

TCGAnalyzeR: An Online Pan-Cancer Tool for Integrative Visualization of Molecular and Clinical Data of Cancer Patients for Cohort and Associated Gene Discovery


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Data
2.2. Pre-Computed Molecular Data Analysis
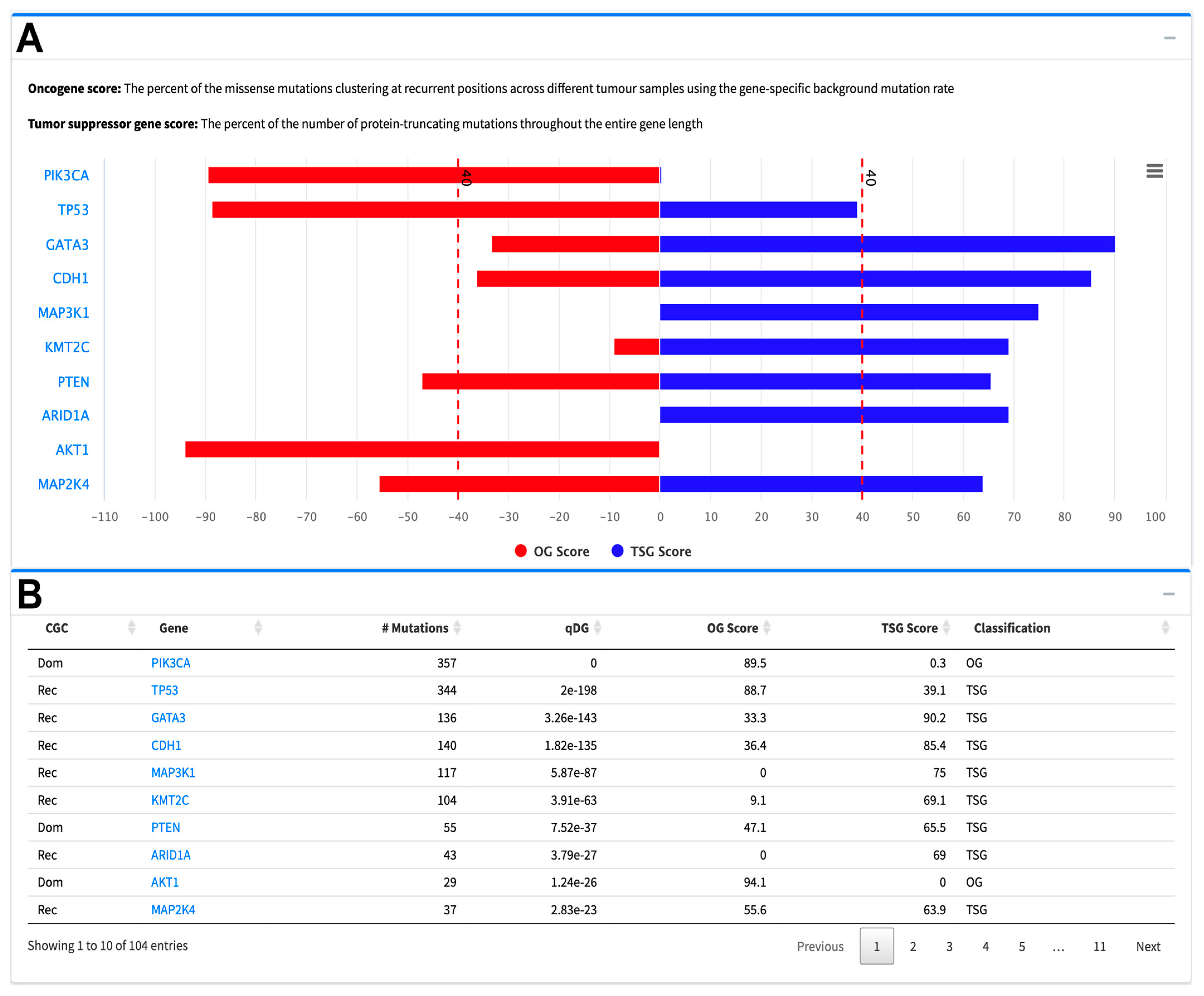
2.2.1. SNV Analysis
2.2.2. CNV Analysis
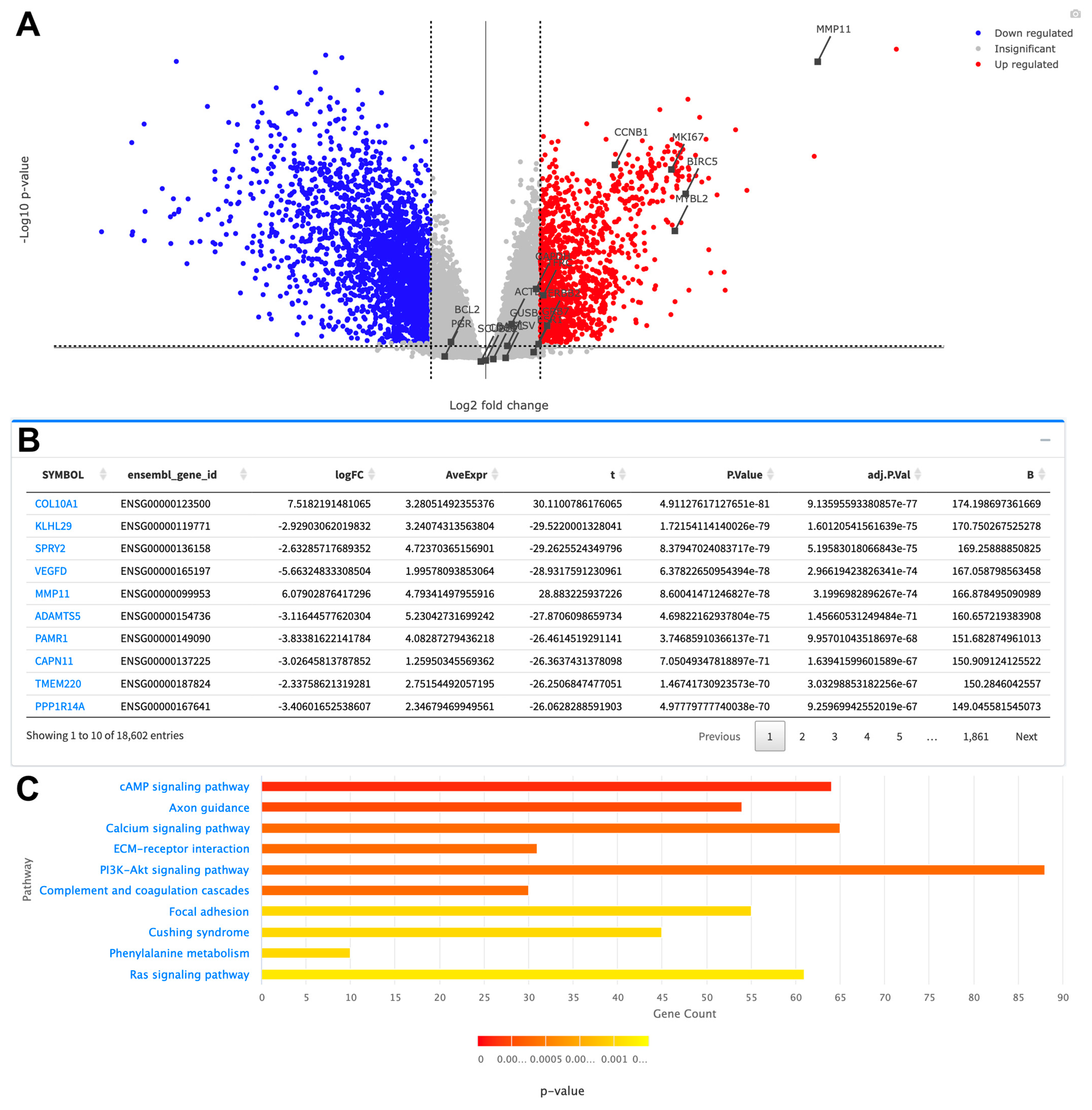
2.2.3. Gene Expression Analysis
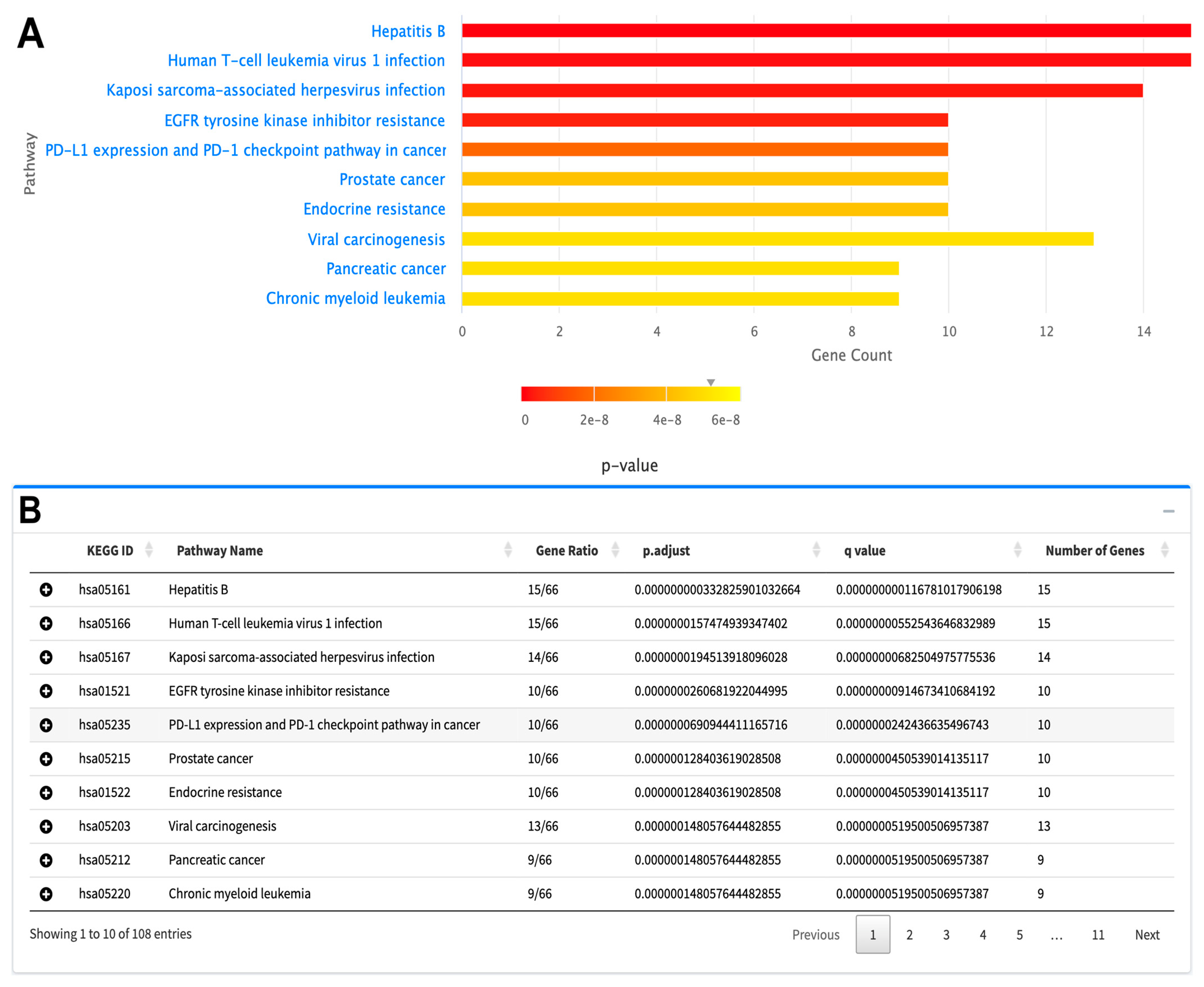
2.2.4. Pathway Enrichment
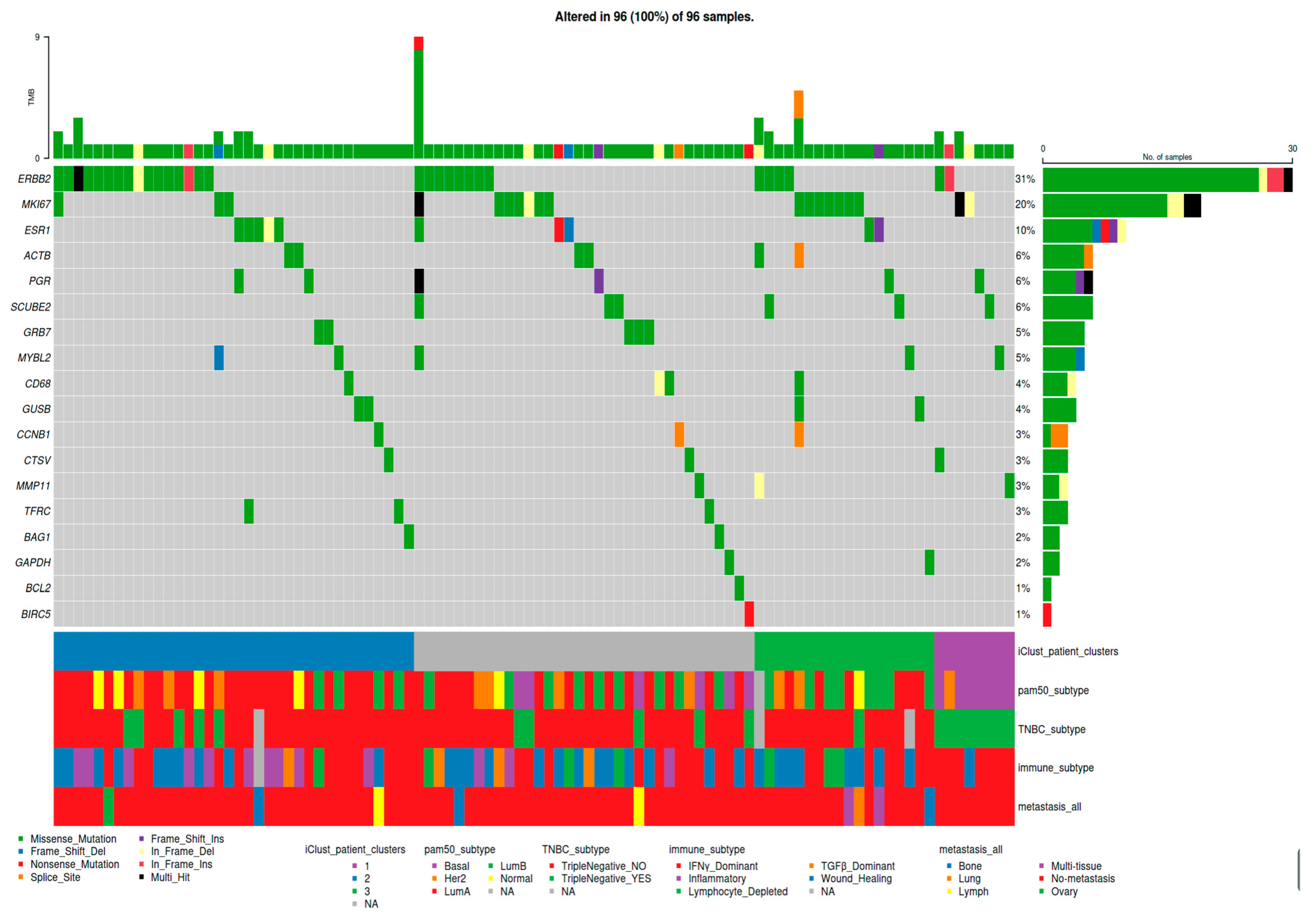
2.2.5. Pre-Computed Patient Clusters and Curated Subcohorts from the Literature
2.2.6. Survival Analysis
2.2.7. Visualization
2.2.8. Performance Optimization
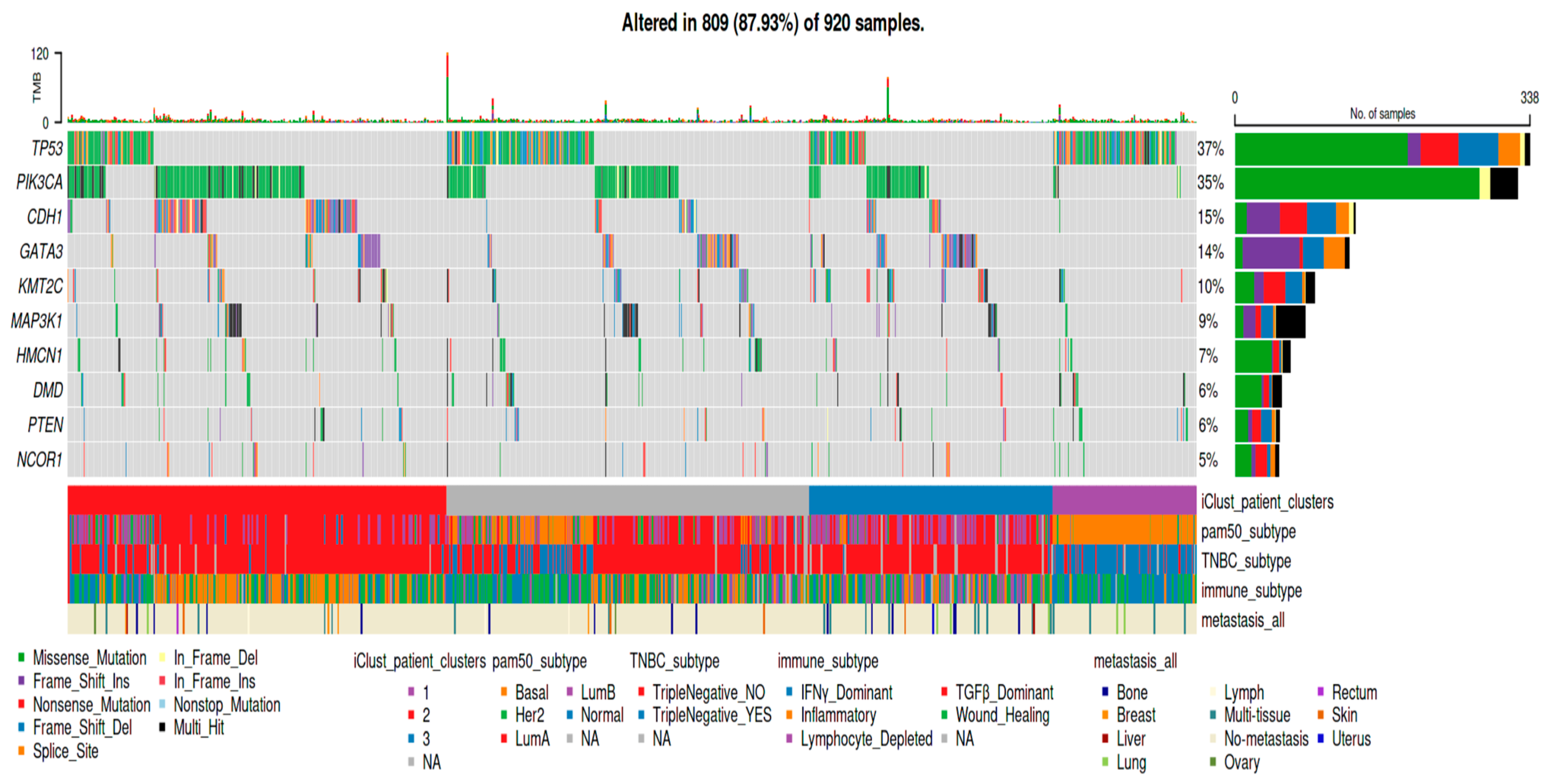
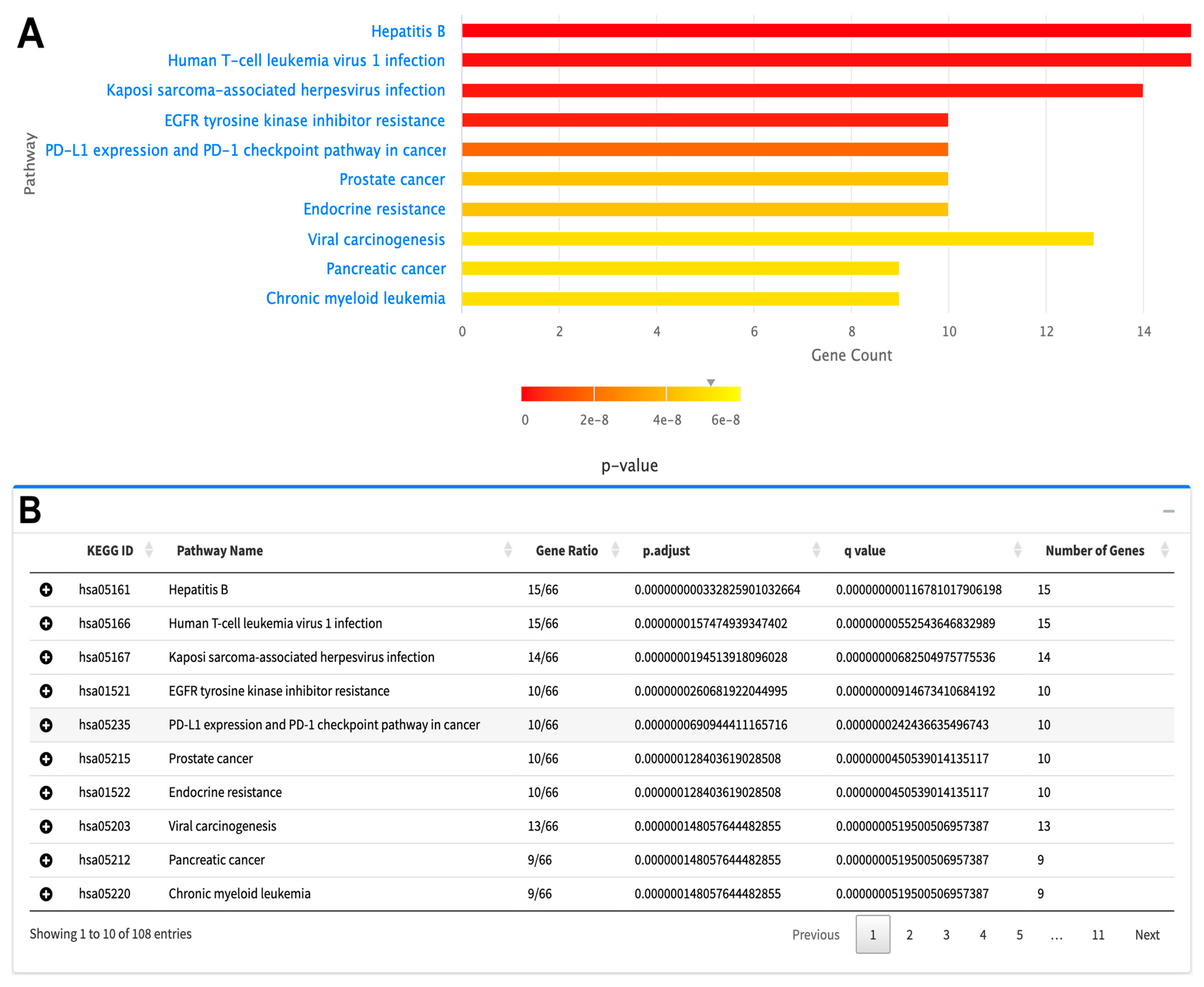
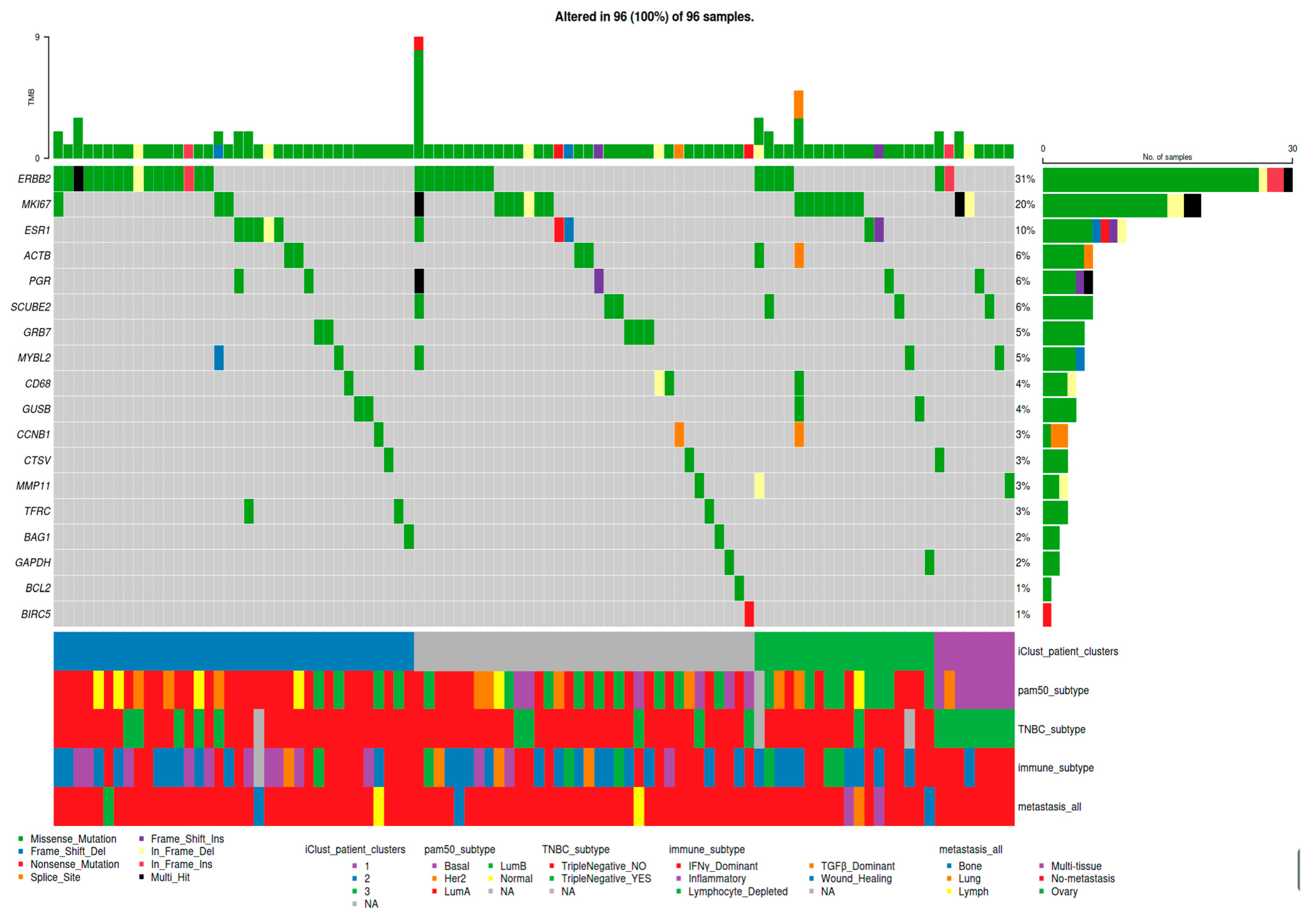
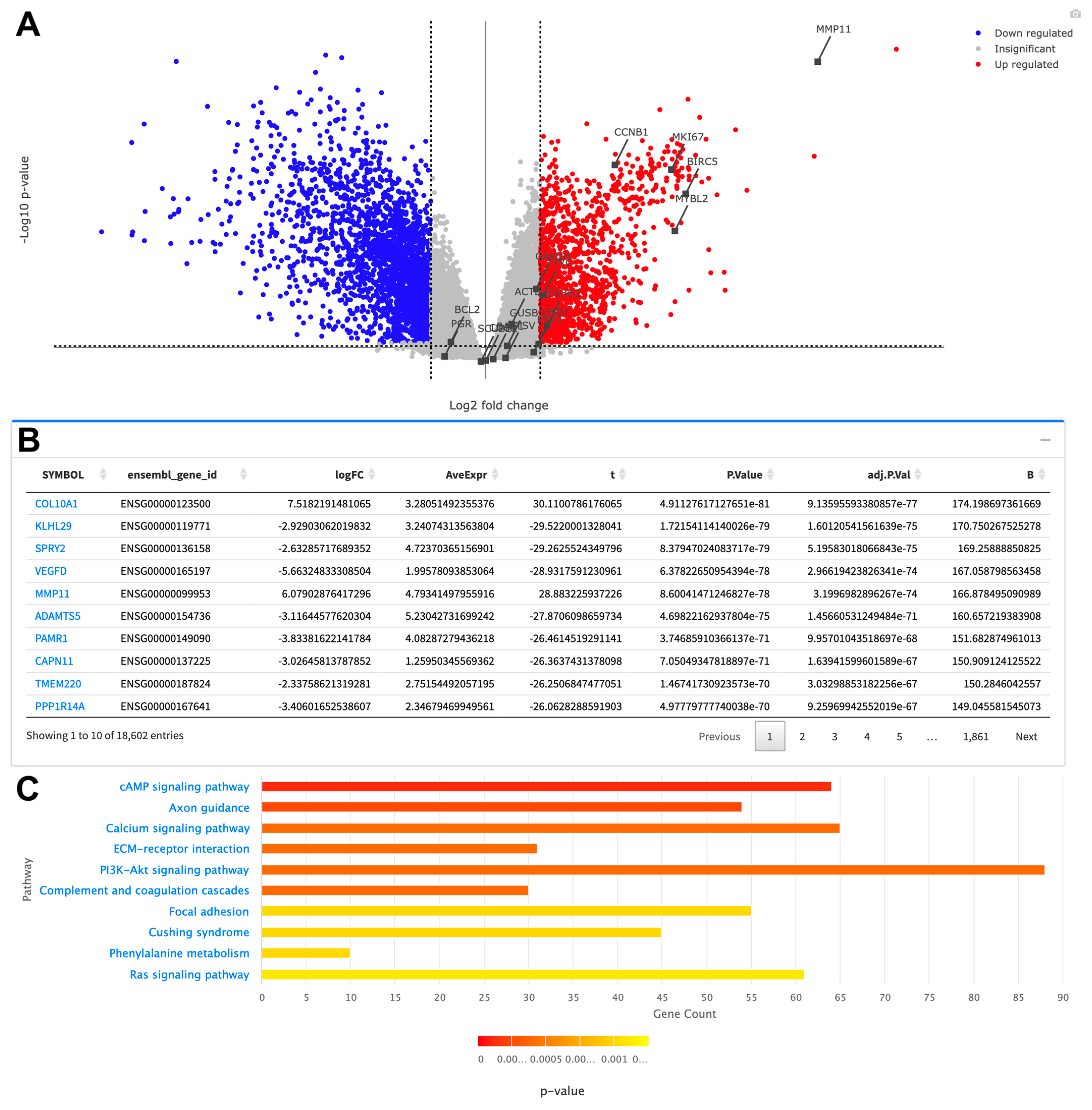
