, 1074–1086. [Google Scholar] [CrossRef]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. LIN28B promotes colon cancer progression and metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef]
- Madison, B.B.; Liu, Q.; Zhong, X.; Hahn, C.M.; Lin, N.; Emmett, M.J.; Stanger, B.Z.; Lee, J.S.; Rustgi, A.K. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev. 2013, 27, 2233–2245. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol. Cancer 2015, 14, 125. [Google Scholar] [CrossRef]
- Sugiura, K.; Masuike, Y.; Suzuki, K.; Shin, A.E.; Sakai, N.; Matsubara, H.; Otsuka, M.; Sims, P.A.; Lengner, C.J.; Rustgi, A.K. LIN28B promotes cell invasion and colorectal cancer metastasis via CLDN1 and NOTCH3. JCI Insight 2023, 8, e167310. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Miska, E.A.; Balasubramanian, S. Identification of small molecule inhibitors of the Lin28-mediated blockage of pre-let-7g processing. Org. Biomol. Chem. 2016, 14, 10208–10216. [Google Scholar] [CrossRef] [PubMed]
- Roos, M.; Pradere, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Byun, W.G.; Koo, J.Y.; Park, H.; Park, S.B. Discovery of a Small-Molecule Inhibitor of Protein–MicroRNA Interaction Using Binding Assay with a Site-Specifically Labeled Lin28. J. Am. Chem. Soc. 2016, 138, 13630–13638. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rowe, R.G.; Jaimes, A.; Yu, C.; Nam, Y.; Pearson, D.S.; Zhang, J.; Xie, X.; Marion, W.; Heffron, G.J.; et al. Small-Molecule Inhibitors Disrupt let-7 Oligouridylation and Release the Selective Blockade of let-7 Processing by LIN28. Cell Rep. 2018, 23, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.A.; Kaur, T.; Kerk, S.A.; Gallagher, E.E.; Sandoval, J.; Garner, A.L. Expansion of cat-ELCCA for the Discovery of Small Molecule Inhibitors of the Pre-let-7–Lin28 RNA–Protein Interaction. ACS Med. Chem. Lett. 2018, 9, 517–521. [Google Scholar] [CrossRef]
- Borgelt, L.; Li, F.; Hommen, P.; Lampe, P.; Hwang, J.; Goebel, G.L.; Sievers, S.; Wu, P. Trisubstituted Pyrrolinones as Small-Molecule Inhibitors Disrupting the Protein–RNA Interaction of LIN28 and Let-7. ACS Med. Chem. Lett. 2021, 12, 893–898. [Google Scholar] [CrossRef]
- Radaeva, M.; Ho, C.H.; Xie, N.; Zhang, S.; Lee, J.; Liu, L.; Lallous, N.; Cherkasov, A.; Dong, X. Discovery of Novel Lin28 Inhibitors to Suppress Cancer Cell Stemness. Cancers 2022, 14, 5687. [Google Scholar] [CrossRef]
- Hommen, P.; Hwang, J.; Huang, F.; Borgelt, L.; Hohnen, L.; Wu, P. Chromenopyrazole–Peptide Conjugates as Small–Molecule Based Inhibitors Disrupting the Protein–RNA Interaction of LIN28-let–7. Chembiochem 2023, 24, e202300376. [Google Scholar] [CrossRef]
- Rosenblum, S.L.; Soueid, D.M.; Giambasu, G.; Vander Roest, S.; Pasternak, A.; DiMauro, E.F.; Simov, V.; Garner, A.L. Live cell screening to identify RNA-binding small molecule inhibitors of the pre-let-7–Lin28 RNA–protein interaction. RSC Med. Chem. 2024, 15, 1539–1546. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Christiansen, J.; Lykke-Andersen, J.; Johnsen, A.H.; Wewer, U.M.; Nielsen, F.C. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol. 1999, 19, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Kristensen, M.A.; Willemoes, M.; Nielsen, F.C.; Christiansen, J. Sequential dimerization of human zipcode-binding protein IMP1 on RNA: A cooperative mechanism providing RNP stability. Nucleic Acids Res. 2004, 32, 4368–4376. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Lederer, M.; Bley, N.; Schleifer, C.; Huttelmaier, S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin. Cancer Biol. 2014, 29, 3–12. [Google Scholar] [CrossRef]
- Hammer, N.A.; Hansen, T.; Byskov, A.G.; Rajpert-De Meyts, E.; Grondahl, M.L.; Bredkjaer, H.E.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Expression of IGF-II mRNA-binding proteins (IMPs) in gonads and testicular cancer. Reproduction 2005, 130, 203–212. [Google Scholar] [CrossRef]
- Hansen, T.V.; Hammer, N.A.; Nielsen, J.; Madsen, M.; Dalbaeck, C.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Dwarfism and impaired gut development in insulin-like growth factor II mRNA-binding protein 1-deficient mice. Mol. Cell. Biol. 2004, 24, 4448–4464. [Google Scholar] [CrossRef]
- Nunez, L.; Buxbaum, A.R.; Katz, Z.B.; Lopez-Jones, M.; Nwokafor, C.; Czaplinski, K.; Pan, F.; Rosenberg, J.; Monday, H.R.; Singer, R.H. Tagged actin mRNA dysregulation in IGF2BP1[−/−] mice. Proc. Natl. Acad. Sci. USA 2022, 119, e2208465119. [Google Scholar] [CrossRef]
- Manieri, N.A.; Drylewicz, M.R.; Miyoshi, H.; Stappenbeck, T.S. Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology 2012, 143, 110–121.e10. [Google Scholar] [CrossRef]
