
Recent Advances in the Molecular Biology of Chronic Lymphocytic Leukemia: How to Define Prognosis and Guide Treatment

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
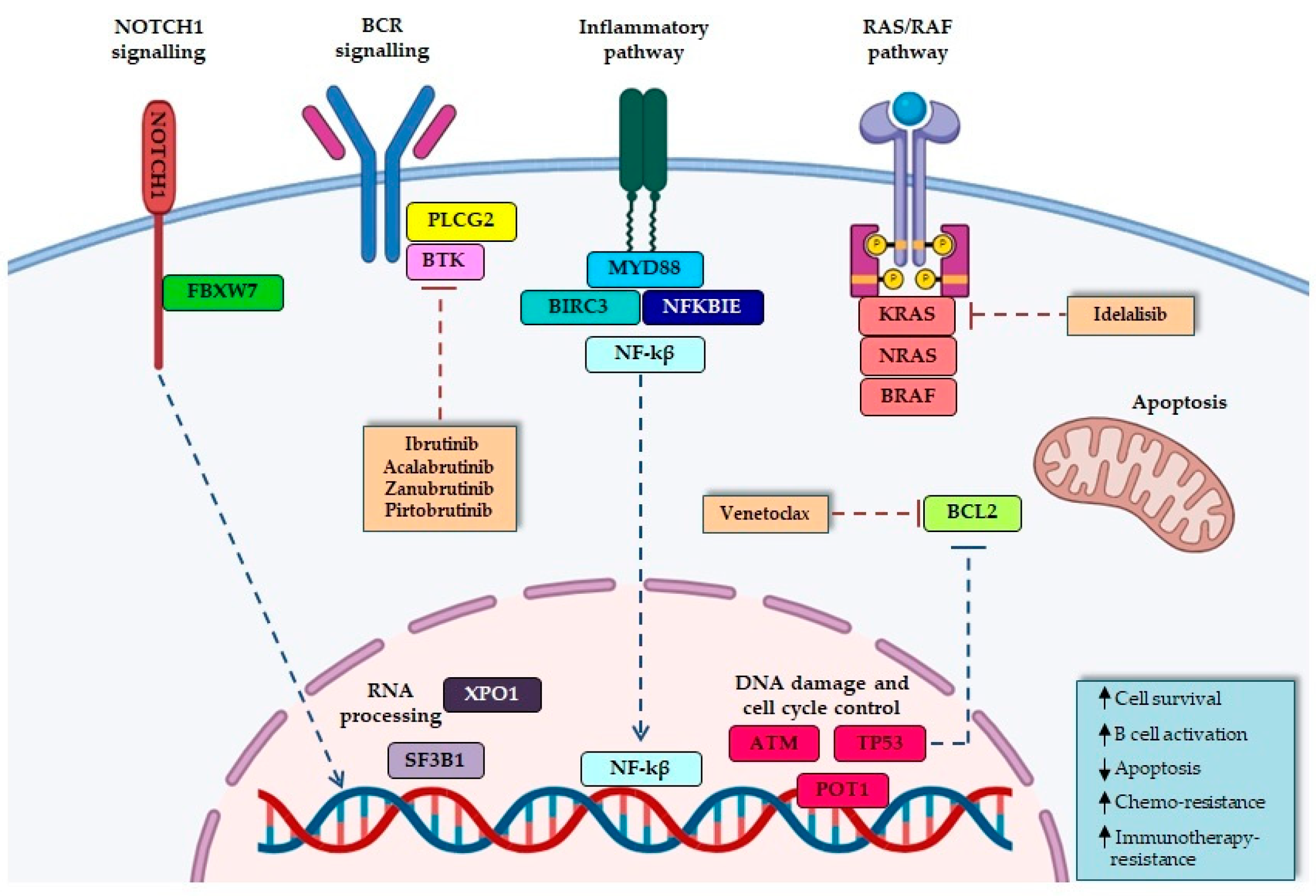
2. How to Define Prognosis
2.1. TP53 Aberrations
2.2. IGVH Mutational Status
2.3. Emerging Prognostic and Predictive Biomarkers
2.3.1. Microenvironment-Dependent Signaling through NOTCH
NOTCH1
FBXW7
2.3.2. RNA Processing
SF3B1
XPO1
2.3.3. DNA Damage and Cell Cycle Control
ATM
POT1
2.3.4. NF-κB Signaling Pathways
BIRC3
NFKBIE
2.3.5. Inflammatory Receptors
MYD88
2.3.6. MAPK–Extracellular Signal-Regulated Kinase
RAS/RAF Genes
3. How to Guide Treatment Choice in First-Line Therapy
3.1. The Role of TP53 Mutations and/or del(17p) in the Choice of First-Line Therapy
3.2. The Role of IGVH Mutational Status in the Choice of First-Line Therapy
4. How to Guide Treatment Choice in Relapsed/Refractory Disease
4.1. The Role of TP53 Mutations and/or del(17p) in the Choice of Second-Line Therapy
4.2. The Role of Prior Lines of Therapy
4.3. Mechanisms of Resistance to Target Therapy and New Drugs
5. The Emerging Role of MRD: Are We Ready for Clinical Use?
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shadman, M. Diagnosis and Treatment of Chronic Lymphocytic Leukemia: A Review. JAMA 2023, 329, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Kajüter, H.; Wellmann, I.; Khil, L.; Jöckel, K.H.; Zhang, C.; Fink, A.M.; Hallek, M.; Stang, A. Survival of patients with chronic lymphocytic leukemia before and after the introduction of chemoimmunotherapy in Germany. Blood Cancer J. 2021, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- van der Straten, L.; Levin, M.D.; Visser, O.; Posthuma, E.F.M.; Doorduijn, J.K.; Kater, A.P.; Dinmohamed, A.G. Survival continues to increase in chronic lymphocytic leukaemia: A population-based analysis among 20 468 patients diagnosed in the Netherlands between 1989 and 2016. Br. J. Haematol. 2020, 189, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; de Oliveira Araujo, I.B.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. Correction: “The 5th edition of The World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms” Leukemia. 2022 Jul;36(7):1720–1748. Leukemia 2023, 37, 1944–1951. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swedlow, S.H.; Anderson, S.H.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, F.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253, Erratum in Blood 2023, 141, 437. [Google Scholar] [CrossRef]
- Marti, G.E.; Rawstron, A.C.; Ghia, P.; Hillmen, P.; Houlston, R.S.; Kay, N.; Schleinitz, T.A.; Caporaso, N.; Consortium, I.F.C. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br. J. Haematol. 2005, 130, 325–332. [Google Scholar] [CrossRef]
- Langerbeins, P.; Zhang, C.; Robrecht, S.; Cramer, P.; Fürstenau, M.; Al-Sawaf, O.; von Tresckow, J.; Fink, A.M.; Kreuzer, K.A.; Vehling-Kaiser, U.; et al. The CLL12 trial: Ibrutinib vs placebo in treatment-naïve, early-stage chronic lymphocytic leukemia. Blood 2022, 139, 177–187. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): A meta-analysis of individual patient data. Lancet Oncol. 2016, 17, 779–790. [Google Scholar] [CrossRef]
- Langerbeins, P.; Giza, A.; Robrecht, S.; Cramer, P.; von Tresckow, J.; Al-Sawaf, O.; Fink, A.M.; Fürstenau, M.; Kutsch, N.; Simon, F.; et al. Reassessing the chronic lymphocytic leukemia International Prognostic Index in the era of targeted therapies. Blood 2024, 143, 2588–2598. [Google Scholar] [CrossRef] [PubMed]
- Braish, J.; Cerchione, C.; Ferrajoli, A. An overview of prognostic markers in patients with CLL. Front. Oncol. 2024, 14, 1371057. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Pavlova, S.; Baliakas, P.; Chatzikonstantinou, T.; Tausch, E.; Catherwood, M.; Rossi, D.; Soussi, T.; Tichy, B.; Kater, A.P.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-2024 update. Leukemia 2024, 38, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jäger, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef]