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sakaguchi, T.; Iketani, A.; Esumi, S.; Esumi, M.; Suzuki, Y.; Ito, K.; Fujiwara, K.; Nishii, Y.; Katsuta, K.; Yasui, H.; et al. Clinical Importance of the Range of Detectable Variants between the Oncomine Dx Target Test and a Conventional Single-Gene Test for EGFR Mutation. Sci. Rep. 2023, 13, 13759. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Brägelmann, J.; Kryukov, I.; Saraiva-Agostinho, N.; Perner, S. FirebrowseR: An R Client to the Broad Institute’s Firehose Pipeline. Database 2017, 2017, baw160. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal--a One-Stop Shop for Cancer Genomics Data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef]
- Adelberger, P.; Eckelt, K.; Bauer, M.J.; Streit, M.; Haslinger, C.; Zichner, T. Coral: A Web-Based Visual Analysis Tool for Creating and Characterizing Cohorts. Bioinformatics 2021, 37, 4559–4561. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Mo, Q.; Shen, R. iClusterPlus: Integrative Clustering of Multi-Type Genomic Data. 2022. Available online: https://bioconductor.org/packages/release/bioc/html/iClusterPlus.html (accessed on 28 December 2023).
- Zengin, T.; Önal-Süzek, T. Analysis of Genomic and Transcriptomic Variations as Prognostic Signature for Lung Adenocarcinoma. BMC Bioinform. 2020, 21, 368. [Google Scholar] [CrossRef] [PubMed]
- Zengin, T.; Önal-Süzek, T. Comprehensive Profiling of Genomic and Transcriptomic Differences between Risk Groups of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. J. Pers. Med. 2021, 11, 154. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Carey, V. BiocOncoTK 2018. Available online: https://www.bioconductor.org/packages/release/bioc/html/BiocOncoTK.html (accessed on 28 December 2023).
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e10. [Google Scholar] [CrossRef]