- Singh, V.; Gowda, C.P.; Singh, V.; Ganapathy, A.S.; Karamchandani, D.M.; Eshelman, M.A.; Yochum, G.S.; Nighot, P.; Spiegelman, V.S. The mRNA-binding protein IGF2BP1 maintains intestinal barrier function by up-regulating occludin expression. J. Biol. Chem. 2020, 295, 8602–8612. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.E.; Noubissi, F.K.; Katti, P.S.; Hahn, C.M.; Davey, S.R.; Lundsmith, E.T.; Klein-Szanto, A.J.; Rhim, A.D.; Spiegelman, V.S.; Rustgi, A.K. IMP1 promotes tumor growth, dissemination and a tumor-initiating cell phenotype in colorectal cancer cell xenografts. Carcinogenesis 2013, 34, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Mongroo, P.S.; Noubissi, F.K.; Cuatrecasas, M.; Kalabis, J.; King, C.E.; Johnstone, C.N.; Bowser, M.J.; Castells, A.; Spiegelman, V.S.; Rustgi, A.K. IMP-1 displays cross-talk with K-Ras and modulates colon cancer cell survival through the novel proapoptotic protein CYFIP2. Cancer Res. 2011, 71, 2172–2182. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Goswami, S.; Sanek, N.A.; Kawakami, K.; Minamoto, T.; Moser, A.; Grinblat, Y.; Spiegelman, V.S. Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 2009, 69, 8572–8578. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Elcheva, I.; Bhatia, N.; Shakoori, A.; Ougolkov, A.; Liu, J.; Minamoto, T.; Ross, J.; Fuchs, S.Y.; Spiegelman, V.S. CRD-BP mediates stabilization of βTrCP1 and c-myc mRNA in response to β-catenin signalling. Nature 2006, 441, 898–901. [Google Scholar] [CrossRef]
- Singh, V.; Walter, V.; Elcheva, I.; Imamura Kawasawa, Y.; Spiegelman, V.S. Global role of IGF2BP1 in controlling the expression of Wnt/β-catenin-regulated genes in colorectal cancer cells. Front. Cell Dev. Biol. 2023, 11, 1236356. [Google Scholar] [CrossRef]
- Biegel, J.M.; Dhamdhere, M.; Gao, S.; Gowda, C.P.; Kawasawa, Y.I.; Spiegelman, V.S. Inhibition of the mRNA-Binding Protein IGF2BP1 Suppresses Proliferation and Sensitizes Neuroblastoma Cells to Chemotherapeutic Agents. Front. Oncol. 2021, 11, 608816. [Google Scholar] [CrossRef]
- Craig, E.A.; Spiegelman, V.S. Inhibition of coding region determinant binding protein sensitizes melanoma cells to chemotherapeutic agents. Pigment. Cell Melanoma Res. 2012, 25, 83–87. [Google Scholar] [CrossRef]
- Kim, T.; Havighurst, T.; Kim, K.; Albertini, M.; Xu, Y.G.; Spiegelman, V.S. Targeting insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in metastatic melanoma to increase efficacy of BRAFV600E inhibitors. Mol. Carcinog. 2018, 57, 678–683. [Google Scholar] [CrossRef]
- Betson, N.; Hajahmed, M.; Gebretsadek, T.; Ndebele, K.; Ahmad, H.A.; Tchounwou, P.B.; Spiegelman, V.S.; Noubissi, F.K. Inhibition of insulin-like growth factor 2 mRNA-binding protein 1 sensitizes colorectal cancer cells to chemotherapeutics. FASEB Bioadv. 2022, 4, 816–829. [Google Scholar] [CrossRef]
- Chen, M.; Tian, B.; Hu, G.; Guo, Y. METTL3-Modulated circUHRF2 Promotes Colorectal Cancer Stemness and Metastasis through Increasing DDX27 mRNA Stability by Recruiting IGF2BP1. Cancers 2023, 15, 3148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Li, K.J.; Feng, J.X.; Liu, G.J.; Feng, Y.L. Blocking the IGF2BP1-promoted glucose metabolism of colon cancer cells via direct de-stabilizing mRNA of the LDHA enhances anticancer effects. Mol. Ther. Nucleic Acids 2021, 23, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.F.; Williams, K.N.; Plesset, J.B.; Headd, J.J.; Mizuno, R.; Chatterji, P.; Lento, A.A.; Klein-Szanto, A.J.; Mick, R.; Hamilton, K.E.; et al. IMP1 3’ UTR shortening enhances metastatic burden in colorectal cancer. Carcinogenesis 2019, 40, 569–579. [Google Scholar] [CrossRef]
- Li, T.; Hu, P.S.; Zuo, Z.; Lin, J.F.; Li, X.; Wu, Q.N.; Chen, Z.H.; Zeng, Z.L.; Wang, F.; Zheng, J.; et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 2019, 18, 112. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, T.; Wu, D.; Min, Z.; Tan, J.; Yu, B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J. Exp. Clin. Cancer Res. 2020, 39, 203. [Google Scholar] [CrossRef]
- Mahapatra, L.; Mao, C.; Andruska, N.; Zhang, C.; Shapiro, D.J. High-throughput fluorescence anisotropy screen for inhibitors of the oncogenic mRNA binding protein, IMP-1. J. Biomol. Screen. 2014, 19, 427–436. [Google Scholar] [CrossRef]
- Mahapatra, L.; Andruska, N.; Mao, C.; Le, J.; Shapiro, D.J. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl. Oncol. 2017, 10, 818–827. [Google Scholar] [CrossRef]
- Wallis, N.; Oberman, F.; Shurrush, K.; Germain, N.; Greenwald, G.; Gershon, T.; Pearl, T.; Abis, G.; Singh, V.; Singh, A.; et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. 2022, 19, 26–43. [Google Scholar] [CrossRef]
- Singh, A.; Singh, V.; Wallis, N.; Abis, G.; Oberman, F.; Wood, T.; Dhamdhere, M.; Gershon, T.; Ramos, A.; Yisraeli, J.; et al. Development of a specific and potent IGF2BP1 inhibitor: A promising therapeutic agent for IGF2BP1-expressing cancers. Eur. J. Med. Chem. 2024, 263, 115940. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Q.; Yang, H.; Zhang, X.W.; Feng, N.; Wang, J.K.; Liu, T.T.; Zeng, K.W.; Tu, P.F. Allosteric Regulation of IGF2BP1 as a Novel Strategy for the Activation of Tumor Immune Microenvironment. ACS Cent. Sci. 2022, 8, 1102–1115. [Google Scholar] [CrossRef]