- Stefaniuk, P.; Onyszczuk, J.; Szymczyk, A.; Podhorecka, M. Therapeutic Options for Patients with TP53 Deficient Chronic Lymphocytic Leukemia: Narrative Review. Cancer Manag. Res. 2021, 13, 1459–1476. [Google Scholar] [CrossRef]
- Malcikova, J.; Pavlova, S.; Kunt Vonkova, B.; Radova, L.; Plevova, K.; Kotaskova, J.; Pal, K.; Dvorackova, B.; Zenatova, M.; Hynst, J.; et al. Low-burden TP53 mutations in CLL: Clinical impact and clonal evolution within the context of different treatment options. Blood 2021, 138, 2670–2685. [Google Scholar] [CrossRef]
- Bomben, R.; Rossi, F.M.; Vit, F.; Bittolo, T.; D’Agaro, T.; Zucchetto, A.; Tissino, E.; Pozzo, F.; Vendramini, E.; Degan, M.; et al. Mutations with Low Variant Allele Frequency Predict Short Survival in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2021, 27, 5566–5575. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Famà, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef]
- Soussi, T.; Baliakas, P. Landscape of TP53 Alterations in Chronic Lymphocytic Leukemia. Front. Oncol. 2022, 12, 808886. [Google Scholar] [CrossRef]
- Blakemore, S.J.; Clifford, R.; Parker, H.; Antoniou, P.; Stec-Dziedzic, E.; Larrayoz, M.; Davis, Z.; Kadalyayil, L.; Colins, A.; Robbe, P.; et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: Data from the randomised UK LRF CLL4 trial. Leukemia 2020, 34, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Stano-Kozubik, K.; Tichy, B.; Kantorova, B.; Pavlova, S.; Tom, N.; Radova, L.; Smardova, J.; Pardy, F.; Doubek, M.; et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015, 29, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carretero, C.; González-Gascón-Y-Marín, I.; Rodríguez-Vicente, A.E.; Quijada-Álamo, M.; Hernández-Rivas, J.; Hernández-Sánchez, M.; Hernández-Rivas, J.M. The Evolving Landscape of Chronic Lymphocytic Leukemia on Diagnosis, Prognosis and Treatment. Diagnostics 2021, 11, 853. [Google Scholar] [CrossRef] [PubMed]
- Agathangelidis, A.; Chatzidimitriou, A.; Chatzikonstantinou, T.; Tresoldi, C.; Davis, Z.; Giudicelli, V.; Kossida, S.; Belessi, C.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: The 2022 update of the recommendations by ERIC, the European Research Initiative on CLL. Leukemia 2022, 36, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Nadeu, F.; Colomer, D.; Campo, E. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205–2217. [Google Scholar] [CrossRef]
- Rammal, S.; Semaan, W.; Aprahamian, N.; Moussallem, R.; Chebly, A. Spotlight on borderline-IGHV mutational status in chronic lymphocytic leukemia. Front. Oncol. 2024, 14, 1430225. [Google Scholar] [CrossRef]
- Angotzi, F.; Cellini, A.; Ruocco, V.; Cavarretta, C.A.; Zatta, I.; Serafin, A.; Pravato, S.; Pagnin, E.; Bonaldi, L.; Frezzato, F.; et al. Chronic Lymphocytic Leukemia (CLL) with Borderline Immunoglobulin Heavy Chain Mutational Status, a Rare Subgroup of CLL with Variable Disease Course. Cancers 2024, 16, 1095. [Google Scholar] [CrossRef]
- Jaramillo, S.; Agathangelidis, A.; Schneider, C.; Bahlo, J.; Robrecht, S.; Tausch, E.; Bloehdorn, J.; Hoechstetter, M.; Fischer, K.; Eichhorst, B.; et al. Prognostic impact of prevalent chronic lymphocytic leukemia stereotyped subsets: Analysis within prospective clinical trials of the German CLL Study Group (GCLLSG). Haematologica 2020, 105, 2598–2607. [Google Scholar] [CrossRef]
- Stamatopoulos, B.; Timbs, A.; Bruce, D.; Smith, T.; Clifford, R.; Robbe, P.; Burns, A.; Vavoulis, D.V.; Lopez, L.; Antoniou, P.; et al. Targeted deep sequencing reveals clinically relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia 2017, 31, 837–845. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef]