- Ramos, M.; Geistlinger, L.; Oh, S.; Schiffer, L.; Azhar, R.; Kodali, H.; de Bruijn, I.; Gao, J.; Carey, V.J.; Morgan, M.; et al. Multiomic Integration of Public Oncology Databases in Bioconductor. JCO Clin. Cancer Inform. 2020, 4, 958–971. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Van den Eynden, J.; Fierro, A.C.; Verbeke, L.P.; Marchal, K. SomInaClust: Detection of Cancer Genes Based on Somatic Mutation Patterns of Inactivation and Clustering. BMC Bioinform. 2015, 16, 125. [Google Scholar] [CrossRef]
- Morganella, S.; Pagnotta, S.M.; Ceccarelli, M. Finding Recurrent Copy Number Alterations Preserving Within-Sample Homogeneity. Bioinformatics 2011, 27, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLOS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mo, Q.; Shen, R.; Guo, C.; Vannucci, M.; Chan, K.S.; Hilsenbeck, S.G. A Fully Bayesian Latent Variable Model for Integrative Clustering Analysis of Multi-Type Omics Data. Biostatistics 2018, 19, 71–86. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in R 2022. Available online: https://cran.r-project.org/web/packages/survival/ (accessed on 28 December 2023).
- Wickham, H.; Hester, J.; Bryan, J. Readr: Read Rectangular Text Data 2022. Available online: https://cran.r-project.org/web/packages/readr/ (accessed on 28 December 2023).
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R Package for Creating Interactive Cluster Heatmaps for Online Publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, B.; Zeng, W.; Zhao, S.; Ge, D. G3viz: An R Package to Interactively Visualize Genetic Mutation Data Using a Lollipop-Diagram. Bioinformatics 2019, 36, 928–929. [Google Scholar] [CrossRef]
- Kunst, J. Highcharter: A Wrapper for the “Highcharts” Library 2022. Available online: https://cran.r-project.org/web/packages/highcharter/ (accessed on 28 December 2023).