- Shang, F.F.; Lu, Q.; Lin, T.; Pu, M.; Xiao, R.; Liu, W.; Deng, H.; Guo, H.; Quan, Z.S.; Ding, C.; et al. Discovery of Triazolyl Derivatives of Cucurbitacin B Targeting IGF2BP1 against Non-Small Cell Lung Cancer. J. Med. Chem. 2023, 66, 12931–12949. [Google Scholar] [CrossRef]
- Dahlem, C.; Abuhaliema, A.; Kessler, S.M.; Krohler, T.; Zoller, B.G.E.; Chanda, S.; Wu, Y.; Both, S.; Muller, F.; Lepikhov, K.; et al. First Small-Molecule Inhibitors Targeting the RNA-Binding Protein IGF2BP2/IMP2 for Cancer Therapy. ACS Chem. Biol. 2022, 17, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Chen, D.; Wang, X.; Li, Y.; Li, Z.; Li, B.; Zhang, Y.; Li, W.; Zhang, J.; Ye, J.; et al. Inhibition of the m6A reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia. Leukemia 2022, 36, 2180–2188. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, Y.; Mao, B.; You, Q.; Guo, X. Targeting insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) for the treatment of cancer. Eur. J. Med. Chem. 2024, 268, 116241. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol. Cell. Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Nakamura, Y.; Yoshida, T.; Shibata, S.; Koike, M.; Takano, H.; Ueda, S.; Uchiyama, Y.; Noda, T.; Okano, H. RNA-binding protein Musashi family: Roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc. Natl. Acad. Sci. USA 2002, 99, 15194–15199. [Google Scholar] [CrossRef]
- Kanemura, Y.; Mori, K.; Sakakibara, S.; Fujikawa, H.; Hayashi, H.; Nakano, A.; Matsumoto, T.; Tamura, K.; Imai, T.; Ohnishi, T.; et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation 2001, 68, 141–152. [Google Scholar] [CrossRef]
- Kawahara, H.; Okada, Y.; Imai, T.; Iwanami, A.; Mischel, P.S.; Okano, H. Musashi1 cooperates in abnormal cell lineage protein 28 (Lin28)-mediated let-7 family microRNA biogenesis in early neural differentiation. J. Biol. Chem. 2011, 286, 16121–16130. [Google Scholar] [CrossRef]
- Kharas, M.G.; Lengner, C.J.; Al-Shahrour, F.; Bullinger, L.; Ball, B.; Zaidi, S.; Morgan, K.; Tam, W.; Paktinat, M.; Okabe, R.; et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 2010, 16, 903–908. [Google Scholar] [CrossRef]
- Sakakibara, S.; Nakamura, Y.; Satoh, H.; Okano, H. RNA-binding protein Musashi2: Developmentally regulated expression in neural precursor cells and subpopulations of neurons in mammalian CNS. J. Neurosci. 2001, 21, 8091–8107. [Google Scholar] [CrossRef]
- Nakamura, M.; Okano, H.; Blendy, J.A.; Montell, C. Musashi, a neural RNA-binding protein required for Drosophila adult external sensory organ development. Neuron 1994, 13, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, K.; Liu, Y.; Zhu, D.; Pan, Y.; Ke, X.; Qu, Y. Small Molecule Palmatine Targeting Musashi-2 in Colorectal Cancer. Front. Pharmacol. 2021, 12, 793449. [Google Scholar] [CrossRef] [PubMed]
- Chiou, G.Y.; Yang, T.W.; Huang, C.C.; Tang, C.Y.; Yen, J.Y.; Tsai, M.C.; Chen, H.Y.; Fadhilah, N.; Lin, C.C.; Jong, Y.J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef] [PubMed]
- Kharin, L.; Bychkov, I.; Karnaukhov, N.; Voloshin, M.; Fazliyeva, R.; Deneka, A.; Frantsiyants, E.; Kit, O.; Golemis, E.; Boumber, Y. Prognostic role and biologic features of Musashi-2 expression in colon polyps and during colorectal cancer progression. PLoS ONE 2021, 16, e0252132. [Google Scholar] [CrossRef]
- Meng, X.; Peng, X.; Ouyang, W.; Li, H.; Na, R.; Zhou, W.; You, X.; Li, Y.; Pu, X.; Zhang, K.; et al. Musashi-2 Deficiency Triggers Colorectal Cancer Ferroptosis by Downregulating the MAPK Signaling Cascade to Inhibit HSPB1 Phosphorylation. Biol. Proced. Online 2023, 25, 32. [Google Scholar] [CrossRef]
- Minuesa, G.; Antczak, C.; Shum, D.; Radu, C.; Bhinder, B.; Li, Y.; Djaballah, H.; Kharas, M.G. A 1536-well fluorescence polarization assay to screen for modulators of the MUSASHI family of RNA-binding proteins. Comb. Chem. High Throughput Screen. 2014, 17, 596–609. [Google Scholar] [CrossRef]
- Minuesa, G.; Albanese, S.K.; Xie, W.; Kazansky, Y.; Worroll, D.; Chow, A.; Schurer, A.; Park, S.M.; Rotsides, C.Z.; Taggart, J.; et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 2691. [Google Scholar] [CrossRef]
- Bai, N.; Adeshina, Y.; Bychkov, I.; Xia, Y.; Gowthaman, R.; Miller, S.A.; Gupta, A.K.; Johnson, D.K.; Lan, L.; Golemis, E.A.; et al. Rationally designed inhibitors of the Musashi protein-RNA interaction by hotspot mimicry. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhu, H.; Hasman, R.A.; Barron, V.A.; Luo, G.; Lou, H. A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Mol. Biol. Cell 2006, 17, 5105–5114. [Google Scholar] [CrossRef]
- Good, P.J. A conserved family of elav-like genes in vertebrates. Proc. Natl. Acad. Sci. USA 1995, 92, 4557–4561. [Google Scholar] [CrossRef]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 2010, 1, 214–229. [Google Scholar] [CrossRef]