- Lee, J.; Wang, Y.L. Prognostic and Predictive Molecular Biomarkers in Chronic Lymphocytic Leukemia. J. Mol. Diagn. 2020, 22, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Jelloul, F.Z.; Yang, R.K.; Wang, P.; Garces, S.; Kanagal-Shamanna, R.; Ok, C.Y.; Loghavi, S.; Routbort, M.J.; Zuo, Z.; Yin, C.C.; et al. Non-coding NOTCH1 mutations in chronic lymphocytic leukemia negatively impact prognosis. Am. J. Hematol. 2022, 97, E100–E102. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef]
- Pagliaro, L.; Cerretani, E.; Vento, F.; Montanaro, A.; Moron Dalla Tor, L.; Simoncini, E.; Giaimo, M.; Gherli, A.; Zamponi, R.; Tartaglione, I.; et al. CAD204520 Targets. Int. J. Mol. Sci. 2024, 25, 766. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Schnaiter, A.; Paschka, P.; Zenz, T.; Rossi, M.; Döhner, K.; Bühler, A.; Böttcher, S.; Ritgen, M.; Kneba, M.; et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: Results from the CLL8 trial. Blood 2014, 123, 3247–3254. [Google Scholar] [CrossRef]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef]
- Close, V.; Close, W.; Kugler, S.J.; Reichenzeller, M.; Yosifov, D.Y.; Bloehdorn, J.; Pan, L.; Tausch, E.; Westhoff, M.A.; Döhner, H.; et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood 2019, 133, 830–839. [Google Scholar] [CrossRef]
- Rossi, D. FBXW7 is a biologically validated cancer driver gene for CLL. Blood 2019, 133, 774–776. [Google Scholar] [CrossRef]
- Mansouri, L.; Thorvaldsdottir, B.; Sutton, L.A.; Karakatsoulis, G.; Meggendorfer, M.; Parker, H.; Nadeu, F.; Brieghel, C.; Laidou, S.; Moia, R.; et al. Different prognostic impact of recurrent gene mutations in chronic lymphocytic leukemia depending on IGHV gene somatic hypermutation status: A study by ERIC in HARMONY. Leukemia 2023, 37, 339–347. [Google Scholar] [CrossRef]
- Yin, S.; Gambe, R.G.; Sun, J.; Martinez, A.Z.; Cartun, Z.J.; Regis, F.F.D.; Wan, Y.; Fan, J.; Brooks, A.N.; Herman, S.E.M.; et al. A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion. Cancer Cell 2019, 35, 283–296.e285. [Google Scholar] [CrossRef]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.S.; Hing, Z.A.; Harrington, B.; Baumhardt, J.; Ozer, H.G.; Lehman, A.; Giacopelli, B.; Beaver, L.; Williams, K.; Skinner, J.N.; et al. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Austen, B.; Powell, J.E.; Alvi, A.; Edwards, I.; Hooper, L.; Starczynski, J.; Taylor, A.M.; Fegan, C.; Moss, P.; Stankovic, T. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 2005, 106, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Crassini, K.; Stevenson, W.S.; Mulligan, S.P.; Best, O.G. Molecular pathogenesis of chronic lymphocytic leukaemia. Br. J. Haematol. 2019, 186, 668–684. [Google Scholar] [CrossRef]
- Lampson, B.L.; Gupta, A.; Tyekucheva, S.; Mashima, K.; Petráčková, A.; Wang, Z.; Wojciechowska, N.; Shaughnessy, C.J.; Baker, P.O.; Fernandes, S.M.; et al. Rare Germline. J. Clin. Oncol. 2023, 41, 1116–1128. [Google Scholar] [CrossRef]
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martínez-Trillos, A.; Villamor, N.; Rodríguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef]
- Rasi, S.; Khiabanian, H.; Ciardullo, C.; Terzi-di-Bergamo, L.; Monti, S.; Spina, V.; Bruscaggin, A.; Cerri, M.; Deambrogi, C.; Martuscelli, L.; et al. Clinical impact of small subclones harboring NOTCH1, SF3B1 or BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2016, 101, e135–e138. [Google Scholar] [CrossRef]
- Bonato, A.; Chakraborty, S.; Bomben, R.; Canarutto, G.; Felician, G.; Martines, C.; Zucchetto, A.; Pozzo, F.; Vujovikj, M.; Polesel, J.; et al. NFKBIE mutations are selected by the tumor microenvironment and contribute to immune escape in chronic lymphocytic leukemia. Leukemia 2024, 38, 1511–1521. [Google Scholar] [CrossRef]