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, S.; Alsaleem, M.; Monteiro, C.J.; Bhardwaj, K.; Joosten, S.E.P.; Fujii, T.; Shirabe, K.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; et al. Targetable ERBB2 Mutation Status Is an Independent Marker of Adverse Prognosis in Estrogen Receptor Positive, ERBB2 Non-Amplified Primary Lobular Breast Carcinoma: A Retrospective in Silico Analysis of Public Datasets. Breast Cancer Res. 2020, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, A.I.; Toss, M.S.; Green, A.R.; Rakha, E.A. The Clinical Significance of Cyclin B1 (CCNB1) in Invasive Breast Cancer with Emphasis on Its Contribution to Lymphovascular Invasion Development. Breast Cancer Res. Treat. 2023, 198, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]

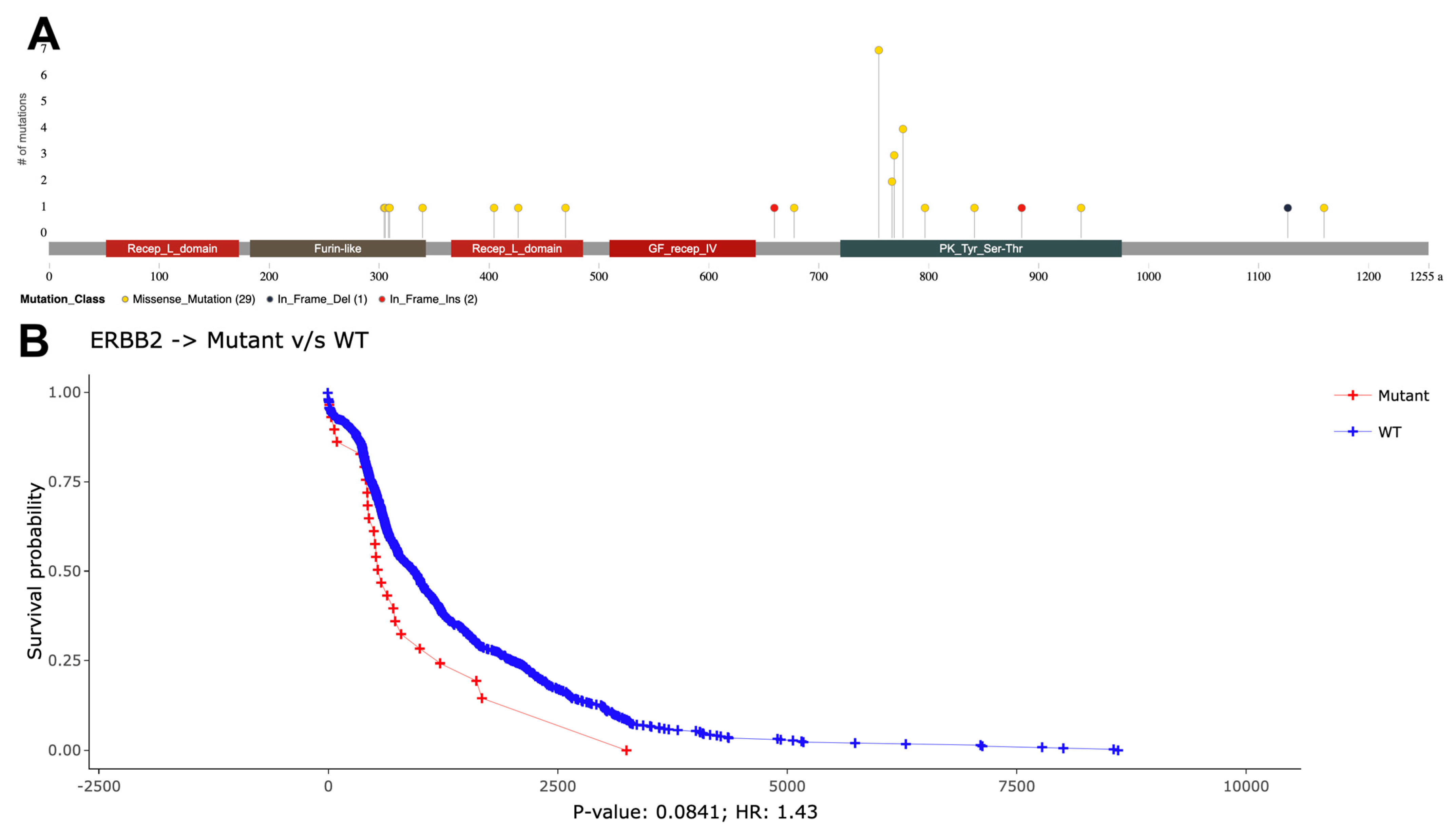

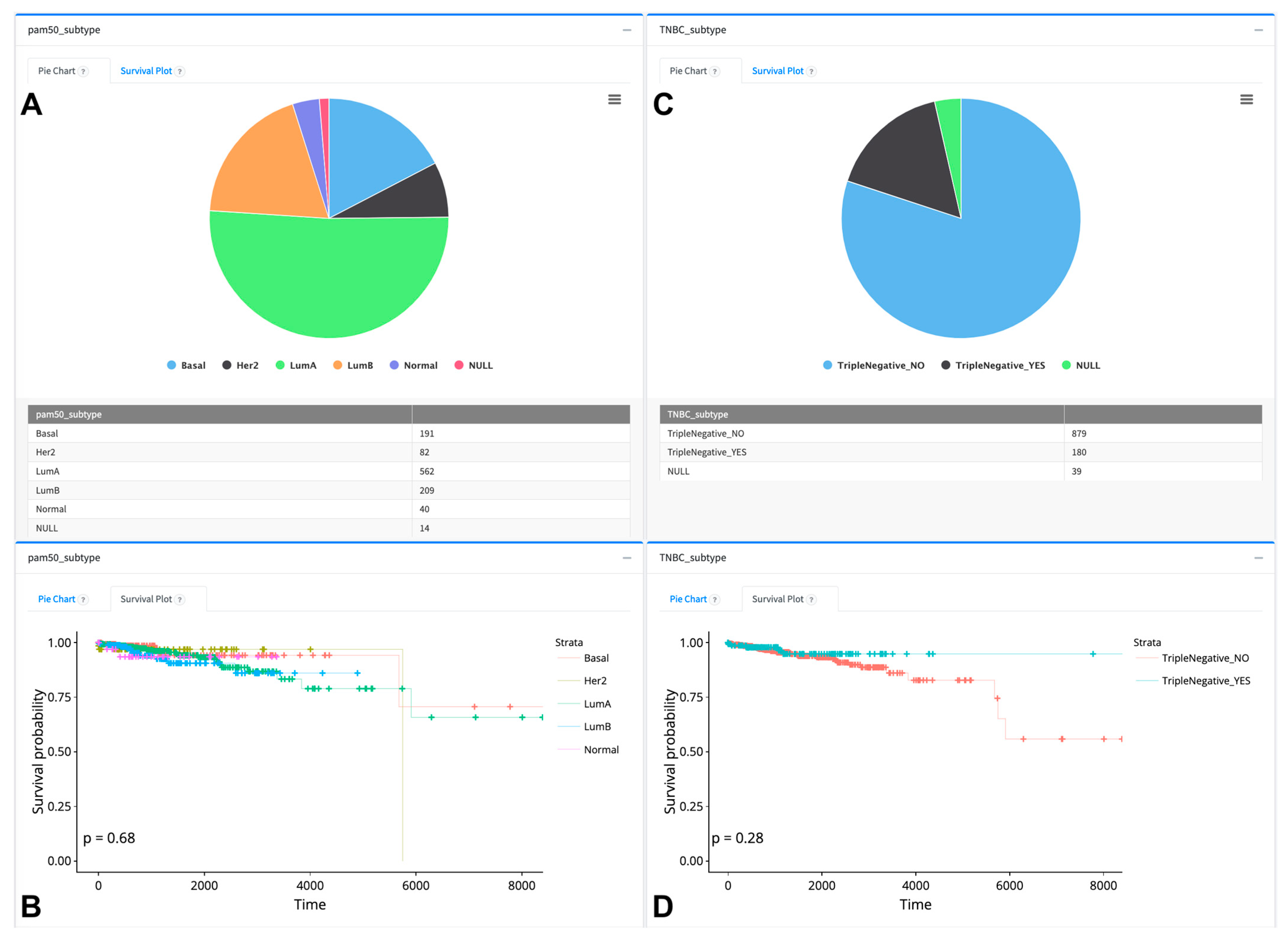
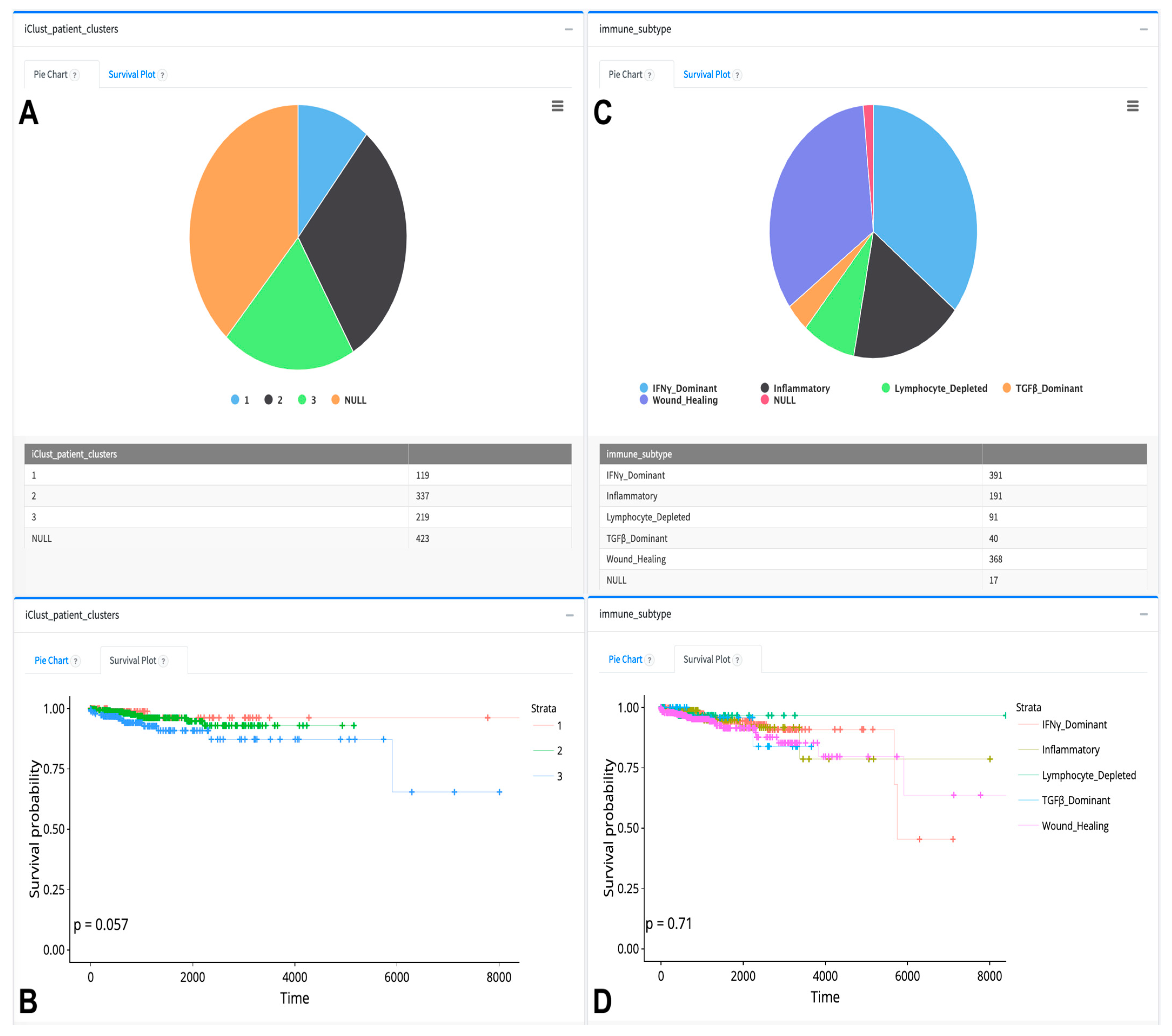


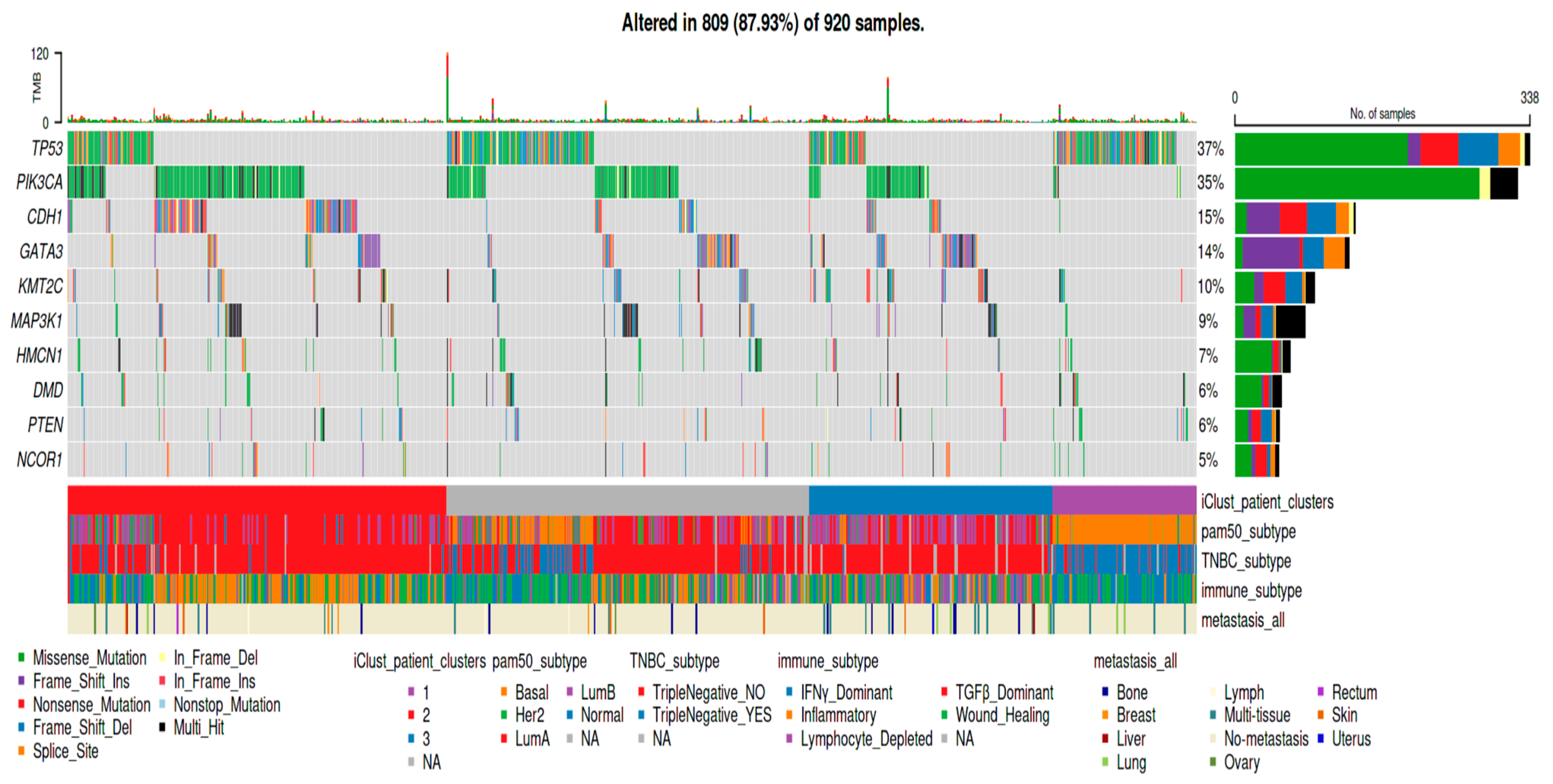{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
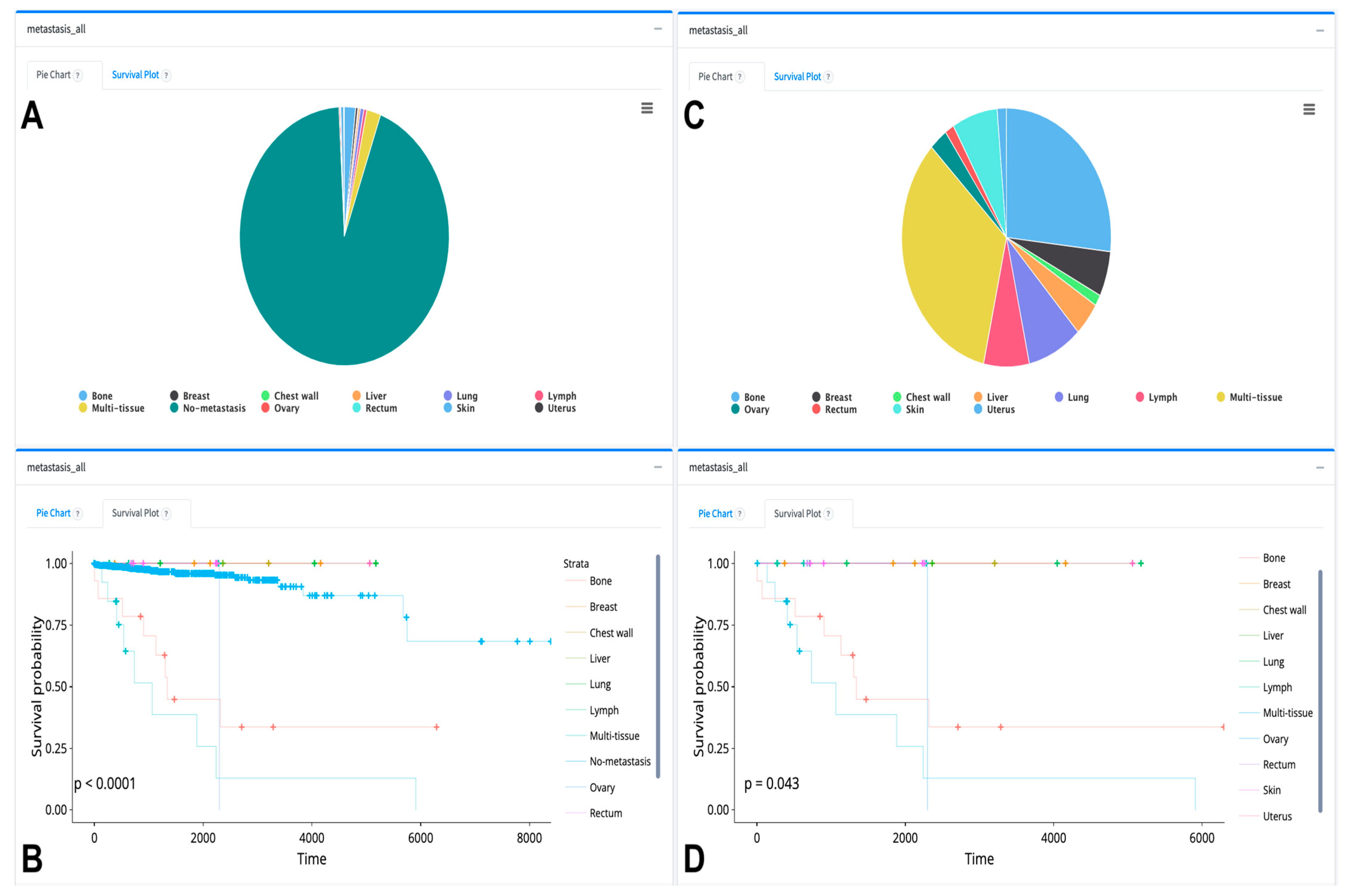{kind=link}