- Liu, L.; Christodoulou-Vafeiadou, E.; Rao, J.N.; Zou, T.; Xiao, L.; Chung, H.K.; Yang, H.; Gorospe, M.; Kontoyiannis, D.; Wang, J.Y. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol. Biol. Cell 2014, 25, 3308–3318. [Google Scholar] [CrossRef]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Koch, I.; von Keyserlingk, N.; Noske, A.; Niesporek, S.; Dietel, M.; Weichert, W. Expression of the ELAV-like protein HuR in human colon cancer: Association with tumor stage and cyclooxygenase-2. Mod. Pathol. 2006, 19, 1261–1269. [Google Scholar] [CrossRef]
- Al-Haidari, A.; Algaber, A.; Madhi, R.; Syk, I.; Thorlacius, H. MiR-155-5p controls colon cancer cell migration via post-transcriptional regulation of Human Antigen R (HuR). Cancer Lett. 2018, 421, 145–151. [Google Scholar] [CrossRef]
- Doller, A.; Winkler, C.; Azrilian, I.; Schulz, S.; Hartmann, S.; Pfeilschifter, J.; Eberhardt, W. High-constitutive HuR phosphorylation at Ser 318 by PKCδ propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 2011, 32, 676–685. [Google Scholar] [CrossRef]
- Yoo, P.S.; Sullivan, C.A.; Kiang, S.; Gao, W.; Uchio, E.M.; Chung, G.G.; Cha, C.H. Tissue microarray analysis of 560 patients with colorectal adenocarcinoma: High expression of HuR predicts poor survival. Ann. Surg. Oncol. 2009, 16, 200–207. [Google Scholar] [CrossRef]
- Meisner, N.C.; Hintersteiner, M.; Mueller, K.; Bauer, R.; Seifert, J.M.; Naegeli, H.U.; Ottl, J.; Oberer, L.; Guenat, C.; Moss, S.; et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 2007, 3, 508–515. [Google Scholar] [CrossRef]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; Tunge, J.A.; et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef]
- Wang, Z.; Bhattacharya, A.; Ivanov, D.N. Identification of Small-Molecule Inhibitors of the HuR/RNA Interaction Using a Fluorescence Polarization Screening Assay Followed by NMR Validation. PLoS ONE 2015, 10, e0138780. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, R.; Mehta, M.; Ahmed, R.; Roy, S.; Xu, L.; Aube, J.; Chen, A.; Zhao, Y.D.; Herman, T.; Ramesh, R.; et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci. Rep. 2017, 7, 9694. [Google Scholar] [CrossRef]
- Wu, X.; Gardashova, G.; Lan, L.; Han, S.; Zhong, C.; Marquez, R.T.; Wei, L.; Wood, S.; Roy, S.; Gowthaman, R.; et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun. Biol. 2020, 3, 193. [Google Scholar] [CrossRef]
- Lal, P.; Cerofolini, L.; D’Agostino, V.G.; Zucal, C.; Fuccio, C.; Bonomo, I.; Dassi, E.; Giuntini, S.; Di Maio, D.; Vishwakarma, V.; et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. 2017, 45, 9514–9527. [Google Scholar] [CrossRef]
- Manzoni, L.; Zucal, C.; Maio, D.D.; D’Agostino, V.G.; Thongon, N.; Bonomo, I.; Lal, P.; Miceli, M.; Baj, V.; Brambilla, M.; et al. Interfering with HuR-RNA Interaction: Design, Synthesis and Biological Characterization of Tanshinone Mimics as Novel, Effective HuR Inhibitors. J. Med. Chem. 2018, 61, 1483–1498. [Google Scholar] [CrossRef]
- Wu, X.; Ramesh, R.; Wang, J.; Zheng, Y.; Armaly, A.M.; Wei, L.; Xing, M.; Roy, S.; Lan, L.; Gao, F.P.; et al. Small Molecules Targeting the RNA-Binding Protein HuR Inhibit Tumor Growth in Xenografts. J. Med. Chem. 2023, 66, 2032–2053. [Google Scholar] [CrossRef]
- Fletcher, A.; Clift, D.; de Vries, E.; Martinez Cuesta, S.; Malcolm, T.; Meghini, F.; Chaerkady, R.; Wang, J.; Chiang, A.; Weng, S.H.S.; et al. A TRIM21-based bioPROTAC highlights the therapeutic benefit of HuR degradation. Nat. Commun. 2023, 14, 7093. [Google Scholar] [CrossRef]
- Qin, W.J.; Shi, J.J.; Chen, R.Y.; Li, C.Y.; Liu, Y.J.; Lu, J.F.; Yang, G.J.; Cao, J.F.; Chen, J. Curriculum vitae of CUG binding protein 1 (CELF1) in homeostasis and diseases: A systematic review. Cell. Mol. Biol. Lett. 2024, 29, 32. [Google Scholar] [CrossRef]
- Edwards, J.M.; Long, J.; de Moor, C.H.; Emsley, J.; Searle, M.S. Structural insights into the targeting of mRNA GU-rich elements by the three RRMs of CELF1. Nucleic Acids Res. 2013, 41, 7153–7166. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Mereau, A.; Osborne, H.B. Mammalian CELF/Bruno-like RNA-binding proteins: Molecular characteristics and biological functions. Biochimie 2006, 88, 515–525. [Google Scholar] [CrossRef]
- Qi, Z.P.; Chen, Z.H.; He, D.L.; Cai, S.L.; Li, B.; Sun, D.; Lv, Z.T.; Xu, E.P.; Shi, Q.; Zhong, Y.S.; et al. RNA binding protein CUGBP1 mediates the liver metastasis of colorectal cancer by regulating the ErbB signal pathway. Transl. Cancer Res. 2021, 10, 3373–3388. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, R.; Guo, W.; Qin, X.; Yang, Z.; Yuan, Z.; Wei, Y.; Mo, C.; Zeng, Z.; Luo, J.; et al. RNA-binding protein CELF1 enhances cell migration, invasion, and chemoresistance by targeting ETS2 in colorectal cancer. Clin. Sci. 2020, 134, 1973–1990. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Sun, X.; Xu, Y.; Tang, B.; Xu, S.; Lu, D.; Ye, Y.; Luo, X.; Diao, X.; Li, F.; et al. Small molecule targeting CELF1 RNA-binding activity to control HSC activation and liver fibrosis. Nucleic Acids Res. 2022, 50, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- SiteMap, Schrödinger, LLC, New York, NY. Available online: http://www.schrodinger.com/ (accessed on 15 September 2024).