- Della-Valle, V.; Roos-Weil, D.; Scourzic, L.; Mouly, E.; Aid, Z.; Darwiche, W.; Lecluse, Y.; Damm, F.; Mémet, S.; Mercher, T.; et al. Nfkbie-deficiency leads to increased susceptibility to develop B-cell lymphoproliferative disorders in aged mice. Blood Cancer J. 2020, 10, 38. [Google Scholar] [CrossRef]
- Shuai, W.; Lin, P.; Strati, P.; Patel, K.P.; Routbort, M.J.; Hu, S.; Wei, P.; Khoury, J.D.; You, M.J.; Loghavi, S.; et al. Clinicopathological characterization of chronic lymphocytic leukemia with MYD88 mutations: L265P and non-L265P mutations are associated with different features. Blood Cancer J. 2020, 10, 86. [Google Scholar] [CrossRef]
- Martínez-Trillos, A.; Navarro, A.; Aymerich, M.; Delgado, J.; López-Guillermo, A.; Campo, E.; Villamor, N. Clinical impact of MYD88 mutations in chronic lymphocytic leukemia. Blood 2016, 127, 1611–1613. [Google Scholar] [CrossRef] [PubMed]
- Giménez, N.; Martínez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Leeksma, A.C.; Taylor, J.; Wu, B.; Gardner, J.R.; He, J.; Nahas, M.; Gonen, M.; Alemayehu, W.G.; Te Raa, D.; Walther, T.; et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia 2019, 33, 390–402. [Google Scholar] [CrossRef]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef]
- Woyach, J.A.; Perez Burbano, G.; Ruppert, A.S.; Miller, C.; Heerema, N.A.; Zhao, W.; Wall, A.; Ding, W.; Bartlett, N.L.; Brander, D.M.; et al. Follow-up from the A041202 study shows continued efficacy of ibrutinib regimens for older adults with CLL. Blood 2024, 143, 1616–1627. [Google Scholar] [CrossRef]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Novak, J.; Strugov, V.; Gill, D.; et al. First-line treatment of chronic lymphocytic leukemia with ibrutinib plus obinutuzumab. Haematologica 2022, 107, 2108–2120. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomised, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef]
- Tam, C.S.; Brown, J.R.; Kahl, B.S.; Ghia, P.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; et al. Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): A randomised, controlled, phase 3 trial. Lancet Oncol. 2022, 23, 1031–1043. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Zhang, C.; Jin, H.Y.; Robrecht, S.; Choi, Y.; Balasubramanian, S.; Kotak, A.; Chang, Y.M.; Fink, A.M.; Tausch, E.; et al. Transcriptomic profiles and 5-year results from the randomized CLL14 study of venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab in chronic lymphocytic leukemia. Nat. Commun. 2023, 14, 6724. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Niemann, C.U.; Kater, A.P.; Fürstenau, M.; von Tresckow, J.; Zhang, C.; Robrecht, S.; Gregor, M.; Juliusson, G.; Thornton, P.; et al. First-Line Venetoclax Combinations in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 388, 1739–1754. [Google Scholar] [CrossRef]
- Fürstenau, M.; Kater, A.P.; Robrecht, S.; von Tresckow, J.; Zhang, C.; Gregor, M.; Thornton, P.; Staber, P.B.; Tadmor, T.; Lindström, V.; et al. First-line venetoclax combinations versus chemoimmunotherapy in fit patients with chronic lymphocytic leukaemia (GAIA/CLL13): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2024, 25, 744–759. [Google Scholar] [CrossRef]
- Kater, A.P.; Owen, C.; Moreno, C.; Follows, G.; Munir, T.; Levin, M.D.; Benjamini, O.; Janssens, A.; Osterborg, A.; Robak, T.; et al. Fixed-Duration Ibrutinib-Venetoclax in Patients with Chronic Lymphocytic Leukemia and Comorbidities. NEJM Evid. 2022, 1, EVIDoa2200006. [Google Scholar] [CrossRef]
- Tam, C.S.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Jacobs, R.; Opat, S.; Barr, P.M.; Tedeschi, A.; Trentin, L.; Bannerji, R.; et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: Primary analysis of the CAPTIVATE FD cohort. Blood 2022, 139, 3278–3289. [Google Scholar] [CrossRef]
- Niemann, C.U.; Munir, T.; Moreno, C.; Owen, C.; Follows, G.A.; Benjamini, O.; Janssens, A.; Levin, M.D.; Robak, T.; Simkovic, M.; et al. Fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 1423–1433. [Google Scholar] [CrossRef]