| TCGA Visualization Tool | Login or Subscription Required for Advanced Features | Survival and Set Operation Diagrams of Selected Patient Subcohorts | Caching of Both User’s Gene and Patient Selection | Number of External TCGA Cohorts from Literature |
|---|---|---|---|---|
| TCGAnalyzeR http://tcganalyzer.mu.edu.tr (accessed on 28 December 2023) | No | Yes | Yes | 123 |
| cBioPortal [5,6] https://www.cbioportal.org/ (accessed on 28 December 2023) | Yes | No | Yes | 0 |
| Xena [8] https://xenabrowser.net/ (accessed on 28 December 2023) | Yes | Yes | No | 0 |
| OncoKB [9] https://www.oncokb.org/ (accessed on 28 December 2023) | Yes | No | No-only genes | 0 |
| ICGC cohort analysis [10] https://dcc.icgc.org/analysis (accessed on 28 December 2023) | No | Yes | Yes | 0 |
| Coral web application [11] https://coral.caleydoapp.org (accessed on 28 December 2023) | Yes | No | No | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zengin, T.; Masud, B.A.; Önal-Süzek, T. TCGAnalyzeR: An Online Pan-Cancer Tool for Integrative Visualization of Molecular and Clinical Data of Cancer Patients for Cohort and Associated Gene Discovery. Cancers 2024, 16, 345. https://doi.org/10.3390/cancers16020345
Zengin T, Masud BA, Önal-Süzek T. TCGAnalyzeR: An Online Pan-Cancer Tool for Integrative Visualization of Molecular and Clinical Data of Cancer Patients for Cohort and Associated Gene Discovery. Cancers. 2024; 16(2):345. https://doi.org/10.3390/cancers16020345
Chicago/Turabian StyleZengin, Talip, Başak Abak Masud, and Tuğba Önal-Süzek. 2024. "TCGAnalyzeR: An Online Pan-Cancer Tool for Integrative Visualization of Molecular and Clinical Data of Cancer Patients for Cohort and Associated Gene Discovery" Cancers 16, no. 2: 345. https://doi.org/10.3390/cancers16020345
APA StyleZengin, T., Masud, B. A., & Önal-Süzek, T. (2024). TCGAnalyzeR: An Online Pan-Cancer Tool for Integrative Visualization of Molecular and Clinical Data of Cancer Patients for Cohort and Associated Gene Discovery. Cancers, 16(2), 345. https://doi.org/10.3390/cancers16020345