- Krishna Rao, V.; Paul, S.; Gulkis, M.; Shen, Z.; Nair, H.; Singh, A.; Li, C.; Sharma, A.K.; Caglayan, M.; Das, C.; et al. Molecular editing of NSC-666719 enabling discovery of benzodithiazinedioxide-guanidines as anticancer agents. RSC Med. Chem. 2024, 15, 937–962. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
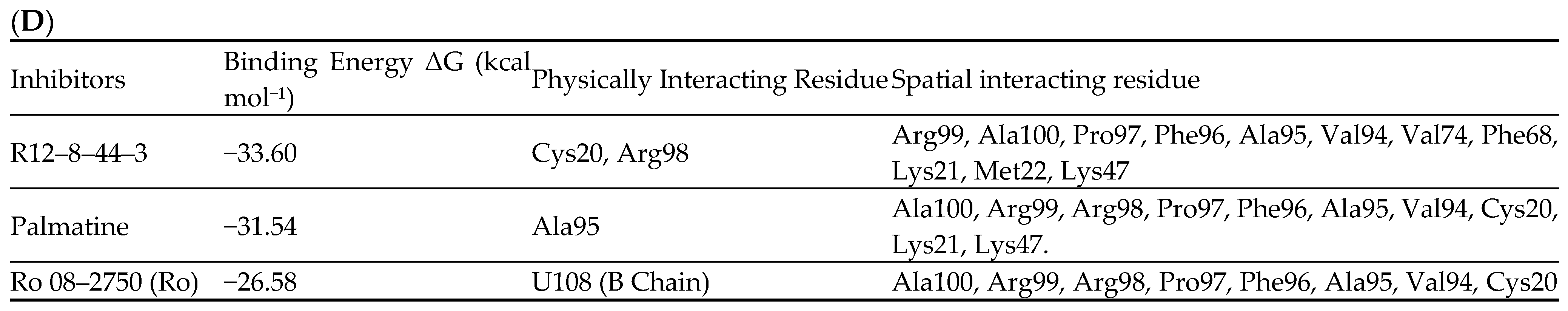
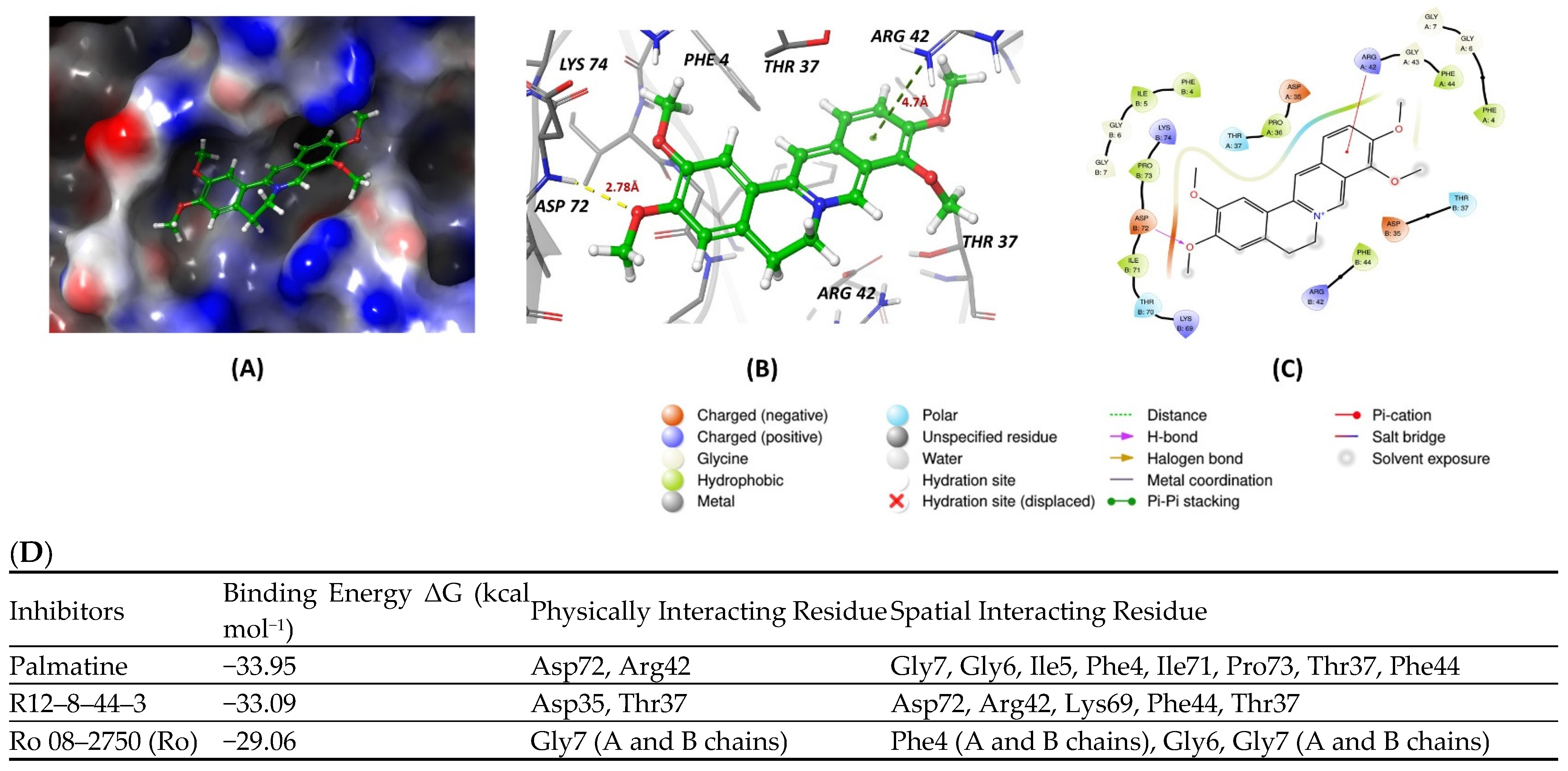
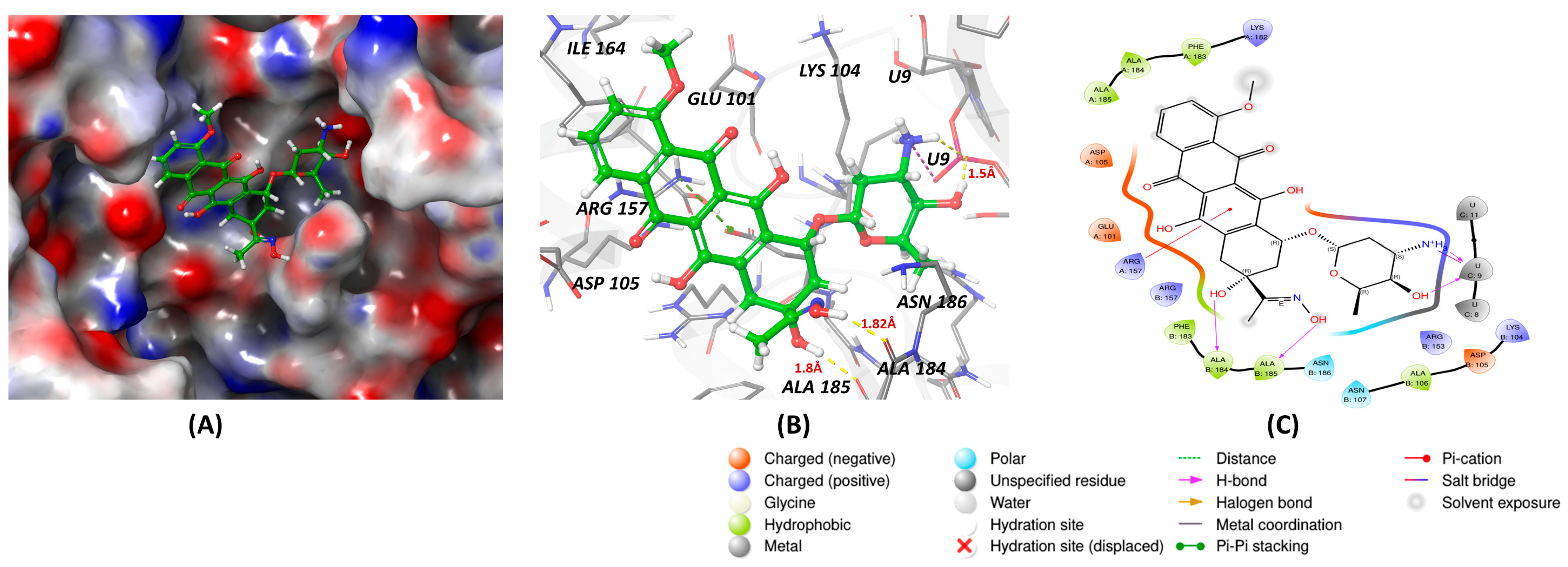
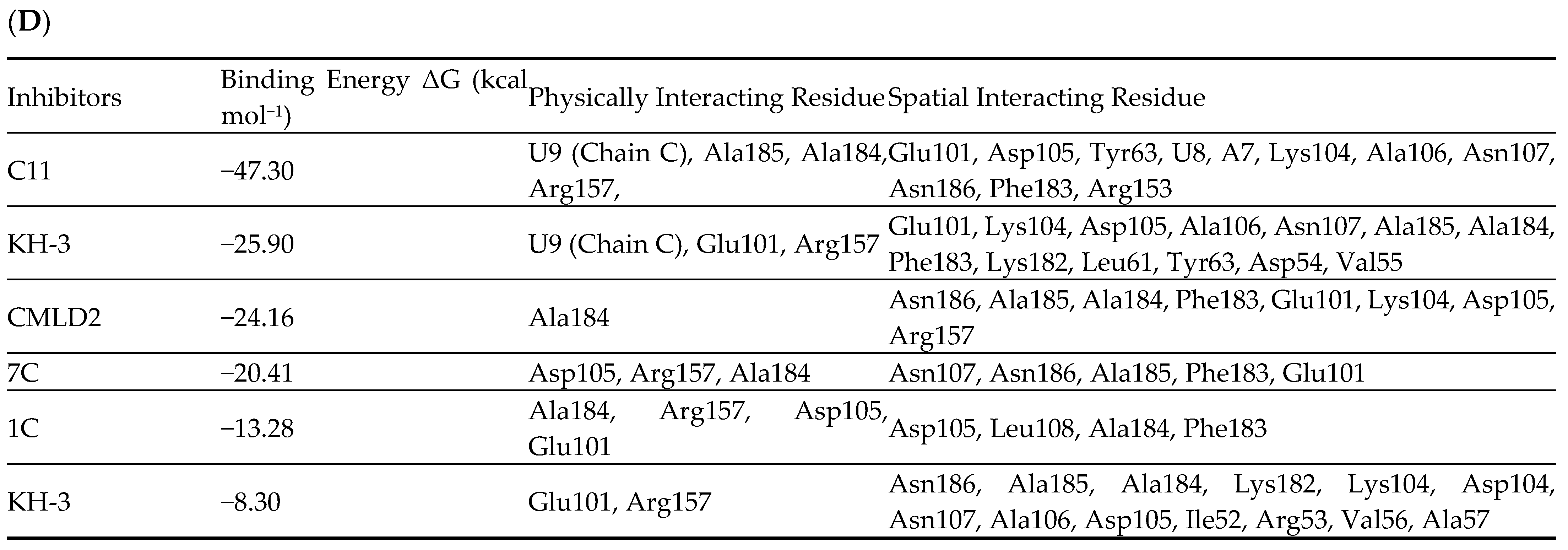
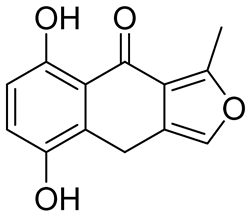
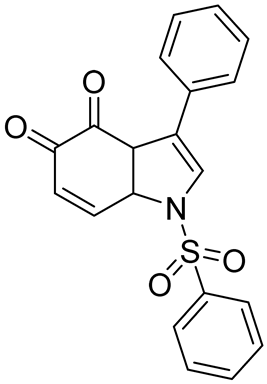
| Structure | Name | KD | Reference |