- Ghia, P.; Allan, J.; Siddiqi, T.; Wierda, W.G.; Tam, C.; Moreno, C.; Tedeschi, A.; Szafer-Glusman, E.; Zhou, C.; Abbazio, C.; et al. P617: Fixed-duration (fd) ibrutinib + venetoclax for first-line treatment of chronic lymphocytic leukemia (cll)/small lymphocytic lymphoma (sll): 4-y follow-up from fd cohort of phase 2 captivate study. HemaSphere 2023, 7, e2822842. [Google Scholar] [CrossRef]
- Eichhorst, B.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Hallek, M.; Jerkeman, M.; Buske, C.; ESMO Guidelines Committee. ESMO Clinical Practice Guideline interim update on new targeted therapies in the first line and at relapse of chronic lymphocytic leukaemia. Ann. Oncol. 2024, 35, 762–768. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Ahn, I.E.; Tian, X.; Wiestner, A. Ibrutinib for Chronic Lymphocytic Leukemia with. N. Engl. J. Med. 2020, 383, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Sivina, M.; Kim, E.; Wierda, W.G.; Ferrajoli, A.; Jain, N.; Thompson, P.; Kantarjian, H.; Keating, M.; Burger, J.A. Ibrutinib induces durable remissions in treatment-naïve patients with CLL and 17p deletion and/or TP53 mutations. Blood 2021, 138, 2589–2592. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.N.; Shanafelt, T.; Wiestner, A.; Moreno, C.; O’Brien, S.M.; Li, J.; Krigsfeld, G.; Dean, J.P.; Ahn, I.E. Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukaemia in patients with TP53 aberrations: A pooled analysis from four clinical trials. Br. J. Haematol. 2022, 196, 947–953. [Google Scholar] [CrossRef]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef]
- Tam, C.S.; Robak, T.; Ghia, P.; Kahl, B.S.; Walker, P.; Janowski, W.; Simpson, D.; Shadman, M.; Ganly, P.S.; Laurenti, L.; et al. Zanubrutinib monotherapy for patients with treatment naïve chronic lymphocytic leukemia and 17p deletion. Haematologica 2021, 106, 2354–2363. [Google Scholar] [CrossRef]
- Allan, J.N.; Flinn, I.W.; Siddiqi, T.; Ghia, P.; Tam, C.S.; Kipps, T.J.; Barr, P.M.; Elinder Camburn, A.; Tedeschi, A.; Badoux, X.C.; et al. Outcomes in Patients with High-Risk Features after Fixed-Duration Ibrutinib plus Venetoclax: Phase II CAPTIVATE Study in First-Line Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2023, 29, 2593–2601. [Google Scholar] [CrossRef]
- Ghia, P.; Wierda, W.G.; Barr, P.M.; Kipps, T.J.; Siddiqi, T.; Allan, J.N.; Hunter, Z.; Zhou, C.; Szoke, A.; Dean, J.P.; et al. Relapse after First-Line Fixed Duration Ibrutinib + Venetoclax: High Response Rates to Ibrutinib Retreatment and Absence of BTK Mutations in Patients with Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL) with up to 5 Years of Follow-up in the Phase 2 Captivate Study. Blood 2023, 142, 633. [Google Scholar]
- Thompson, P.A.; Bazinet, A.; Wierda, W.G.; Tam, C.S.; O’Brien, S.M.; Saha, S.; Peterson, C.B.; Plunkett, W.; Keating, M.J. Sustained remissions in CLL after frontline FCR treatment with very-long-term follow-up. Blood 2023, 142, 1784–1788. [Google Scholar] [CrossRef]
- Fischer, K.; Bahlo, J.; Fink, A.M.; Goede, V.; Herling, C.D.; Cramer, P.; Langerbeins, P.; von Tresckow, J.; Engelke, A.; Maurer, C.; et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: Updated results of the CLL8 trial. Blood 2016, 127, 208–215. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Hanson, C.A.; Paietta, E.M.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Long-term outcomes for ibrutinib-rituximab and chemoimmunotherapy in CLL: Updated results of the E1912 trial. Blood 2022, 140, 112–120. [Google Scholar] [CrossRef]