|---|---|---|---|
 | 6896009 | Not known | [66] |
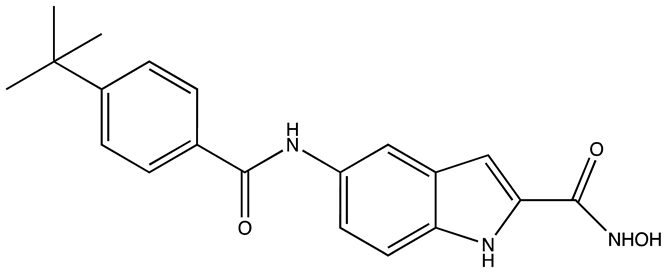
 | BTYNB | Not Known | [67] |
 | 7773 | 17 µM | [68] |
 | AVJ16 | 1.4 µM | [69] |
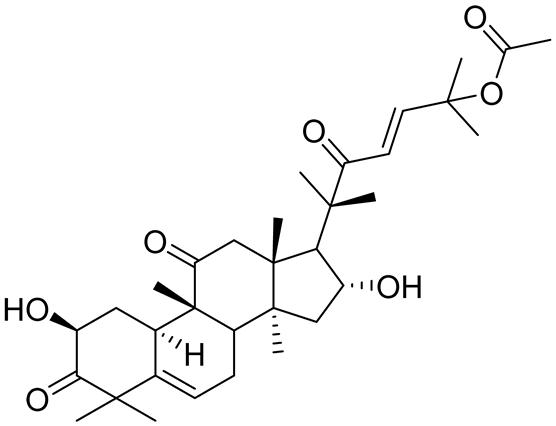 | CuB | 1.2 µM | [70] |
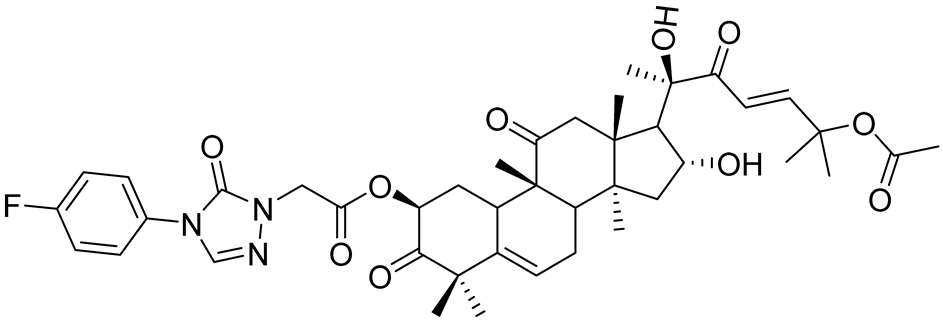 | A11 | 2.88 nM | [71] |
| Structure | Name | IC50 (µM) | Reference |
|---|---|---|---|
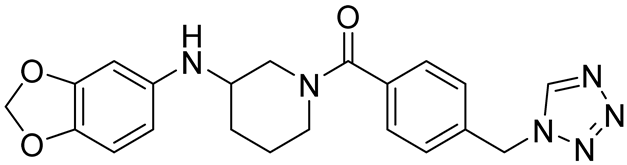
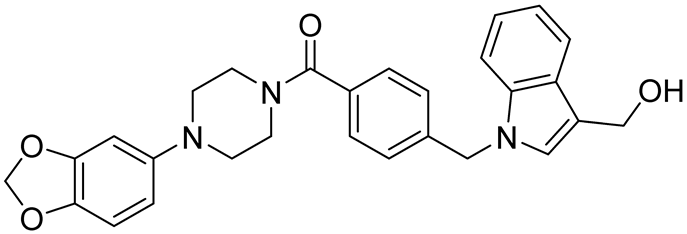
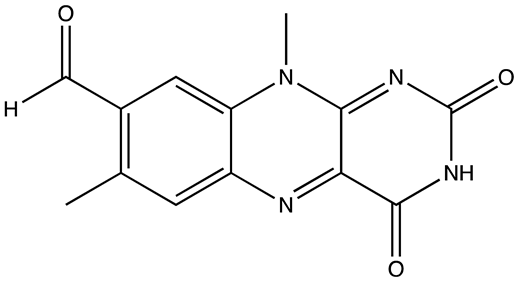 | Ro 08–2750 (Ro) | 2.7 | [87] |
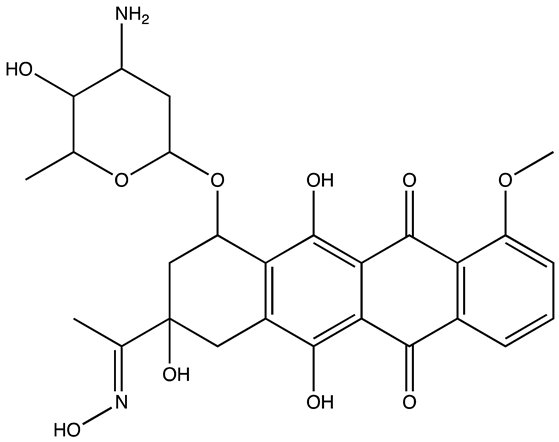
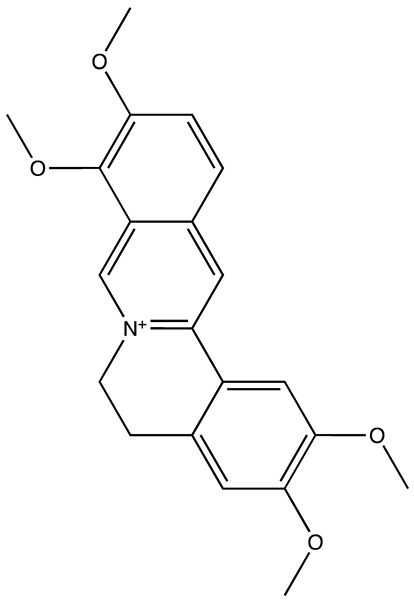 | Palmatine | N/A | [82] |