- Hillmen, P.; Pitchford, A.; Bloor, A.; Broom, A.; Young, M.; Kennedy, B.; Walewska, R.; Furtado, M.; Preston, G.; Neilson, J.R.; et al. Ibrutinib and rituximab versus fludarabine, cyclophosphamide, and rituximab for patients with previously untreated chronic lymphocytic leukaemia (FLAIR): Interim analysis of a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Munir, T.; Cairns, D.A.; Bloor, A.; Allsup, D.; Cwynarski, K.; Pettitt, A.; Paneesha, S.; Fox, C.P.; Eyre, T.A.; Forconi, F.; et al. Chronic Lymphocytic Leukemia Therapy Guided by Measurable Residual Disease. N. Engl. J. Med. 2024, 390, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Opat, S.; Tedeschi, A.; Badoux, X.C.; Kuss, B.J.; Jackson, S.; Moreno, C.; et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J. Clin. Oncol. 2021, 39, 3853–3865. [Google Scholar] [CrossRef] [PubMed]
- Odetola, O.; Ma, S. Relapsed/Refractory Chronic Lymphocytic Leukemia (CLL). Curr. Hematol. Malig. Rep. 2023, 18, 130–143. [Google Scholar] [CrossRef]
- Condoluci, A.; Rossi, D. Biology and Treatment of Richter Transformation. Front. Oncol. 2022, 12, 829983. [Google Scholar] [CrossRef]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.F.; D’Rozario, J.; Owen, C.J.; Assouline, S.; Lamanna, N.; Robak, T.; de la Serna, J.; Jaeger, U.; et al. Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood 2022, 140, 839–850. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef]
- Kater, A.; Harrup, R.; Kipps, T.; Eichhorst, B.; Owen, C.J.; Assouline, S.; Lamanna, N.; Robak, T.; De La Serna, J.; Jaeger, U.; et al. S201 final 7-year follow up and retreatment substudy analysis of murano: Venetoclaxrituximab (venr)-treated patients with relapsed/refractory chronic lymphocytic leukemia (R/R CLL). HemaSphere 2023, 7, e492813f. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illés, A.; Kay, N.; et al. Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef]
- Rogers, K.A.; Thompson, P.A.; Allan, J.N.; Coleman, M.; Sharman, J.P.; Cheson, B.D.; Jones, D.; Izumi, R.; Frigault, M.M.; Quah, C.; et al. Phase II study of acalabrutinib in ibrutinib-intolerant patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica 2021, 106, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Eichhorst, B.; Hillmen, P.; Jurczak, W.; Kaźmierczak, M.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Zhou, K.; et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 388, 319–332. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Jones, J.A.; Coutre, S.E.; Mato, A.R.; Hillmen, P.; Tam, C.; Österborg, A.; Siddiqi, T.; Thirman, M.J.; Furman, R.R.; et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): A phase 2, open-label, multicentre study. Lancet Oncol. 2016, 17, 1409–1418. [Google Scholar] [CrossRef]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Šimkovič, M.; Kriachok, I.; Illés, Á.; de la Serna, J.; Dolan, S.; Campbell, P.; et al. Acalabrutinib Versus Investigator’s Choice in Relapsed/Refractory Chronic Lymphocytic Leukemia: Final ASCEND Trial Results. Hemasphere 2022, 6, e801. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Tausch, E.; Roberts, A.W.; Davids, M.S.; Eichhorst, B.; Hallek, M.; Hillmen, P.; Schneider, C.; Schetelig, J.; Böttcher, S.; et al. Six-year follow-up and subgroup analyses of a phase 2 trial of venetoclax for del(17p) chronic lymphocytic leukemia. Blood Adv. 2024, 8, 1992–2004. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Gill, S.; Hexner, E.O.; Schuster, S.; Nasta, S.; Loren, A.; Svoboda, J.; Stadtmauer, E.; Landsburg, D.J.; Mato, A.; et al. Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: First-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef]
- Nakhoda, S.; Vistarop, A.; Wang, Y.L. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 2023, 200, 137–149. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef]
- Quinquenel, A.; Fornecker, L.M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Lama, T.G.; Kyung, D.; O’Brien, S. Mechanisms of ibrutinib resistance in chronic lymphocytic leukemia and alternative treatment strategies. Expert. Rev. Hematol. 2020, 13, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Sedlarikova, L.; Petrackova, A.; Papajik, T.; Turcsanyi, P.; Kriegova, E. Resistance-Associated Mutations in Chronic Lymphocytic Leukemia Patients Treated With Novel Agents. Front. Oncol. 2020, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Galanina, N.; Guo, A.; Lee, J.; Kadri, S.; Van Slambrouck, C.; Long, B.; Wang, W.; Ming, M.; Furtado, L.V.; et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 2016, 7, 68833–68841. [Google Scholar] [CrossRef]
- Bonfiglio, S.; Sutton, L.A.; Ljungström, V.; Capasso, A.; Pandzic, T.; Weström, S.; Foroughi-Asl, H.; Skaftason, A.; Gellerbring, A.; Lyander, A.; et al. BTK and PLCG2 remain unmutated in one-third of patients with CLL relapsing on ibrutinib. Blood Adv. 2023, 7, 2794–2806. [Google Scholar] [CrossRef]
- Khalsa, J.K.; Cha, J.; Utro, F.; Naeem, A.; Murali, I.; Kuang, Y.; Vasquez, K.; Li, L.; Tyekucheva, S.; Fernandes, S.M.; et al. Genetic events associated with venetoclax resistance in CLL identified by whole-exome sequencing of patient samples. Blood 2023, 142, 421–433. [Google Scholar] [CrossRef]
- Jain, N.; Croner, L.J.; Allan, J.N.; Siddiqi, T.; Tedeschi, A.; Badoux, X.C.; Eckert, K.; Cheung, L.W.K.; Mukherjee, A.; Dean, J.P.; et al. Absence of BTK, BCL2, and PLCG2 Mutations in Chronic Lymphocytic Leukemia Relapsing after First-Line Treatment with Fixed-Duration Ibrutinib plus Venetoclax. Clin. Cancer Res. 2024, 30, 498–505. [Google Scholar] [CrossRef]
- Woyach, J.; Stephens, D.M.; Flinn, I.W.; Bhat, S.; Savage, R.E.; Chai, F.; Eathiraj, S.; Granlund, L.; Szuszkiewicz, L.A.; Schwartz, B.; et al. Final Results of Phase 1, Dose Escalation Study Evaluating ARQ 531 in Patients with Relapsed or Refractory B-Cell Lymphoid Malignancies. Blood 2019, 134, 4298. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.; Valeryevich, D.A.; Patel, M.R.; Tees, M.T.; Flinn, I.W.; Ai, W.Z.; Patel, K.; Wang, M.; O’Brien, S.M.; Nandakumar, S.; et al. A first-in-human phase 1 trial of NX-2127, a first-in-class oral BTK degrader with IMiD-like activity, in patients with relapsed and refractory B-cell malignancies. J. Clin. Oncol. 2022, 40, TPS7581. [Google Scholar] [CrossRef]
- Hillmen, P.; Rawstron, A.C.; Brock, K.; Muñoz-Vicente, S.; Yates, F.J.; Bishop, R.; Boucher, R.; MacDonald, D.; Fegan, C.; McCaig, A.; et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J. Clin. Oncol. 2019, 37, 2722–2729. [Google Scholar] [CrossRef]
- Rogers, K.A.; Huang, Y.; Ruppert, A.S.; Awan, F.T.; Heerema, N.A.; Hoffman, C.; Lozanski, G.; Maddocks, K.J.; Moran, M.E.; Reid, M.A.; et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood 2018, 132, 1568–1572. [Google Scholar] [CrossRef]
- Wierda, W.G.; Rawstron, A.; Cymbalista, F.; Badoux, X.; Rossi, D.; Brown, J.R.; Egle, A.; Abello, V.; Cervera Ceballos, E.; Herishanu, Y.; et al. Measurable residual disease in chronic lymphocytic leukemia: Expert review and consensus recommendations. Leukemia 2021, 35, 3059–3072. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Böttcher, S.; Letestu, R.; Villamor, N.; Fazi, C.; Kartsios, H.; de Tute, R.M.; Shingles, J.; Ritgen, M.; Moreno, C.; et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia 2013, 27, 142–149. [Google Scholar] [CrossRef]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: A multivariate analysis from the randomized GCLLSG CLL8 trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef]
- Romero, D. Benefit with MRD-guided treatment in CLL. Nat. Rev. Clin. Oncol. 2024, 21, 84. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Lopez, C.A.; Barrientos, J.C. MRD-directed therapy in CLL: Ready for prime time? Hematology Am Soc Hematol. Educ Program. 2023, 2023, 413–420. [Google Scholar] [CrossRef]
- Nadeu, F.; Clot, G.; Delgado, J.; Martín-García, D.; Baumann, T.; Salaverria, I.; Beà, S.; Pinyol, M.; Jares, P.; Navarro, A.; et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia 2018, 32, 645–653. [Google Scholar] [CrossRef]