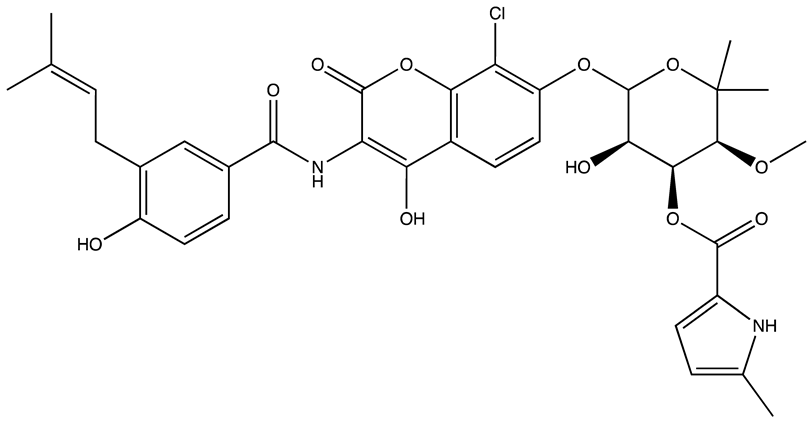
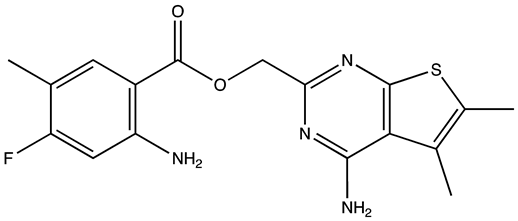 | R12–8–44–3 | 9.7 | [88] |
| Structure | Name | IC50 (µM) | Reference |
|---|---|---|---|
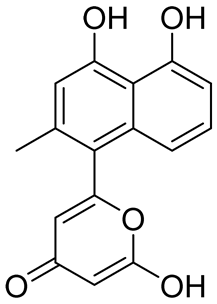
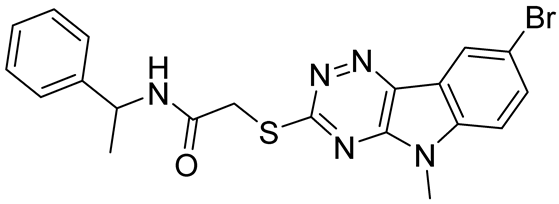
 | Compound 27 | 22.99 | [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, V.; Singh, A.; Liu, A.J.; Fuchs, S.Y.; Sharma, A.K.; Spiegelman, V.S. RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer. Cancers 2024, 16, 3502. https://doi.org/10.3390/cancers16203502
Singh V, Singh A, Liu AJ, Fuchs SY, Sharma AK, Spiegelman VS. RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer. Cancers. 2024; 16(20):3502. https://doi.org/10.3390/cancers16203502
Chicago/Turabian StyleSingh, Vikash, Amandeep Singh, Alvin John Liu, Serge Y. Fuchs, Arun K. Sharma, and Vladimir S. Spiegelman. 2024. "RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer" Cancers 16, no. 20: 3502. https://doi.org/10.3390/cancers16203502
APA StyleSingh, V., Singh, A., Liu, A. J., Fuchs, S. Y., Sharma, A. K., & Spiegelman, V. S. (2024). RNA Binding Proteins as Potential Therapeutic Targets in Colorectal Cancer. Cancers, 16(20), 3502. https://doi.org/10.3390/cancers16203502