- Fisher, A.; Goradia, H.; Martinez-Calle, N.; Patten, P.; Munir, T. The evolving use of measurable residual disease in chronic lymphocytic leukemia clinical trials. Front. Oncol. 2023, 13, 1130617. [Google Scholar] [CrossRef] [PubMed]
- Benintende, G.; Pozzo, F.; Innocenti, I.; Autore, F.; Fresa, A.; D’Arena, G.; Gattei, V.; Lurenti, L. Measurable residual disease in chronic lymphocytic leukemia. Front. Oncol. 2023, 13, 1112616. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Biomarker | Detection Method * | Clinical Significance in Prognosis |
|---|---|---|
| Unmutated IGHV | Sanger, NGS | Shorter TTFT and poorer response to CIT Mandatory in pre-treatment evaluation (stable status during disease course) |
| Del(17p)/TP53 mutation | FISH + Sanger, NGS | Resistance to CIT and rapid disease progression Mandatory in pre-treatment evaluation |
| Complex Karyotype | Conventional Karyotyping | Unfavorable outcome after CIT, independently of TP53 alterations; Controversial role after novel targeted agents |
| NOTCH1 mutation | Sanger, NGS | Worse outcome and poor response to rituximab treatment |
| FBXW7 mutation | Sanger, NGS | Poorer PFS and OS in patients with early-stage disease |
| SF3B1 mutation | Sanger, NGS | Poor prognosis |
| XPO1 mutation | Sanger, NGS | Inferior PFS and OS. Independent prognostic variables |
| Del(11q)/ATM mutation | FISH + Sanger, NGS | Shorter TTFT but better response to BTK inhibitors in the presence of del(11q) Germline mutations could be detected |
| POT1 mutation | Sanger, NGS | Poor OS Germline mutations could be detected |
| BIRC3 mutation | Sanger, NGS | Unfavorable prognosis, but not confirmed across literature; probable predictive role |
| NFKBIE mutation | Sanger, NGS | Reduced response to ibrutinib treatment |
| MYD88 mutation | Sanger, NGS | Good prognosis |
| RAS/RAF mutation | Sanger, NGS | BRAF: adverse OS NRAS/KRAS: no adverse OS |
| Trial | Arms | Phase | Total Participants | MRD As a Primary Outcome | MRD as a Secondary Outcome | MRD Stopping Rules | MRD Assessment |
|---|---|---|---|---|---|---|---|
| MURANO | BR vs. VR | 3 | 389 | No | YES | No | FC, ASO |
| CLARITY | IV single arm | 2 | 54 | YES | No | YES | FC |
| CLL14 | ClbO vs. VO | 3 | 432 | No | YES | No | FC, ASO, NGS |
| CAPTIVATE MRD | IV then I or placebo if U-MRD4 | 2 | 54 | No | YES for the IV vs. placebo | No | FC |
| CAPTIVATE FD | IV | 2 | 159 | No | YES | No | FC |
| GLOW | IV vs. ClbO | 3 | 211 | No | YES | No | NGS, FC |
| FLAIR | FCR, IR, I, IV | 3 | 771 | YES for IV vs. 1 or 1R | YES for FCR vs. IR | YES for the I arms | FC |
| CLL13 | CIT vs. VR vs. VO vs. VIO | 3 | 926 | YES | No | No | FC |
| GALACTIC | O consolidation | 2 | 48 | YES | No | No | FC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcari, A.; Morello, L.; Borotti, E.; Ronda, E.; Rossi, A.; Vallisa, D. Recent Advances in the Molecular Biology of Chronic Lymphocytic Leukemia: How to Define Prognosis and Guide Treatment. Cancers 2024, 16, 3483. https://doi.org/10.3390/cancers16203483
Arcari A, Morello L, Borotti E, Ronda E, Rossi A, Vallisa D. Recent Advances in the Molecular Biology of Chronic Lymphocytic Leukemia: How to Define Prognosis and Guide Treatment. Cancers. 2024; 16(20):3483. https://doi.org/10.3390/cancers16203483
Chicago/Turabian StyleArcari, Annalisa, Lucia Morello, Elena Borotti, Elena Ronda, Angela Rossi, and Daniele Vallisa. 2024. "Recent Advances in the Molecular Biology of Chronic Lymphocytic Leukemia: How to Define Prognosis and Guide Treatment" Cancers 16, no. 20: 3483. https://doi.org/10.3390/cancers16203483
APA StyleArcari, A., Morello, L., Borotti, E., Ronda, E., Rossi, A., & Vallisa, D. (2024). Recent Advances in the Molecular Biology of Chronic Lymphocytic Leukemia: How to Define Prognosis and Guide Treatment. Cancers, 16(20), 3483. https://doi.org/10.3390/cancers16203483